

Abstract
Nitric oxide is critical to immunity, but its role in the development of the immune system is unknown. Here, we show that S-nitrosoglutathione reductase (GSNOR), a protein key to the control of protein S-nitrosylation, is important for the development of lymphocytes. Genetic deletion of GSNOR in mice results in significant decrease in both T and B lymphocytes in the periphery. In thymus, GSNOR deficiency causes excessive protein S-nitrosylation, increases apoptosis, and reduces the number of CD4 single-positive thymocytes. Lymphopenia andincrease in S-nitrosylation and apoptosis in GSNOR-deficient mice are largely abolished by genetic deletion of inducible nitric oxide synthase (iNOS). Furthermore, the protection of lymphocyte development by GSNOR is apparently intrinsic to hematopoietic cells. Thus GSNOR, likely through regulation of S-nitrosylation and apoptosis, physiologically plays aprotective role in the development of the immune system.
Introduction
NO, best established as a major mediator of the immune response, is increasingly recognized as an important regulator of the immune system as well (1). Inducible nitric oxide synthase has been detected in many cells of the immune system, although it is absent in most or all normal lymphocytes (1). During immune responses, activated T cells, via interferon-γ and perhaps other factors, induce expression of iNOS in immunosuppressive cells including macrophages (2, 3) and mesenchymal stem cells (4, 5). NO or other reactive nitrogen species from iNOS in these cells during immune responses inhibits proliferation of T cells (2-5). NO bioactivity from iNOS has also been shown to contribute to elimination of activated effector T cells in the contraction phase of immune responses, probably through increasing T cell apoptosis (6, 7). Constitutive expression of iNOS is detectable in thymus of immunologically naïve mice (8, 9), although development of thymocytes and peripheral lymphocytes appears to be normal in iNOS-deficient (iNOS−/−) mice (10, 11). Despite extensive study of NO in immunology, a role for NO important to the development of the immune system has not been shown.
NO bioactivity originating from NO synthases involves a number of reactive nitrogen species, including S-nitrosothiol (SNO) (12). S-nitrosylation, the covalent attachment of NO to cysteine sulfur that forms SNO, has emerged as a major mechanism through which NO modifies protein functions to exert control over a wide range of biological processes (13). S-nitrosoglutathione (GSNO), the main non-protein SNO in cells, exists in equilibrium with protein SNOs (14, 15). The ubiquitously expressed GSNO reductase (also known as alcohol dehydrogenase class III) degrades GSNO and, through the equilibrium between GSNO and S-nitrosylated proteins, regulates cellular levels of protein S-nitrosylation (14-18). Genetic studies with targeted deletion of GSNOR gene have established that NO bioactivity and function are controlled not only at the level of synthesis by NOS but also through enzymatic degradation by GSNOR (14-19). Studies of GSNOR-deficient (GSNOR−/−) mice show that endogenous SNOs in systemic inflammation can cause apoptosis of lymphocytes in thymus, spleen, and lymph nodes, indicating that endogenously formed SNOs may impair lymphocyte survival (15).
During our study of GSNOR−/− mice, we noticed that despite being reared in a specific-pathogen-free facility, the animals sometimes suffered lung infection by opportunistic pathogens, thus appearing to be immunodeficient. In the present study, we show that GSNOR deficiency, likely through SNO originating from iNOS, impairs lymphocyte development and causes lymphopenia in naïve GSNOR−/− mice.
Materials and Methods
Animals
GSNOR−/− mice (15), after backcrossing 10 times to C57BL/6, were bred with iNOS−/− and eNOS−/− mice (the Jackson Laboratory) to obtain GSNOR−/−iNOS−/− and GSNOR−/−eNOS−/− mice, respectively. Both GSNOR−/−iNOS−/− and GSNOR−/−eNOS−/− mice are fertile, although litter sizes of GSNOR−/−eNOS−/− mice are often smaller. B6.SJL-Ptprca (C57BL/6-Ly5.1) mice are obtained from the National Cancer Institute, and TCRα−/− (20) mice are from the Jackson Laboratory. All mice were maintained on normal mouse chow (5058 PicoLab Mouse Diet 20) in a specific-pathogen-free facility at the University of California, San Francisco (UCSF). Lung inflammation was analyzed by counting inflammatory cells in airway lining fluid (17). Lung infection by the opportunistic pathogen Pasteurella pneumotropica was detected through PCR analysis by UC Davis Comparative Pathology Laboratory. Rederivation of GSNOR−/− mice through in vitro fertilization and embryo transfer was carried out by Murine Cryopreservation and Recovery Laboratory of UC Davis. The experimental protocol was approved by the Institutional Animal Care and Use Committee of UCSF.
Flow Cytometry
Complete blood count (CBC) was performed by the UC Davis Comparative Pathology Laboratory. Single-cell suspensions were incubated with anti-CD16/CD32 blocking Ab (2.4G2) for 5 min at room temperature and then labeled with the following antibodies: APC-Cy7-conjugated anti-CD4, FITC-conjugated anti-CD8a, PerCP-Cy5.5-conjugated anti-CD11b, APC- or FITC-conjugated anti-CD19, APC-conjugated anti-CD44, PE-conjugated anti-CD62L, APC- or FITC-conjugated anti-GR1, PE-conjugated anti-Ly5.1, and PerCP-Cy5.5-conjugated anti-Ly5.2. Flow cytometry was carried out using a BD Biosciences LSR-II with live cells gated by DAPI exclusion and lymphocytes gated on size and granularity on the basis of forward and side scatter.
Mixed bone marrow chimeras
Bone marrow from GSNOR−/− mice (Ly5.2) was mixed at a 1:1 ratio with bone marrow from GSNOR-wildtype C57BL/6-Ly5.1 mice, and 2×106 cells were injected into lethally irradiated (2 × 600 rad) GSNOR−/− or wild-type (Ly5.2) mice. Chimeras were provided with 2 g/L neomycin sulfate and 100 mg/L polymyxin B in drinking water. Mice were analyzed at 16-20 weeks after reconstitution.
Competitive reconstitution of CD4 cells
CD4 cells purified from spleen and mesenteric lymph node of GSNOR−/− mice (Ly5.2) were mixed at 1:1 ratio with those purified from C57BL/6-Ly5.1 mice and injected intravenously into TCRα−/− mice (5 × 106 cells per recipient). Mice were analyzed at 4 and 9 days after adoptive transfer.
TUNEL assay
Frozen tissue sections were prepared and assayed with In Situ Cell Death Detection Kit (Fluorescein) from Roche Applied Science, following the manufacturer’s protocol.
Selective enrichment and mass spectrometric identification of S-nitrosylated proteins
Thymuses from 3-4 mice were combined and homogenized in a lysis solution [250 mM HEPES, pH 7.7, 1 mM diethylenetriamine pentaacetic acid (DTPA), 0.1 mM neocuproine, 1% Triton X-100, and protease inhibitors] on ice. The homogenate was centrifuged at 13,000 x g for 30 min, at 4°C and the supernatant was collected. The amount of protein S-nitrosothiols in the thymus extract was measured with the tri-iodide method as described previously (21). An aliquot of the thymus extract, supplemented with 100 mM mannitol and 5 mM methyl methanethiosulfonate (MMTS), was illuminated with a conventional UV-transilluminator for 5 min on ice. Another aliquot of the extract was incubated with 1 mM ascorbate (Asc)/0.1mM CuSO4 for 30min at 37°C. UV photolysis and Cu/Asc treatment eliminate S-nitrosylation and were used to generate negative controls. Four mg of the UV-exposed, Cu/Asc-treated, or untreated protein extracts were precipitated with 3 volumes of acetone at −20°C for 20 min, pelleted by centrifugation, and washed twice with 1 mL of −20°C acetone. The proteins were solubilized in blocking solution (250 mM HEPES, pH 7.7, containing 1 mM DTPA, 0.1 mM neocuproine, 2.5% SDS, and 20 mM MMTS) and incubated at 50°C for 30 min, with vortexing every 4-5 min to block free thiols. After excess MMTS was removed by repeated (2x) acetone precipitation, the blocked proteins were resuspended in 250 mM MES, 1 mM DTPA, pH 6.0, and 1% SDS. The proteins were loaded onto columns containing organomercury-resin (MRC), which was generated by conjugation of ρ-amino-phenylmercuric acetate to N-hydroxysuccinimide-activated Affi-Gel 10 agarose beads and was activated with 0.1 M NaHCO3 (pH 8.8). While S-nitrosylated proteins bound to MRC resin covalently through the SNO-mercury reaction (22), unbound proteins were removed by extensive washing with 50 mM Tris-HCl, pH 7.4, 300 mM NaCl, 0.05 % SDS, and 0.1 % Triton X-100. The bound proteins were eluted with 50 mM β-mercaptoethanol in TBS buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaCl) and then subjected to GeLC-MS/MS analysis (21). Alternatively, to identify the specific site(s) of modification, the bound proteins were digested in column (1 μg of trypsin in 0.1M ammonium bicarbonate, pH 7.4) in the dark at room temperature for 16h. After extensive wash with ammonium bicarbonate, mild performic acid treatment (3% in water at room temperature for 30 min) was used to release the bound peptides and oxidize cysteine thiols to sulfonic acid (23). Under the workflow employed, 100% of cysteine-containing peptides were detected with the sulfonic acid modification. The recovered peptides were analyzed by LC-MS/MS (21). Evaluation of SEQUEST peptide sequence assignments and protein identification were as described previously (21). Sequence-to-spectrum assignments from two biological replicates were combined in SCAFFOLD (21). The false identification rate (peptides identified in the negative controls) was <3%.
Statistical analysis
The data were analyzed with the two-tailed Student’s t-test.
Results
Susceptibility to lung infection by opportunistic pathogens in GSNOR−/− mice
When first generated and studied, GSNOR−/− mice were free from lung infection and inflammation (17). However, lung inflammation, indicative of infection, was recently detected in unchallenged GSNOR−/− mice that were reared in a specific-pathogen-free facility (Supplemental Fig. 1). We also detected lung infection by the opportunistic pathogen Pasteurella pneumotropica in one of the three GSNOR−/− mice studied. Lung inflammation was not detectable in GSNOR−/− mice after rederivation to get rid of opportunistic pathogens. Thus GSNOR−/− mice appear to be immunodeficient.
Lymphopenia in GSNOR−/− mice
Although total counts of blood leukocytes, as with erythrocytes and platelets, are comparable between GSNOR−/− and wildtype mice (Fig. 1A) (15), flow cytometric analyses showed significantly fewer CD4 and CD8 T cells in the blood of GSNOR−/− mice than in wildtype controls (Fig. 1B and 1C). Decrease in T cells, particularly CD4 cells, was also observed in the spleen of GSNOR−/− mice (Fig. 2C). While B cell counts were slightly higher in the blood of GSNOR−/− mice than in wildtype controls (Fig. 1), B cells were significantly fewer in the spleen of GSNOR−/− mice (Fig. 2C), suggesting overall decrease of peripheral B cells in the knockout mice. Antibody production after immunization nevertheless appeared unimpaired in GSNOR−/− mice (Supplemental Fig. 2). In contrast to lymphocytes, myeloid cells including CD11b+ cells were not decreased in either blood or spleen of GSNOR−/− mice (Fig. 1 and 2). Thus, GSNOR deletion in mice results in lymphopenia.
FIGURE 1.

Decrease in T lymphocytes in blood of GSNOR−/− mice. A, Numbers of white blood cells (WBC), red blood cells (RBC), and platelets in wildtype (WT; open) and GSNOR−/− (KO; filled) mice. The data (mean ± SE) are from age-matched 3 WT and 4 KO mice. B, Percentage of CD4 (CD4+CD8−CD19−) T cells, CD8 (CD4−CD8+CD19−) T cells, CD19 (CD4−CD8−CD19+) B cells, and CD11b (CD11b+CD4−CD8−CD19−) cells in WBC of wildtype and GSNOR−/− mice. The data are mean (± SE) from age-matched (5-12 wk) WT (n=10-14) and KO (n=13-17) mice. **, P < 0.01, WT vs. KO. C, Representative flow cytometric analysis of total viable WBC from wildtype and GSNOR−/− mice.
FIGURE 2.

Decrease in lymphocytes in the spleen of GSNOR−/− mice. Weight (A), total leukocyte counts (B), and CD4, CD8, CD19, and CD11b cell counts (C) in spleens of wildtype (WT) and GSNOR−/− (KO) mice. D, Flow cytometric analysis of gated CD4 T cells. The data are mean (± SE) from WT (n = 4-9) and KO (n = 4-11) mice. **, P < 0.01.
We employed antibodies to surface markers CD44 and CD62L to further analyze CD4 T cells, the population decreased most prominently in GSNOR−/− mice. Whereas the number of CD62L− CD44high CD4 cells, the memory or memory-like cells, was comparable between GSNOR−/− and wildtype mice, CD62L+ cells, in particular the CD62L+CD44low naive cells, were significantly fewer in the spleen of GSNOR−/− mice than in wildtype mice (Fig. 2D). Thus GSNOR deficiency appears to cause decrease in the population of naive CD4 T cells.
Deficiency of CD4 single-positive thymocytes in GSNOR−/− mice
We investigated whether lymphopenia in GSNOR−/− mice results from defective proliferation or abnormal death of the peripheral lymphocytes. Following adoptive transfer of equivalent numbers of GSNOR−/− and wildtype CD4 cells into T cell receptor α subunit-deficient (TCRα−/−) mice, competitive repopulation of the cells resulted in a 1:1 ratio of GSNOR−/− to wildtype CD4 cells in recipient mice (Fig. 3 and data not shown), indicating normal growth of the GSNOR−/− cells. In addition, data from a number of other analyses, including proliferation assay with carboxyfluorescein succinimidyl ester (CFSE) in vitro and terminal deoxynucleotidyl transferase mediated dUTP nick-end-labeling (TUNEL) assay of spleen and lymph node, revealed no defects in GSNOR−/− T cells (Supplemental Fig. 3, 4, and data not shown). Thus, lymphopenia in GSNOR−/− mice is unlikely to result from defects in peripheral lymphocytes.
Figure 3.
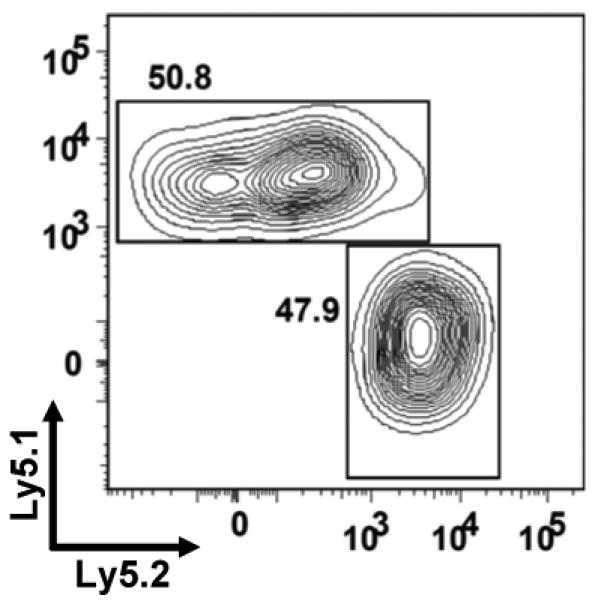
Competitive repopulation in TCRα−/− mice by peripheral CD4 T cells from GSNOR−/− and wildtype mice. CD4 cells purified from GSNOR−/− mice (Ly5.2) were mixed at 1:1 ratio with those purified from GSNOR-wildtype mice (Ly5.1) and injected intravenously into TCRα−/− mice (5×106 cells per recipient). Shown is representative analysis of CD4 T cells in spleen of a recipient 4 days after adoptive transfer. Similar results of 1:1 ratio of GSNOR−/− to wildtype CD4 cells in spleen, blood and lymph nodes were obtained at both 4 and 9 days after adoptive transfer in 2 independent experiments.
We analyzed thymocyte populations to determine whether lymphopenia in GSNOR−/− mice results from diminished production of lymphocytes from thymus. The total number of thymocytes in GSNOR−/− mice was modestly decreased (Fig 4A). Whereas the proportions of double-positive (DP) and CD8 single-positive (SP) thymocytes in GSNOR−/− mice were comparable to those of wildtype mice, the proportion of CD4 SP thymocytes was significantly lower in GSNOR−/− than in wildtype mice (Fig. 4). Accordingly DP and CD8 SP cell counts were affected modestly in GSNOR−/− mice, but the number of CD4 SP thymocytes was markedly decreased (Supplemental Fig. 5). Thus, the development of CD4 SP thymocytes is significantly affected by GSNOR deficiency.
FIGURE 4.

Decrease in CD4 single-positive cells in thymus of GSNOR−/− mice. A, Total thymocytes in wildtype, GSNOR−/−, and GSNOR−/−iNOS−/− (DKO) mice. WT vs. KO: P < 0.05. B, Representative flow cytometric analysis of thymocytes. C, Percentage of CD4 single-positive thymocytes. WT vs. KO: P < 0.001.
Lymphopenia in GSNOR−/− mice is rescued by genetic deletion of iNOS
To further establish the role of NO in disrupting lymphocyte development in GSNOR−/− mice and to identify the source of NO, we analyzed lymphocytes in mice deficient in both GSNOR and iNOS or endothelial NOS (eNOS). While lymphopenia was not rescued in GSNOR−/−eNOS−/− mice (data not shown), deletion of iNOS in GSNOR−/− mice led to significant recovery of populations of CD4 SP cells in thymus (Fig. 4), CD4 T cells in blood (Fig. 5A) and spleen (Fig. 5B), and B cells in spleen (Fig. 5B). Thus, lymphocyte development is apparently disrupted in GSNOR−/− mice by SNO originating from iNOS.
FIGURE 5.

Deficiency of lymphocytes in GSNOR−/− background is largely rescued by additional deletion of the iNOS Gene. Peripheral blood (A) and spleen (B) from 4-8 GSNOR−/−iNOS−/− mice. *, P < 0.05, and **, P < 0.01, DKO vs. KO.
Increase in apoptosis in thymus of GSNOR−/− mice
We employed the TUNEL assay to analyze apoptosis in thymuses of GSNOR−/− and control mice. The level of apoptosis in GSNOR−/− thymus was significantly higher than that in wildtype controls (Fig. 6), indicating that the reduction of thymocytes may result from elevated apoptosis in GSNOR−/− mice. Furthermore, the increase in apoptosis in GSNOR−/− thymus was completely abolished in GSNOR−/−iNOS−/− mice. Thus elevated apoptosis in GSNOR−/− thymus is likely caused by iNOS-derived SNO, which is abolished by GSNOR in wildtype mice.
FIGURE 6.
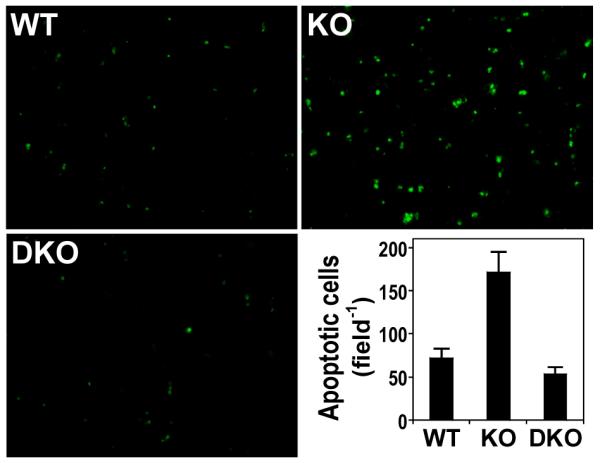
Increase in apoptosis in thymus of GSNOR−/− mice. Apoptotic cells in wildtype (n=6), GSNOR−/− (n=6), and GSNOR−/−iNOS−/− (n=3) mice were detected by TUNEL assay. The number of apoptotic cells in GSNOR−/− mice is significantly higher than in wildtype (P < 0.01) and GSNOR−/−iNOS−/− (P < 0.01) mice.
Protein S-nitrosylation is increased in thymus of GSNOR−/− mice
Quantitative analysis indicated a significant increase in the level of protein SNOs in the thymus of GSNOR−/− mice as compared to wildtype or GSNOR−/−iNOS−/− mice (Supplemental Fig. 6A). The level of protein SNOs in the GSNOR−/−iNOS−/− thymus was almost 90% lower than that in GSNOR−/− thymus, indicating the necessity of iNOS-derived NO for the formation of protein SNOs. To identify endogenously S-nitrosylated proteins in thymus, we applied a novel proteomic approach, which employs the direct reaction of SNOs with mercury to form a thiol-mercury bond (22). In this assay, S-nitrosylated proteins are captured onto a column of organomercury resin (MRC) through the SNO-mercury reaction, released by β-mercaptoethanol, and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). Captured proteins were also subjected to in-column trypsin digestion to isolate the SNO peptides, and then analyzed by LC-MS/MS to identify the site(s) of S-nitrosylation. UV photolysis and copper-ascorbate treatment, which abolish S-nitrosylation, were included in control experiments to avoid false identification of SNOs. With this method we identified 96 sites of cysteine S-nitrosylation in 83 proteins in wildtype thymus and 160 sites in 132 proteins in GSNOR−/− thymus (Supplemental Table I and II). Thus the number of S-nitrosylated targets increased under GSNOR deficiency. Importantly, only 11 sites of S-nitrosylation in 10 proteins were detected in GSNOR−/−iNOS−/− thymus (Supplemental Table III) demonstrating that the vast majority of the in vivo S-nitrosoproteome in the GSNOR−/− thymus was dependent on iNOS-derived NO. The S-nitrosylated proteins in thymus included a few proteins important to apoptosis. Caspase-6, a mediator of apoptosis (24), was S-nitrosylated at 2 cysteine residues (cys146, cys 271) in GSNOR−/− thymus, but S-nitrosylated caspase-6 was not detected in wildtype or GSNOR−/−iNOS−/− thymus (Supplemental Table I-III, Supplemental Fig. 6B and 6C). Similarly to caspase-6, S-nitrosylation of voltage-dependent anion channel-1, a protein implicated in the control of mitochondria-mediated apoptosis (25), was detected only in GSNOR−/− thymus (Supplemental Table I-III). In addition, S-nitrosylated GAPDH was detected in wildtype, GSNOR−/−, and GSNOR−/−iNOS−/− thymus (Fig. 7 and Supplemental Table I-III). However, the intensity of the ion derived from S-nitrosylated GAPDH was much higher in GSNOR−/− than in wildtype and GSNOR−/−iNOS−/− thymus (Fig. 7A). Taken together our data suggest that in thymus GSNOR is important for preventing excessive protein S-nitrosylation from iNOS activity.
FIGURE 7.

S-nitrosylation of GAPDH is greatly increased in thymus of GSNOR−/− mice. A, Extracted ion chromatographs of a peptide at m/z, from wildtype, GSNOR−/−, and GSNOR−/− iNOS−/− thymuses. The m/z value corresponds to the expected m/z value for the doubly charged SNO peptide VPTPNVSVVDLTCR from GAPDH. B, Establishment of the sequence and sulfonation site of the peptide in (A) from GSNOR−/− thymus by tandem mass spectrometry. Xcorr: SEQUEST cross correlation score at ppb mass error. The modified GAPDH peptide is undetectable when the samples are first exposed to UV or treated with copper-ascorbate to destroy SNO. The data are representative of 2 independent experiments, each of which contains thymuses from 3-4 mice.
Lymphopenia in GSNOR−/− mice is rescued by wildtype bone marrow
When bone marrow of irradiated GSNOR−/− mouse was reconstituted with mixed bone marrow cells from wildtype and GSNOR−/− mice, T cell deficiency in thymus and peripheral tissues was rescued (Fig. 8). Thus, the function of GSNOR in hemopoeitic cells is apparently sufficient for normal development of T cells, and GSNOR in non-hemopoeitic cells is not required.
FIGURE 8.

Lymphocyte population in GSNOR−/− mice is restored by wildtype bone marrow. Bone marrow cells of GSNOR-wildtype mice (Ly5.1) were mixed with the same number of bone marrow cells from GSNOR−/− mice (Ly5.2) and injected into lethally irradiated GSNOR−/− mice. A, Right, representative flow cytometric analysis of CD4 and CD8 T cells in blood of a GSNOR−/− mouse 16 weeks after bone marrow transfer; Left, Frequency of CD4 cells (mean + SE) in blood of wildtype, GSNOR−/−, and the bone marrow (BM)-reconstituted GSNOR−/− mice (KO + BM; n=7). P < 0.001, KO vs. KO + BM. B, Left, representative analysis of CD4 and Gr1 cells in blood of BM-reconstituted GSNOR−/− mice; Right, Frequency of cells (mean + SD) derived from GSNOR−/− bone marrow (KO BM) in blood (n=7) and lymph node (n=2-6) of mixed bone marrow chimeras. C, Deficiency of CD4 SP thymocytes is rescued by wildtype bone marrow. Shown is a representative of 2 independent experiments.
Following competitive reconstitution in GSNOR−/− mice with equal numbers of GSNOR−/− and wildtype bone marrow cells, equal numbers of Gr1+ granulocytes were derived from the two donor sources (Fig. 8B). Thus, bone marrow in recipient mice appeared to be reconstituted by wildtype and GSNOR−/− hemopoeitic stem cells equally well, and development of Gr1+ granulocytes from GSNOR−/− stem cells was comparable to that from wildtype stem cells. In contrast, both CD4 SP cells in thymus (Fig. 8C) and CD4 T cells in peripheral tissues (Fig. 8B and data not shown) of the reconstituted mice were derived virtually completely from wildtype mice, indicating that development of CD4 T cells is severely compromised by GSNOR deficiency. Surprisingly, DP and CD8 SP thymocytes and CD19+ B cells in the reconstituted mice were derived mostly from wildtype mice (Fig. 8 and data not shown). In addition, selective development of T and B cells from wildtype bone marrow was also observed when competitive reconstitution experiments were performed in wildtype recipient mice (data not shown). Thus, GSNOR deficiency in hemopoietic cells, at least in the competitive condition, significantly impaired development of B and T lymphocytes.
Discussion
The present study shows the unexpected result that normal development of lymphocytes, and hence of the adaptive immune system, depends on the function of GSNOR in the regulation of endogenous SNOs. We find that genetic deletion of GSNOR causes an increase in protein S-nitrosylation and cell apoptosis in thymus and widespread lymphopenia. The defects in the immune system are largely prevented by further deletion of iNOS, indicating iNOS as the origin of immunosuppressive SNOs. Further, our results suggest that lymphocyte development may depend on functional GSNOR in hemopoietic cells.
Our study suggests that GSONR deficiency causes apoptosis through excessive protein S-nitrosylation from iNOS in thymus of immunologically unchallenged mice. Expression of iNOS has been described in dendritic cells and other stromal cells in thymus of immunologically naïve mice (8, 9, 26). Thymic expression of iNOS may be increased by stimulation of TCR signaling with anti-CD3 antibody (8) or superantigen Staphylococcal enterotoxin B (9). Elevated iNOS activity can cause apoptosis of thymocytes (8, 27). We showed previously that thymocyte apoptosis caused by elevated iNOS activity in immune response was greatly increased by GSONR deficiency (15). We find in the present study that GSONR deficiency increases apoptosis from iNOS in thymus of immunologically naïve mice. In these animals GSONR deficiency also results in increase in thymic content of total protein SNOs, the abundance of S-nitrosylated proteins, and the level of S-nitrosylation of proteins. When excessive protein S-nitrosylation is abolished by iNOS deletion, increase of thymic apoptosis is prevented, further supporting a causative role of dysregulated S-nitrosylation in apoptosis. Proteins with elevated levels of S-nitrosylation in GSNOR−/− thymus include a few involved in the control of apoptosis.
While caspase-6 is an important execution caspase in apoptosis (24), voltage-dependent anion channel-1 is considered important for the control of mitochondria-mediated apoptosis (25). S-nitrosylation, which can modulate enzymatic activity, channel conductivity, and protein-protein interaction (13), might modulate the function of caspase-6 or voltage-dependent anion channel-1 in apoptosis. S-nitrosylation of GAPDH has been showed to promote nuclear accumulation of GAPDH and to cause apoptosis in macrophages and neurons (28). It is thus possible that the marked increase of GAPDH S-nitrosylation in GSNOR−/− thymus may promote apoptosis of thymocytes. Because apoptosis is a key mechanism for the control of the development and number of lymphocytes in thymus (29), increased apoptosis in thymus of GSNOR−/− mice may cause reduced thymic output of T cells and consequently lymphopenia in periphery.
T cell lymphopenia in GSNOR−/− mice may result largely from diminished thymic output. Reduction of CD4 and CD8 SP thymocytes in GSNOR−/− mice is associated with reduction of CD4 and CD8 T cells in periphery. In fact the decreases of CD4 and CD8 SP cells in thymus are quantitatively comparable to those of CD4 and CD8 cells respectively in secondary lymphoid organs. Diminished thymic output typically has little effect on the number of memory T cells but often significantly decreases the number of naive T cells (30, 31), in part because proliferation of naive T cells induced by lymphopenia converts them to memory-like T cells (32). Reduction of the naive T cell population with no change in the number of memory phenotype cells in GSNOR−/− mice thus provides further evidence suggesting reduced thymic output as the major cause of T cell lymphopenia. Alternatively T cell lymphopenia in GSNOR−/− mice might result from increase in lymphocyte death in periphery. However, apoptosis in peripheral lymphoid tissues appears unchanged from GSNOR deficiency. Nor is T cell lymphopenia likely to result from impaired proliferation of T cells in periphery, because lymphopenia-induced proliferation of T cells is unlikely to produce naive phenotype T cells (32), and because proliferative responses of T cells from GSNOR−/− mice are similar to those of wildtype control.
GSNOR appears to be particularly important to the development of CD4 SP thymocytes. Both CD4 and CD8 SP thymocytes develop from DP cells following positive selection. Because GSNOR deficiency causes no change in the proportions of DP and CD8 SP cells in thymocytes, but a significant reduction in the proportion of CD4 SP cells, GSNOR deficiency does not appear to affect the development of CD8 SP cells from DP cells or CD4-versus-CD8 lineage choice. Instead, GSNOR deficiency may impair the generation of CD4 SP cells from DP cells or the survival of CD4 SP lineage cells. A number of proteins important to CD4 SP lineage have been identified in thymocytes, although most of the proteins also play important roles in the development of DP or CD8 SP cells (33, 34). Conditional deletion of GATA-3 or c-Myb in DP thymocytes through CD4-Cre causes minimal changes in the percentage of DP and CD8 SP cells but significantly reduces the percentage of CD4 SP cells (35, 36). In addition, CD83 deficiency in thymic epithelial cells has little effect on the development of DP or CD8 SP thymocytes but significantly reduces the number of CD4 SP cells (37). It remains to be determined if deficiency of GSNOR and GATA-3, c-Myb, or CD83 inhibits a common step or pathway of CD4 SP development.
Our results, particularly those from bone marrow chimeras, suggest that GSNOR may be important to the development of both T and B cells. GSNOR−/− bone marrow stem cells in the competitive reconstitution experiment only gave rise to minimal DP, CD4 SP, and CD8 SP thymocytes, suggesting that GSNOR is important for T cell development not only at the stage of CD4 SP cells but also at an earlier stage. This is consistent with reduction of total thymocytes in GSNOR−/− mice. In addition, the marked deficiency of most T and B cells, but no deficiency of granulocytes, from GSNOR−/− bone marrow stem cells may suggest defective development of common lymphoid progenitors (38) of GSNOR−/− mice. The immunosuppressive effect could be a side effect of NO, or it might represent dysregulated NO signaling in lymphocyte progenitors. Alternatively, the inhibitory mechanisms in T and B lineage cells might be unrelated. We reported earlier that GSNOR, through its control of S-nitrosylation, is essential for the survival of lymphocytes in the inflammatory response (15). Our current findings demonstrate that GSNOR is also important for the development of lymphocytes, establishing a broad function for GSNOR in the immune system.
Supplementary Material
Acknowledgements
We would like to thank Drs. Yan Zhang, Yanan Zhu, and Cliff Mcarthur for technical assistance; Drs. Anthony DeFranco and Christopher Allen for helpful comments. The authors have no conflicting financial interests.
Footnotes
This work was supported by the Sandler Family Supporting Foundation (L.L and R.M.L), NIH CA122359 (L.L), NIH HL54926 (H.I), and HHMI (R.M.L).
- GSNO
- S-nitrosoglutathione
- GSNOR
- S-nitrosoglutathione reductase
- iNOS
- inducible nitric oxide synthase
- NO
- nitric oxide
- SNO
- S-nitrosothiol.
References
- 1.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 2.Albina JE, Abate JA, Henry WL., Jr. Nitric oxide production is required for murine resident peritoneal macrophages to suppress mitogen-stimulated T cell proliferation. Role of IFN-gamma in the induction of the nitric oxide-synthesizing pathway. J Immunol. 1991;147:144–148. [PubMed] [Google Scholar]
- 3.Mills CD. Molecular basis of “suppressor” macrophages. Arginine metabolism via the nitric oxide synthetase pathway. J Immunol. 1991;146:2719–2723. [PubMed] [Google Scholar]
- 4.Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, Muroi K, Ozawa K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood. 2007;109:228–234. doi: 10.1182/blood-2006-02-002246. [DOI] [PubMed] [Google Scholar]
- 5.Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, Zhao RC, Shi Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–150. doi: 10.1016/j.stem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 6.Dalton DK, Haynes L, Chu CQ, Swain SL, Wittmer S. Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J Exp Med. 2000;192:117–122. doi: 10.1084/jem.192.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vig M, Srivastava S, Kandpal U, Sade H, Lewis V, Sarin A, George A, Bal V, Durdik JM, Rath S. Inducible nitric oxide synthase in T cells regulates T cell death and immune memory. J Clin Invest. 2004;113:1734–1742. doi: 10.1172/JCI20225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tai XG, Toyo-oka K, Yamamoto N, Yashiro Y, Mu J, Hamaoka T, Fujiwara H. Expression of an inducible type of nitric oxide (NO) synthase in the thymus and involvement of NO in deletion of TCR-stimulated double-positive thymocytes. J Immunol. 1997;158:4696–4703. [PubMed] [Google Scholar]
- 9.Virag L, Hasko G, Salzman AL, Szabo C. NADPH diaphorase histochemistry detects inducible nitric oxide synthetase activity in the thymus of naive and staphylococcal enterotoxin B-stimulated mice. J Histochem Cytochem. 1998;46:787–791. doi: 10.1177/002215549804600701. [DOI] [PubMed] [Google Scholar]
- 10.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 11.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 12.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 13.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 14.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 15.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 16.Feechan A, Kwon E, Yun BW, Wang Y, Pallas JA, Loake GJ. A central role for S-nitrosothiols in plant disease resistance. Proc Natl Acad Sci U S A. 2005;102:8054–8059. doi: 10.1073/pnas.0501456102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Protection from Experimental Asthma by an Endogenous Bronchodilator. Science. 2005;308:1618–1621. doi: 10.1126/science.1108228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci Transl Med. 2:19ra13. doi: 10.1126/scitranslmed.3000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML, et al. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature. 1992;360:225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- 21.Greco TM, Hodara R, Parastatidis I, Heijnen HF, Dennehy MK, Liebler DC, Ischiropoulos H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:7420–7425. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst. 1958;83:670–672. [Google Scholar]
- 23.Pesavento JJ, Garcia BA, Streeky JA, Kelleher NL, Mizzen CA. Mild performic acid oxidation enhances chromatographic and top down mass spectrometric analyses of histones. Mol Cell Proteomics. 2007;6:1510–1526. doi: 10.1074/mcp.M600404-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 25.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Aiello S, Noris M, Piccinini G, Tomasoni S, Casiraghi F, Bonazzola S, Mister M, Sayegh MH, Remuzzi G. Thymic dendritic cells express inducible nitric oxide synthase and generate nitric oxide in response to self- and alloantigens. J Immunol. 2000;164:4649–4658. doi: 10.4049/jimmunol.164.9.4649. [DOI] [PubMed] [Google Scholar]
- 27.Fehsel K, Kroncke KD, Meyer KL, Huber H, Wahn V, Kolb-Bachofen V. Nitric oxide induces apoptosis in mouse thymocytes. J Immunol. 1995;155:2858–2865. [PubMed] [Google Scholar]
- 28.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 29.Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 30.Bourgeois C, Hao Z, Rajewsky K, Potocnik AJ, Stockinger B. Ablation of thymic export causes accelerated decay of naive CD4 T cells in the periphery because of activation by environmental antigen. Proc Natl Acad Sci U S A. 2008;105:8691–8696. doi: 10.1073/pnas.0803732105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voehringer D, Liang HE, Locksley RM. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J Immunol. 2008;180:4742–4753. doi: 10.4049/jimmunol.180.7.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jameson SC. Maintaining the norm: T-cell homeostasis. Nat Rev Immunol. 2002;2:547–556. doi: 10.1038/nri853. [DOI] [PubMed] [Google Scholar]
- 33.Ho IC, Tai TS, Pai SY. GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat Rev Immunol. 2009;9:125–135. doi: 10.1038/nri2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothenberg EV, Taghon T. Molecular genetics of T cell development. Annu Rev Immunol. 2005;23:601–649. doi: 10.1146/annurev.immunol.23.021704.115737. [DOI] [PubMed] [Google Scholar]
- 35.Pai SY, Truitt ML, Ting CN, Leiden JM, Glimcher LH, Ho IC. Critical roles for transcription factor GATA-3 in thymocyte development. Immunity. 2003;19:863–875. doi: 10.1016/s1074-7613(03)00328-5. [DOI] [PubMed] [Google Scholar]
- 36.Maurice D, Hooper J, Lang G, Weston K. c-Myb regulates lineage choice in developing thymocytes via its target gene Gata3. Embo J. 2007;26:3629–3640. doi: 10.1038/sj.emboj.7601801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujimoto Y, Tu L, Miller AS, Bock C, Fujimoto M, Doyle C, Steeber DA, Tedder TF. CD83 expression influences CD4+ T cell development in the thymus. Cell. 2002;108:755–767. doi: 10.1016/s0092-8674(02)00673-6. [DOI] [PubMed] [Google Scholar]
- 38.Laiosa CV, Stadtfeld M, Graf T. Determinants of lymphoid-myeloid lineage diversification. Annu Rev Immunol. 2006;24:705–738. doi: 10.1146/annurev.immunol.24.021605.090742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.