

Abstract
Objectives
Macrophage migration inhibitory factor (MIF) promotes leukocyte recruitment and antagonizes the anti-inflammatory effects of glucocorticoids (GC). The aim of this study was to examine whether interaction between MIF and GC underlies the ability of MIF to promote leukocyte-endothelial cell interactions.
Methods
Intravital microscopy was used to assess leukocyte-endothelial cell interactions in wild-type and MIF−/− mice following treatment with LPS, the GC dexamethasone, and inhibition of endogenous GC using the GC receptor antagonist, RU486.
Results
Dexamethasone reduced LPS-induced leukocyte interactions in wild-type mice to levels similar to those observed in MIF−/− mice not treated with dexamethasone, whereas in MIF−/− mice, leukocyte interactions were not further inhibited by dexamethasone. RU486 increased LPS-induced leukocyte adhesion and emigration to a similar extent in both wild-type and MIF−/− mice, indicating that endogenous GC exert a similar inhibitory effect on leukocyte trafficking in wild-type and MIF−/− mice. Both MIF deficiency and RU486 treatment reduced VCAM-1 expression, while neither treatment modulated expression of ICAM-1 or chemokines CCL2, KC and MIP-2.
Conclusions
These results suggest that endogenous MIF and GC regulate leukocyte-endothelial cell interactions in vivo reciprocally but through predominantly independent mechanisms, and that the anti-inflammatory effect of MIF deficiency is comparable to that of exogenous GC.
Keywords: cell trafficking, cytokines, adhesion molecules, inflammation
INTRODUCTION
Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine which acts as a pro-inflammatory mediator in a variety of inflammatory responses (8, 15, 43). MIF expression is increased in inflammatory conditions, and in experimental models of inflammatory disease (20, 31, 34, 35, 44, 50). Moreover, inhibition or deletion of MIF results in significant attenuation of responses in models of sepsis, arthritis, lupus, and other forms of inflammation (4, 6, 9, 14, 26, 35, 40, 53, 57, 66). One possible pathway whereby MIF may contribute to these inflammatory responses is via leukocyte recruitment. We have previously observed that in MIF-deficient (MIF−/−) mice, leukocyte-endothelial cell interactions and leukocyte entry into tissues are reduced during inflammatory responses (21). Various mechanisms have been invoked to explain this finding. We have demonstrated that application of MIF to the microvasculature induces leukocyte recruitment via release of CCL2 (22). Similarly, recent work has demonstrated the ability of MIF to act in a ‘chemokine-like’ fashion, inducing rapid arrest of leukocytes under flow conditions, in part via structural similarities of MIF with chemokines such as IL-8 (5, 64). However, an additional potential explanation for the observed reduction in leukocyte recruitment in MIF−/− mice may stem from the ability of MIF to counter-regulate the physiological anti-inflammatory effects of endogenous glucocorticoids (GC) (19).
GC are endogenous anti-inflammatory hormones which regulate inflammation by inhibiting production of pro-inflammatory cytokines such as TNF and IL-1, as well as adhesion molecule expression and cell migration in response to inflammatory stressors (12, 25, 67). Their key role in regulating inflammation has been demonstrated by studies in which in vivo blockade of endogenous GC results in exacerbation of inflammatory responses, and increased leukocyte-endothelial cell interactions (16, 33, 60). However, in contrast to pro-inflammatory cytokines such as TNF and IL-1 which are suppressed by GC, MIF is regulated by GC in a distinct fashion, in that low physiologic concentrations of GC induce MIF expression (7). Moreover, MIF has the capability of overriding the anti-inflammatory effects of GC. This has been shown both in vitro, where MIF reverses the inhibitory effects of GC on production of pro-inflammatory cytokines, and in vivo, where treatment with recombinant MIF inhibits the anti-inflammatory effects of exogenous GC in endotoxemia and experimental arthritis (7, 57). These findings support the hypothesis that MIF acts to counter-regulate the anti-inflammatory effects of endogenous GC. However, whether the GC-antagonist function of MIF is central to its promotion of leukocyte trafficking in vivo is unknown.
Further evidence of an interaction between MIF and GC comes from recent in vitro studies in which the absence of MIF resulted in an increased sensitivity to GC (1, 55). In addition, MIF−/− mice undergoing a model of multiple sclerosis have been observed to have elevated corticosterone levels at the height of the immune response, indicating that MIF inhibits systemic release of endogenous GC under inflammatory conditions (53). These findings, and the observations that physiologic GC and endogenous MIF have opposing effects on leukocyte trafficking, raise the possibility that the ability of these molecules to counter-regulate the other’s actions underlies their effects on leukocyte recruitment. Fingerle-Rowson et al examined this issue and showed that MIF could antagonize the GC-induced stress-associated redistribution of lymphocytes out of the vasculature (17). However, whether this function of MIF extends to leukocyte recruitment induced by acute inflammation has not been investigated. Therefore the aim of this study was to test the hypothesis that MIF promotes leukocyte-endothelial cell interactions via antagonism of the effects of GC on these processes. We reasoned that this hypothesis could best be tested in a system in which the effects of MIF and GC could be studied independently. This was achieved by comparing wild-type and MIF−/− mice in the presence and absence of GC. These aims were achieved using intravital microscopy to examine leukocyte-endothelial cell interactions in inflamed post-capillary venules in wild-type and MIF−/− mice, following treatment with exogenous GC or inhibition of endogenous GC using GC receptor antagonism.
MATERIALS AND METHODS
Animals
MIF−/− mice on a 129/Sv × C57BL/6 background were used as previously described (6). Wild-type mice of the same background were bred from MIF+/+ littermates and used as controls. Confirmatory experiments were performed using MIF−/− mice generated on a pure C57BL/6 background (18), via comparison to wild-type C57BL/6 mice. Experiments were approved by the Animal Ethics Committee of Monash University.
Reagents and Antibodies
RU486 (mifepristone), dexamethasone (DEX) and LPS (from Escherichia coli serotype 0127.B8) were purchased from Sigma-Aldrich, St Louis, MO. Recombinant MIF (rMIF) was produced in an Escherichia coli expression system and had undetectable endotoxin concentrations as previously reported (22). Antibodies used in vivo were: 6C7.1, a rat mAb against murine VCAM-1 (generously provided by Dr. Britta Englehardt, Theodor Kocher Institute, Bern, Switzerland, and Dr. Dietmar Vestweber Max-Planck-Institut, Muenster, Germany); and rat anti-keyhole limpet hemocyanin (KLH) (BD Biosciences, San Diego, CA) as control Ab for adhesion molecule expression experiments.
Administration of LPS, rMIF, RU486 and Dexamethasone
To induce a local inflammatory response, LPS (10 or 100 ng in 200 µl saline) was administered to wild-type or MIF−/− mice via an intrascrotal (i.s.) injection adjacent to the cremaster muscle as previously described (21). Four hrs later the cremaster muscle was prepared for intravital microscopy. In some experiments, rMIF (1 µg in 200 µl saline) was administered to the cremaster muscle (opposite side to LPS-injected cremaster) of MIF−/− mice via i.s. injection (22). To assess the effects of exogenous GC, DEX (0.2 mg/kg in 200 µl saline) was given via i.p. injection 1 hour prior to LPS. This dose has been observed previously to reduce inflammation in an antigen-induced arthritis model (57). To examine the effects of endogenous GC, mice were pre-treated with RU486 (20 mg/kg in 0.5% carboxymethyl cellulose) via i.p. injection 18 hours and 1 hour prior to LPS treatment.
Intravital Microscopy
The mouse cremaster muscle was prepared for intravital microscopy as described previously (38). Briefly, mice were anesthetized by i.p. injection of ketamine hydrochloride (150 mg/kg; Pfizer Australia, West Ryde, NSW, Australia) and xylazine hydrochloride (10 mg/kg; Bayer Pharmaceuticals, Pymble, NSW, Australia). The left jugular vein was cannulated for administration of additional anesthetic and antibodies. The mouse was then placed on a thermo-controlled heating pad regulating the core temperature to 37°C. The cremaster muscle was dissected free of surrounding tissues and exteriorized onto an optically-clear viewing pedestal. The muscle was cut longitudinally with a cautery and held flat against the pedestal by attaching silk sutures to the edges of the tissue. The muscle was then superfused with bicarbonate-buffered saline (pH 7.4, 37°C), and covered with a coverslip held in place with vacuum grease.
An intravital microscope (Axioplan 2 Imaging; Carl Zeiss Australia) with a X20 objective lens (20X/0.40 NA) and X10 eye piece was used to observe the cremasteric microcirculation. A colour video camera (Sony SSC-DC50AP, Carl Zeiss Australia) was used to project the images onto a calibrated monitor (Sony PVM-20N5E) and the images were recorded for playback analysis using a videocassette recorder (Panasonic NV-HS950, Klapp Electronics, Prahran, Vic., Australia). Up to three venules (25–40 µm in diameter) were selected for recording in each experiment. Leukocyte rolling flux, rolling velocity, adhesion and emigration, and venular diameter were quantitated off-line using playback analysis, as previously described (38, 49). The centreline red blood cell velocity (VRBC) of the vessel was measured using an optical doppler velocimeter (Microcirculation Research Institute, Texas A&M University, College Station, TX), and the mean red blood cell velocity (Vmean) was determined as VRBC/1.6. Venular wall shear rate (γ) was calculated based on the Newtonian definition: γ = 8(Vmean/Dv) (38). Leukocyte rolling flux, rolling velocity, and adhesion and microvascular shear rate were measured 4.5 hrs after LPS treatment, whereas leukocyte emigration was determined in multiple postcapillary venules 5 hrs after LPS.
Real time PCR analysis of chemokine and adhesion molecule mRNA
In order to generate larger tissue samples than those available from the cremaster muscle, analysis of CCL2, RANTES, KC & MIP-2, ICAM-1 & VCAM-1 and MIF mRNA expression was performed in the gastrocnemius muscle. Wild-type and MIF−/− mice were injected in the gastrocnemius muscle with LPS or vehicle after treatment with vehicle or RU486. After 4 hrs, muscles were harvested and total RNA was extracted using TRIzol reagent (Gibco BRL), according to the manufacturer’s protocol. 0.5–1 µg of total RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen, Mount Waverley, Victoria, Australia) and oligo-(dT)20. PCR amplification was performed on a Rotor-Gene 3000 (Corbett Research, Mortlake, NSW, Australia). The primer-specific nucleotide sequences used were: CCL2 (MCP-1) (5'-CCCCAAGAAGGAATGGGTCC-3' and 5'-GGTTGTGGAAAAGGTAGTGG-3'); KC (5'-GGGTGTTGTGCGAAAAGA-3' and 5'-CAAAATGTCCAAGGGAAG-3'); MIP-2 (5’-TTTCTGGGGAGAGGGTGAGTTG-3’ and 3’-GCCATCCGACTGCATCTATTTG 5’); RANTES (5’-GCAAGTGCTCCAATCTTGCA-3’ and 5’-CTTCTCTGGGTTGGCACACA −3’); ICAM-1 (5'-GCCTTGGTAGAGGTGACTGAG-3' and 5'-GACCGGAGCTGAAAAGTTGTA-3'); VCAM-1 (5’-CCTCACTTGCAGCACTACGGGCT-3’ and 5’-TTTTCCAATATCCTCAATGACGGG-3’); MIF (5'-TGACTTTTAGCGGCACGAAC-3’ and 5’-GACTCAAGCGAAGGTGGAAC-3’); and 18s (5'-GTAACCCGTTGAACCCCATTC-3' and 5'-GCCTCACTAAACCATCCAATCG-3') (Invitrogen). Amplification was carried out in a total volume of 10 µL containing cDNA dilutions, primers (3 pmol) and DNA Master SYBR Green I kit (Sigma-Aldrich). Control reactions for product identification consisted of analyzing the melting peaks (°C) and determining the length of the PCR products (bp) by agarose gel electrophoresis. Amplification efficiency was controlled by the use of an internal control (house keeping gene) and external standards. The amount of target mRNA expression was calculated and normalized to 18s expression.
CCL2 and KC ELISA
Gastrocnemius muscles from wild-type and MIF−/− mice treated with LPS/vehicle or LPS/RU486 were obtained as above. Liquid nitrogen-frozen tissues were disrupted using a mortar and pestle followed by homogenization with a QIAshredder homogenizer (Qiagen). CCL2 and KC protein concentrations were measured in supernatants by ELISA (R&D Systems, via Bio-Scientific, Gymea, NSW, Australia).
Quantitation of VCAM-1 expression in the cremaster muscle
VCAM-1 expression in the cremasteric microvasculature of LPS/vehicle and LPS/RU486-treated wild-type and MIF−/− mice was quantitated using a previously described method (22, 48). Briefly, Alexa Fluor 488 (Molecular Probes, Eugene, OR)-conjugated anti-VCAM-1 Ab (6C7.1, 90 µg) and Alexa Fluor 594-conjugated anti-KLH (20 µg, as control non-binding Ab) were administered i.v. to anesthetized mice, 4 hrs after LPS treatment. Antibodies were allowed to circulate for 5 minutes then the mouse was exsanguinated, and the cremaster muscle surgically exposed in the same manner as for intravital microscopy. Vascular distribution of 6C7.1Alexa 488 and anti-KLHAlexa 594 were assessed by fluorescence microscopy. Images were visualized using a SIT video camera (Dage-MTI VE-1000) on pre-defined gain and black level settings, and image analysis performed on captured images using Scion Image. The length of vessel positive for 6C7.1Alexa 488 staining and the average staining intensity were quantified. Occasional vessels containing detectable Alexa-594-associated fluorescence were excluded from analysis. Final data are shown as arbitrary units, representing the product of the average 6C7.1Alexa 488-associated fluorescence intensity multiplied by the length of positive vessel (48).
Statistical Analysis
All data are presented as mean ± sem. Student’s t-tests were used to compare experimental groups, with a value of p < 0.05 deemed significant. In some instances, when data were not normally distributed, Mann-Whitney non-parametric rank tests were used to compare groups.
RESULTS
Reversal of the effects of MIF deficiency on LPS-induced leukocyte recruitment by exogenous MIF
We previously reported the effect of endogenous MIF on in vivo leukocyte recruitment (21). In the present study, we tested whether the effects of MIF deficiency could be reversed via administration of exogenous MIF. As previously described, LPS-induced adhesion and emigration were reduced in MIF−/− mice relative to wild-type mice. Local administration of rMIF to MIF−/− mice restored adhesion and emigration to wild-type levels (Fig. 1). To examine the possibility of the background strain contributing to the difference observed between MIF−/− and wild-type mice on the mixed B6/129Sv background, additional experiments were performed using MIF−/− mice on the C57BL/6 background. C57BL/6 MIF−/− mice also showed significant reductions in adhesion and emigration relative to wild-type C57BL/6 mice (adhesion: WT, 23.9 ± 5.4 vs MIF−/−, 9.3 ± 2.2 cells/100 µm, p < 0.05; emigration: WT, 34.4 ± 2.7 vs MIF−/−, 6.1 ± 1.7 cells/field µm, p < 0.001).
Figure 1. Exogenous MIF reverses effect of MIF-deficiency on leukocyte trafficking.
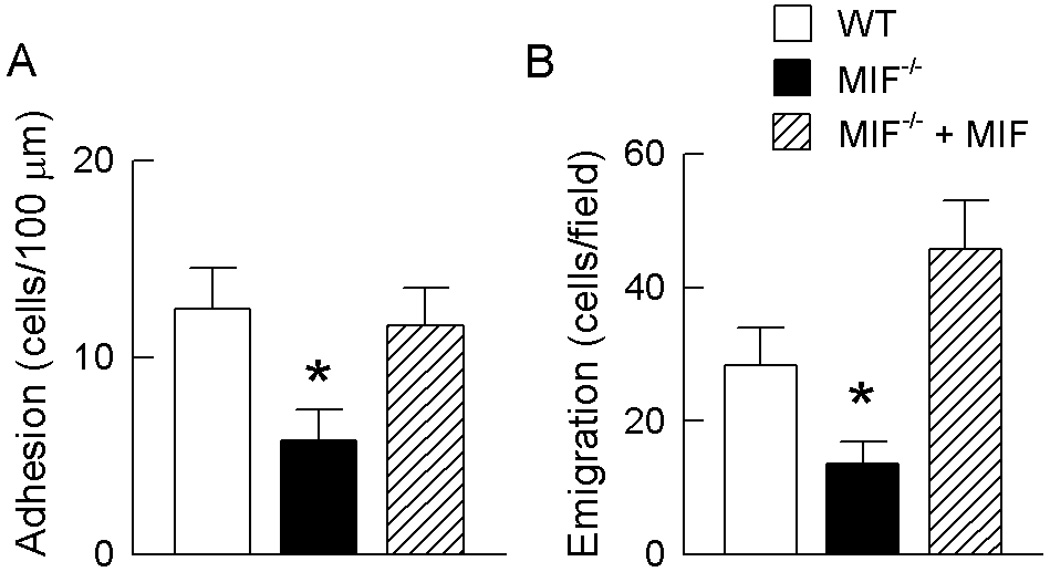
Leukocyte adhesion (A) and emigration (B) in cremasteric post-capillary venules of LPS-treated (10 ng) wild-type mice, MIF−/− mice and MIF−/− mice which received rMIF (1 ug). Adhesion data were collected 4.5 hrs post LPS whereas emigration data were collected 5 hours post-LPS. N=6 mice/gp. * p < 0.05 vs LPS-treated wild-type mice.
Effects of exogenous administration of dexamethasone on leukocyte recruitment in wild-type and MIF−/− mice
Although exogenous MIF has been shown to counteract the inhibitory effects of GC in vivo, whether endogenous MIF has this capability has not been demonstrated in vivo. Therefore, we compared the inhibitory effect of DEX on LPS-induced leukocyte trafficking in wild-type and MIF−/− mice. In wild-type mice, DEX treatment resulted in significant reductions in LPS-induced leukocyte rolling flux, adhesion and emigration relative to mice treated with LPS alone (Fig. 2A–C). Mice receiving DEX treatment also displayed reduced circulating leukocyte counts, raising the possibility that the reduction in interactions may have been due in part to changes in cell counts (Table 1). In MIF−/− mice, DEX treatment also reduced leukocyte rolling flux and circulating leukocyte counts (Fig. 2A & Table 1), as seen in wild-type mice treated with DEX. In contrast, DEX treatment of MIF−/− mice did not reduce leukocyte adhesion and emigration from the already low levels observed in MIF−/− mice treated with LPS alone (Fig. 2B & C).
Figure 2. Differential effect of dexamethasone on leukocyte trafficking in wild-type and MIF−/− mice.
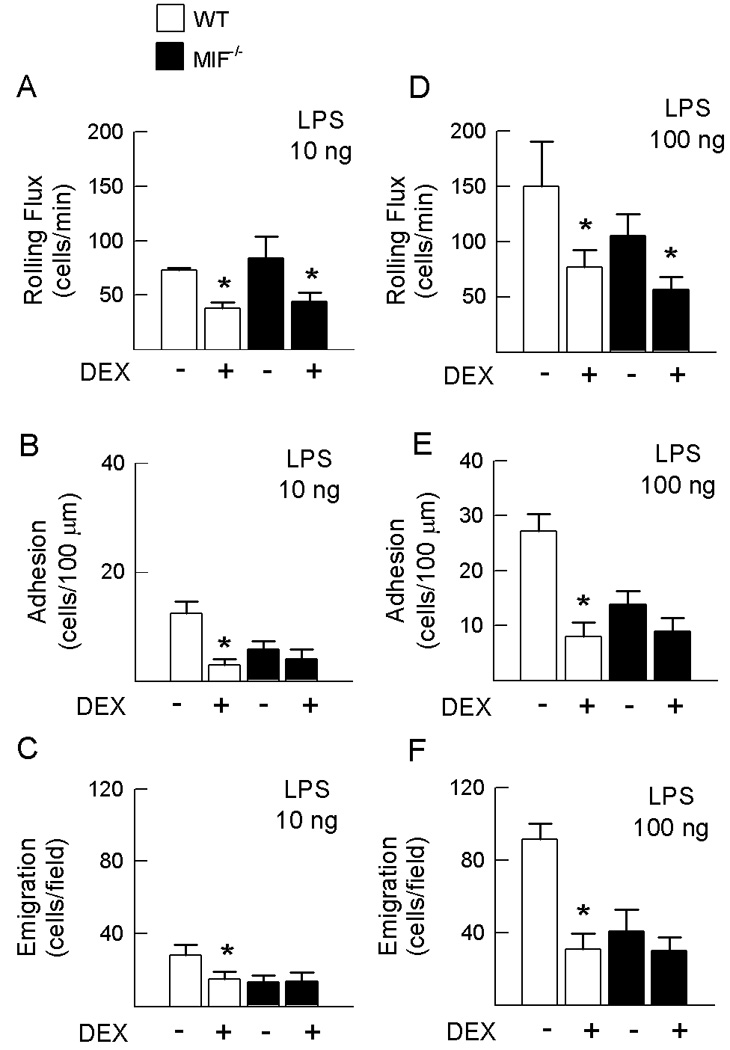
Effect of DEX (0.2 mg/kg, i.p.) on LPS-induced leukocyte rolling flux, adhesion and emigration in cremasteric post-capillary venules of wild-type and MIF−/− mice. Responses were examined at two doses of LPS: A, B & C show parameters from wild-type and MIF−/− mice treated with 10 ng LPS, in the presence or absence of DEX pretreatment. D, E & F show data from similarly-treated wild-type and MIF−/− mice treated with 100 ng LPS. N=6 mice/gp. * p < 0.05 for mice treated with LPS & DEX vs. mice treated with LPS alone in the corresponding strain.
Table 1.
Hemodynamic parameters and circulating leukocyte counts in wild-type and MIF−/− mice treated with LPS (10 ng), DEX and RU486*
| Shear rate, (s−1) |
Leukocyte count (×10−6/mL) |
|
|---|---|---|
| Wild-type mice | ||
| LPS (n=6) | 456 ± 38 | 5.7 ± 0.6 |
| LPS + DEX (n=6) | 407 ± 66 | 2.4 ± 0.2† |
| LPS + RU486 (n=6) | 336 ± 10† | 8.8 ± 1.1† |
| MIF−/− mice | ||
| LPS (n=6) | 586 ± 75 | 7.9 ± 1.1 |
| LPS + DEX (n=6) | 520 ± 60 | 3.9 ± 0.8† |
| LPS + RU486 (n=8) | 388 ± 36† | 11 ± 1.3† |
Data are shown as mean ± sem. LPS = lipopolysaccharide. DEX = dexamethasone.
p < 0.05 versus LPS alone treatment.
One possible explanation for the inability to observe an effect of DEX on leukocyte adhesion and emigration in MIF−/− mice is that the numbers of adherent and emigrated leukocytes in MIF−/− mice were already low. We therefore examined wild-type and MIF−/− mice treated with a higher dose of LPS (100 ng). Compared to mice treated with 10 ng LPS, both wild-type and MIF−/− mice treated with 100 ng LPS exhibited 2–3 fold increases in leukocyte rolling flux, adhesion and emigration (Table 2). Comparison of WT and MIF−/− mice treated with this higher dose of LPS revealed that leukocyte adhesion and emigration remained significantly reduced in MIF−/− mice (Table 2). MIF−/− mice treated with 100 ng LPS exhibited significantly increased circulating leukocyte counts compared to similarly treated wild-type mice, indicating that reduced interactions in the absence of MIF were not due to reduced leukocyte numbers (Table 3). In contrast, MIF−/− mice treated with 100 ng LPS exhibited significantly elevated shear rates compared to wild-type animals receiving the same treatment (Table 3), suggesting this as a possible contributor to the reductions in leukocyte-endothelial cell interactions in MIF−/− mice.
Table 2.
Comparison of leukocyte-endothelial cell interactions in wild-type and MIF−/− mice treated with either 10 ng or 100 ng LPS*
| Leukocyte rolling flux (cells/min) |
Leukocyte Adhesion (cells/100 µm) |
Leukocyte Emigration (cells/field) |
|
|---|---|---|---|
| Wild-type | |||
| 10 ng LPS (n=6) | 73.3 ± 1.7 | 12.5 ± 2.1 | 28.4 ± 5.5 |
| 100 ng LPS (n=6) | 150 ± 40.6† | 27.4 ± 0.6† | 91.6 ± 8.7† |
| MIF−/− | |||
| 10 ng LPS (n=6) | 83.8 ± 20.5 | 5.8 ± 1.6 | 13.5 ± 3.3 |
| 100 ng LPS (n=6) | 105.5 ± 19.3 | 13.9 ± 2.4†** | 41.0 ± 12.0†** |
Data represent mean ± sem of at least 6 mice/group. Rolling and adhesion data were collected 4.5 hrs after LPS administration, whereas emigration data were collected at the end of the experiment (5 hrs after LPS).
denotes p < 0.05 vs 10 ng LPS treatment in corresponding strain.
denotes p < 0.05 for MIF−/− vs wild-type mice, in 100 ng LPS-treated groups.
Table 3.
Hemodynamic parameters and circulating leukocyte counts in wild-type and MIF−/− mice treated with LPS (100 ng) and DEX*
| Shear rate (s−1) |
Leukocyte count (×10−6/mL) |
|
|---|---|---|
| Wild-type mice | ||
| LPS (n=6) | 462 ± 39 | 4.7 ± 0.6 |
| LPS + DEX (n=5) | 498 ± 51 | 1.7 ± 0.2† |
| MIF−/− mice | ||
| LPS (n=6) | 710 ± 61τ | 9.9 ± 0.6τ |
| DEX (n=6) | 705 ± 92 | 4.9 ± 0.8† |
Data are shown as mean ± sem.
denotes p < 0.05 versus wild-type mice receiving the same treatment.
denotes p < 0.05 versus LPS alone treatment.
We next examined the effect of DEX on leukocyte-endothelial cell interactions induced by this higher dose of LPS (Fig. 2D–F). As seen at the lower LPS dose, DEX treatment significantly inhibited rolling flux, adhesion, and emigration in wild-type mice treated with 100 ng LPS (Fig. 2D–F), and reduced circulating leukocyte counts (Table 3). In MIF−/− mice treated with 100 ng LPS, DEX treatment did not further reduce adhesion or emigration, but did significantly inhibit leukocyte rolling flux (Fig. 2D–F), mirroring the effects of DEX in MIF−/− mice treated with the lower dose of LPS. Furthermore, even in response to the higher dose of LPS, MIF deficiency inhibited LPS-induced leukocyte adhesion and emigration to a similar degree as DEX treatment in wild-type mice.
RU486 causes increased leukocyte recruitment in both wild-type and MIF−/− mice
The aim of the next series of experiments was to determine whether the ability of endogenous MIF to promote leukocyte-endothelial cell interactions is dependent on antagonism of the inhibitory effects of endogenous GC. This was achieved by examining the effect of MIF deficiency in the presence and absence of GC receptor inhibition, via RU486. In both wild-type and MIF−/− mice, RU486 caused no alteration in LPS-induced leukocyte flux relative to mice treated with LPS alone (data not shown). In contrast, RU486 caused a significant increase in LPS-induced leukocyte adhesion and emigration in both wild-type (Fig. 3A & B) and MIF−/− mice (Fig. 3C & D). Furthermore, the magnitude of effect of GC receptor inhibition was similar in wild-type and MIF −/− mice. RU486 treatment also induced reductions in microvascular shear stress and increased circulating leukocyte counts (Table 1), which may have contributed to the increase in leukocyte-endothelial interactions observed. These findings indicate that the ability of endogenous GC to regulate leukocyte adhesion and emigration is comparable in the presence and absence of MIF.
Figure 3. GC receptor inhibition increases recruitment in both wild-type and MIF−/− mice.
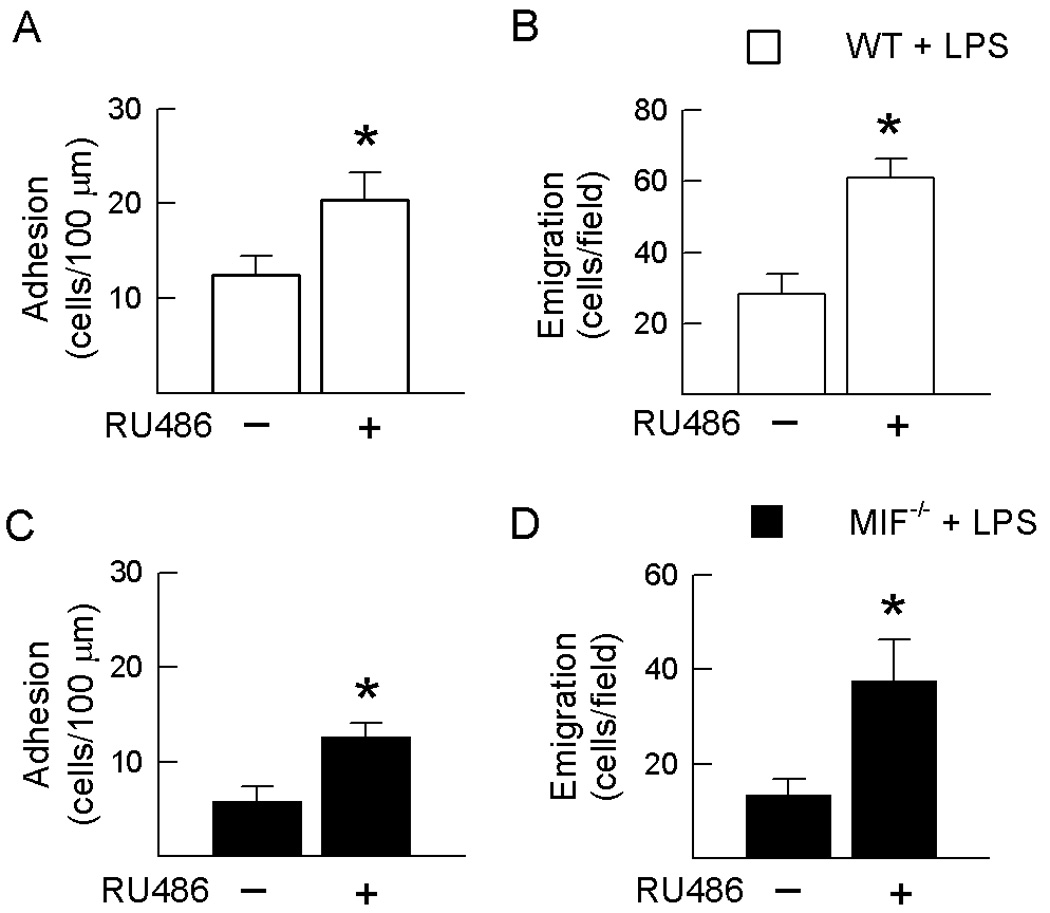
Effect of GC receptor inhibition via RU486 on leukocyte adhesion (A, C) and emigration (B, D) in LPS-treated wild-type mice (A, B) and LPS and MIF−/− mice (C, D). Mice were treated with LPS (10 ng) treatment then adhesion data were collected 4.5 hrs post LPS whereas emigration data were collected 5 hours post-LPS. N=6–8/gp. * p < 0.05 for mice treated with LPS & RU486 vs. mice treated with LPS alone in the corresponding strain.
Further analysis of results from wild-type and MIF−/− mice treated with RU486 demonstrated that, under conditions of GC inhibition, the inhibitory effects of MIF deficiency on leukocyte-endothelial cell interactions were comparable to those observed in GC-intact mice. In the RU486-treated groups, significant reductions in adhesion (wild-type − 20.4 ± 2.9 vs. MIF−/− 12.6 ± 1.5 cells/100 µm, p = 0.011) and emigration (wild-type − 60.9 ± 5.4 vs. MIF−/− 37 ± 8.9 cells/field, p = 0.03) were observed in MIF−/− mice relative to wild-type mice. These findings indicate that endogenous MIF can promote leukocyte adhesion and emigration regardless of the presence or absence of endogenous GC. Together, these data indicate that the opposing effects of endogenous MIF and endogenous GC on leukocyte adhesion and emigration are independent.
Roles of chemokines and adhesion molecules in regulation of leukocyte recruitment by endogenous GC and MIF
The observation that endogenous GC and MIF can independently regulate leukocyte recruitment raises the possibility that GC and MIF impact on distinct recruitment mechanisms. We therefore sought to determine the mechanisms responsible for the effects of endogenous MIF and GC on LPS-induced leukocyte recruitment by examining the expression of chemokines and adhesion molecules. We first assessed the effects of endogenous GC and MIF on the chemokines CCL2 (MCP-1) and KC, both of which have been demonstrated to be regulated by both MIF and GC (22, 26, 29, 37, 61), as well as MIP-2 and RANTES. Under these conditions, LPS induced varying levels of increase in chemokine mRNA (Fig. 4). However, in LPS-treated mice, chemokine mRNA expression did not differ significantly between wild-type and MIF−/− mice, except in the case of RANTES which was reduced in LPS-treated MIF−/− mice (Fig. 4F). Similarly, in most cases RU486 treatment did not further increase chemokine mRNA above that in wild-type and MIF−/− mice treated with LPS and vehicle. Chemokine expression was examined in further detail via measurement of KC and CCL2 protein in LPS-treated muscles. These data indicated that muscle chemokine content in LPS-treated mice was not altered by MIF genotype or RU486 treatment (Fig. 4B & D). These findings suggest that these chemokines are not major targets of either MIF or endogenous GC in LPS-induced responses.
Figure 4. MIF deficiency and GC receptor inhibition do not alter chemokine expression.

A, C, E, F: Effect of GC inhibition on LPS-induced chemokine mRNA expression in muscle in the presence and absence of endogenous MIF. Wild-type and MIF−/− mice were treated with vehicle alone, LPS (10 ng, 4 hrs) + vehicle, or with the GC receptor antagonist, RU486 prior to LPS treatment. Four hrs later, mRNA for CCL2 (A), KC (C), MIP-2 (E) and RANTES (F) were quantitated using real-time PCR and expressed relative to 18s mRNA. Data are shown as mean ± sem of n=4–8 separate experiments. * denotes p < 0.05 vs vehicle. B, D. Effect of GC inhibition on LPS-induced CCL2 (B) and KC (D) protein expression in muscle in the presence and absence of endogenous MIF. Wild-type and MIF−/− mice were treated with LPS (10 ng, 4 hrs) + vehicle, or with RU486 prior to LPS treatment. CCL2 and KC protein expression were quantitated in homogenised muscle samples using ELISA, and expressed as pg chemokine/mg total protein. Data from individual experiments are shown and horizontal line indicates group mean (n=9−12 separate experiments).
We next examined the expression of the endothelial adhesion molecules ICAM-1 and VCAM-1 in the presence and absence of MIF, and in GC-intact or RU486-treated mice, given the established ability of LPS to increase expression of these molecules. Examination of ICAM-1 expression in the gastrocnemius muscle using real-time PCR demonstrated that LPS induced a significant increase in ICAM-1 mRNA in both wild-type and MIF−/− mice (Fig. 5). No difference in ICAM-1 expression was observed between wild-type and MIF−/− mice. Similarly, ICAM-1 mRNA expression was unaffected by treatment with RU486 in either strain of mice (Fig. 5). These data suggest that regulation of LPS-induced leukocyte recruitment by endogenous GC and MIF is not due to modulation of ICAM-1 expression.
Figure 5. ICAM-1 and VCAM expression in wild-type and MIF−/− mice, and in the presence and absence of RU486.

A, B. Roles of endogenous MIF and GC in regulating LPS-induced expression of mRNA for ICAM-1 (A) and VCAM-1 (B) in muscle. Wild-type and MIF−/− mice were treated with vehicle alone, LPS (10 ng, 4 hrs) + vehicle, or with RU486 prior to LPS treatment. ICAM-1 & VCAM-1 mRNA were quantitated using real-time PCR and expressed relative to 18s mRNA. Data are shown as mean ± sem of n = 5–8. * denotes p<0.05 vs vehicle. C. Roles of endogenous MIF and GC in regulating LPS-induced VCAM-1 expression in the cremasteric microvasculature. Wild-type and MIF−/− mice were treated with LPS (10 ng) following pretreatment with RU486, or vehicle. Four hrs later, VCAM-1 expression was assessed using fluorescence microscopy to quantitate deposition of an Alexa 488-conjugated anti-VCAM-1 Ab. Data are shown for wild-type and MIF−/− mice following treatment with vehicle or RU486 (n=6/group). * p = 0.013 for MIF−/− vs wild-type vehicle-treated mice. ** p = 0.018 for effect of RU486 in LPS-treated wild-type mice.
LPS also induced significant increases in VCAM-1 mRNA expression in the same samples, to a greater extent in wild-type versus MIF−/− mice (Fig. 5B). However, in contrast to ICAM-1, RU486 suppressed LPS-induced upregulation of VCAM-1 mRNA. This was an unexpected finding given the observed effect of RU486 on leukocyte trafficking. Therefore we also examined VCAM-1 expression by quantitating intravascular VCAM-1 protein in vivo using fluorochrome-conjugated anti-VCAM-1 Ab (Fig. 5C). In LPS-treated wild-type mice, VCAM-1 protein expression was approximately 5 fold greater than that we previously observed in untreated mice using this approach (22). In LPS-treated MIF−/− mice, VCAM-1 expression was significantly reduced compared to that in wild-type mice, providing a potential mechanism for the effect of endogenous MIF in promoting leukocyte adhesion (Fig. 5C). However, in wild-type mice RU486 treatment was associated with a significant reduction in LPS-induced VCAM-1 expression, an effect which was less apparent in MIF−/− mice (Fig. 5C), findings which corresponded with those for mRNA expression (Fig 5B). Given our observation of elevated leukocyte adhesion in response to RU486 (Fig. 3), this result argues against a role for regulation of VCAM-1 expression in mediating the effects of endogenous GC.
DISCUSSION
Endogenous GC and MIF are key molecules in the regulation of the inflammatory response in vivo. GC have potent anti-inflammatory and immunosuppressive effects including inhibition of pro-inflammatory cytokine production and adhesion molecule expression (12, 67). In contrast, MIF has a key role in promoting the development of inflammatory diseases via its effects on cytokine and mediator expression (4, 6, 35, 51, 57). Although its expression can be induced by GC, a key function of MIF is its ability to antagonize the anti-inflammatory effects of GC both in vitro and in vivo (7, 57). More recently, MIF has been shown to promote a hallmark of inflammation, leukocyte recruitment (21). The ability of MIF to counteract the anti-inflammatory effects of GC raises the possibility that reduced leukocyte recruitment seen in MIF−/− mice is due to enhanced effects of endogenous GC in the absence of MIF. The findings we report here demonstrate that endogenous MIF and GC have opposing but independent effects on leukocyte trafficking, arguing against an interaction between these molecules in their regulation of inflammatory responses. Furthermore, these findings provide support for the potential anti-inflammatory effectiveness of MIF inhibition, as the reduction in leukocyte-endothelial cell interactions observed in MIF−/− mice was comparable to that induced by administration of exogenous GC.
Previous in vivo studies have shown that DEX treatment attenuates LPS-induced leukocyte-endothelial cell interactions (3, 13). This was supported by the results of the present study, in which pretreatment of wild-type mice with DEX caused reductions in LPS-induced leukocyte rolling flux, adhesion and emigration. However, in MIF−/− mice, DEX was unable to further reduce leukocyte-endothelial cell interactions across a range of LPS doses. This may indicate that the mechanisms whereby exogenous GC inhibit leukocyte recruitment overlap with those altered by MIF deficiency. These results would argue that MIF inhibition provides a robust anti-inflammatory effect which is comparable to that of GC. However, this finding suggests that under conditions of MIF inhibition, there may be no added benefit of exogenous GC treatment.
Further, these findings could be interpreted to indicate that the independence or otherwise of the actions of MIF and GC depends on the source of the GC. While endogenous GC, as demonstrated by RU486 treatment, and MIF are each able to mediate effects independently of each other, exogenous GC, i.e. DEX, only works effectively in MIF-expressing mice. The reason for this apparent divergence between the effects of endogenous and exogenous GC is unclear. However, it may reflect dose-dependent effects on activation of the GC receptor by GC present at either physiological (endogenous GC) or pharmacological (DEX) levels. Or alternatively, the exogenous version of GC may have a different pharmacokinetic profile in the circulation than that of endogenous corticosterone.
Conversely, GC receptor blockade via RU486 caused significant increases in leukocyte adhesion and emigration in both wild-type and MIF−/− mice, indicating that the anti-inflammatory effects of endogenous GC are MIF-independent. Moreover, MIF−/− mice were consistently found to have reduced leukocyte-endothelial cell interactions, regardless of GC receptor blockade, indicating that reductions in leukocyte recruitment in MIF−/− mice were independent of endogenous GC. One potential explanation for this observation is that MIF and GC regulate recruitment via distinct mechanisms. GC have been shown to inhibit expression of many molecular pathways with roles in leukocyte trafficking, including adhesion molecules and chemokines, as well as leukocyte motility (12, 25, 67). Expression of the adhesion molecule P-selectin is particularly relevant to the model of inflammation used in this study, as we have observed that this molecule is responsible for greater than 90% of the rolling interactions observed in this model (21). P-selectin expression has been shown to be inhibited by both endogenous and exogenous GC (32, 45). In contrast, our previous studies in MIF−/− mice demonstrated that reductions in leukocyte trafficking in these animals were not associated with any effect on endothelial P-selectin expression or leukocyte P-selectin binding activity (21). Together, these findings indicate that GC but not MIF have the ability to modulate the P-selectin pathway.
In contrast, the present study provides the first demonstration of the ability of endogenous MIF to regulate endothelial VCAM-1 expression. MIF−/− mice showed significantly reduced VCAM-1 expression following LPS treatment, proving a plausible mechanism whereby MIF deficiency results in reduced leukocyte recruitment. In contrast, a comparable effect on ICAM-1 expression was not observed. Interestingly, we have previously reported that exposure to exogenous MIF does not increase VCAM-1 expression in the uninflamed microvasculature, under conditions where it is expressed constitutively at functional levels (21). Taken together with the present findings, these data demonstrate that endogenous MIF plays a role in enabling maximal upregulation of VCAM-1 in response to LPS stimulation, but that exogenous MIF alone is incapable of increasing endothelial VCAM-1 expression.
In regards to the effects of exogenous GC on adhesion molecule expression, DEX has been shown to inhibit ICAM-1 expression in vivo (45). Similarly, expression of VCAM-1 in vivo has been shown to be reduced by exogenous GC, therefore we anticipated increased expression following GC receptor blockade (45). Unexpectedly, RU486 treatment, while resulting in increased leukocyte-endothelial cell interactions, was associated with reduced VCAM-1 expression in the cremasteric microvasculature in both wild-type and MIF−/− mice, findings supported by reduced mRNA levels in the gastrocnemius muscle. The explanation for this finding, which suggests that endogenous GC act to promote VCAM-1 expression, is unclear. However it suggests that the effects of endogenous GC do not always mimic the actions of exogenous GC. The alternative explanation, that partial agonist effects of RU486 underlie the unexpected reduction in VCAM-1 expression in RU486-treated mice, is not supported by numerous in vivo studies in which RU486 has a clear net GC-antagonist effect, resulting in the promotion of inflammation (24, 36, 42, 52).
The breadth of the anti-inflammatory effects of GC has been related to GC inhibition of multiple signaling pathways. GC inhibit activation of the NF-κB pathway, and through the induction of MAPK phosphatase-1 (MKP-1), inhibit MAPK activation, with resultant reductions in expression of cytokines, chemokines and adhesion molecules (1, 11, 12, 46, 55, 62). Both of these signaling pathways are known to promote multiple aspects of leukocyte recruitment (23, 39, 54, 65). In contrast, the pro-inflammatory effects of MIF are predominantly associated with activation of MAPK family members including ERK and p38 (41, 58). Correspondingly, cells derived from MIF−/− mice exhibit decreased MAPK activation (1). There is minimal evidence, in contrast, that MIF directly activates the NF-κB pathway (30), but MIF has recently been demonstrated to inhibit GC-induced MKP-1 expression (1, 55). These divergences between the signaling pathways regulated by MIF and GC may explain the present observations of independent actions of these mediators.
We also investigated the ability of both MIF and endogenous GC to regulate recruitment via an effect on the expression of the chemokines KC, CCL2, MIP-2 and RANTES. Expression of CCL2 and RANTES, but not MIP-2, has been previously reported to be inhibited by exogenous GC, i.e. DEX, in vivo (2, 10, 56), whereas results of experiments assessing the sensitivity of KC expression to DEX have been variable (56, 59). In the present study, the effect of LPS on chemokine expression was comparable both in wildtype and MIF−/− mice, and in the presence and absence of GC receptor blockade, with the exception of RANTES, which was reduced in MIF−/− mice. These data suggest that, at least under the conditions used in the present studies, regulation of expression of KC, CCL2 or MIP-2 does not underlie the effects of MIF and endogenous GC on leukocyte recruitment.
Comparison of microvascular responses in wild-type and MIF−/− mice treated with the higher dose of LPS (100 ng) revealed significantly elevated shear rates in MIF−/− mice (Table 3). It is possible that the anti-adhesive forces provided by the elevated shear rate contributed to the reduced leukocyte-endothelial cell interactions seen in the mutant strain. MIF has been proposed as a critical regulator of vascular tone in septic shock (63), potentially via its ability to promote production of the vasodilators nitric oxide and PGE2 (27, 58). Despite this, a comparable significant elevation in shear was not observed at the lower dose of LPS used (10 ng, Table 1) indicating that altered shear alone is insufficient to explain the consistent reduction in leukocyte trafficking observed in MIF−/− mice.
GC remain the mainstay therapeutic modality in some forms of chronic inflammation, particularly, rheumatoid arthritis. While the present studies show clear independence of MIF and endogenous GC during a relatively short term (4–5 hrs) form of inflammation, whether this applies under more extended inflammatory conditions remains unclear. There is evidence that the glucocorticoid receptor, through which GC mediate their effects, is reduced in patients treated with GC, and in animals undergoing a 24 hr sepsis response and GC treatment (28, 47). These observations would indicate that the milieu of extended inflammation and ongoing exposure to therapeutic levels of exogenous GC has the potential to alter GC-mediated responses, and potentially the interaction with the MIF pathway.
In conclusion, the current study indicates that endogenous MIF and GC have powerful and reciprocal effects on leukocyte trafficking, wherein LPS-induced leukocyte recruitment is suppressed by endogenous GC and promoted by MIF. The demonstration that the effects of endogenous GC are independent of the presence or absence of MIF, and that the effects of endogenous MIF are independent of the effects of endogenous GC, disproves the hypothesis that the effects of MIF are limited to the antagonism of GC. Moreover, the observation that the magnitude of the effects of MIF deficiency was similar to that of exogenous GC administration also argues in favour of the potential effectiveness of anti-MIF therapeutic strategies in inflammatory disease.
ACKNOWLEDGEMENTS
Sources of support This study was supported by a Program Grant from the National Health & Medical Research Council (NHMRC) Australia (#334067), and an RO1 grant from the NIH (#AR51807-01). MJH is an NHMRC Senior Research Fellow.
The authors would like to thank Dr. Gunter Fingerle-Rowson (University Hospital Cologne, Cologne, Germany) for generous provision of the C57BL/6 background MIF−/− mice.
REFERENCES
- 1.Aeberli D, Yang YH, Mansell A, Santos L, Leech M, Morand EF. Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett. 2006;580:974–981. doi: 10.1016/j.febslet.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 2.Ajuebor MN, Gibbs L, Flower RJ, Das AM, Perretti M. Investigation of the functional role played by the chemokine monocyte chemoattractant protein-1 in interleukin-1-induced murine peritonitis. Br J Pharmacol. 1998;125:319–326. doi: 10.1038/sj.bjp.0702071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allcock GH, Allegra M, Flower RJ, Perretti M. Neutrophil accumulation induced by bacterial lipopolysaccharide: effects of dexamethasone and annexin 1. Clin. Exp. Immunol. 2001;123:62–67. doi: 10.1046/j.1365-2249.2001.01370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 5.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 6.Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J. Exp. Med. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 8.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognised source of macrophage migration inhibitory factor. J. Exp. Med. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calandra T, Echtenacher B, Le Roy D, Pugin J, Metz CN, Hultner L, Heumann D, Mannel D, Bucala R, Glauser MP. Protection from septic shock by neutralization of macrophage inhibitory factor. Nat. Med. 2000;6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 10.Castellani ML, Shanmugham LN, Petrarca C, Simeonidou I, Frydas S, De Colli M, Vecchiet J, Falasca K, Tete S, Salini V, Conti P. Expression and secretion of RANTES (CCL5) in granulomatous calcified tissue before and after lipopolysaccharide treatment in vivo. Calcif Tissue Int. 2007;80:60–67. doi: 10.1007/s00223-006-0115-2. [DOI] [PubMed] [Google Scholar]
- 11.Crinelli R, Antonelli A, Bianchi M, Gentilini L, Scaramucci S, Magnani M. Selective inhibition of NF-κB activation and TNF-α production in macrophages by red blood cell-mediated delivery of dexamethasone. Blood Cells, Molecules, and Diseases. 2000;26:211–222. doi: 10.1006/bcmd.2000.0298. [DOI] [PubMed] [Google Scholar]
- 12.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: The glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9991–9995. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davenpeck KL, Zagorski J, Schleimer RP, Bochner BS. Lipopolysaccharide-induced leukocyte rolling and adhesion in the rat mesenteric microcirculation: regulation by glucocorticoids and role of cytokines. J. Immunol. 1998;161:6861–6870. [PubMed] [Google Scholar]
- 14.Denkinger CM, Denkinger M, Kort JJ, Metz C, Forsthuber TG. In vivo blockade of macrophage migration inhibitory factor ameliorates acute experimental autoimmune encephalomyelitis by impairing the homing of encephalitogenic T cells to the central nervous system. J. Immunol. 2003;170:1274–1282. doi: 10.4049/jimmunol.170.3.1274. [DOI] [PubMed] [Google Scholar]
- 15.Donnelly SC, Bucala R. Macrophage migration inhibitory factor: a regulator of glucocorticoid activity with a critical role in inflammatory disease. Mol. Med. Today. 1997;3:502–507. doi: 10.1016/S1357-4310(97)01133-7. [DOI] [PubMed] [Google Scholar]
- 16.Farsky SP, Sannomiya P, Garcia-Leme J. Secreted glucocorticoids regulate leukocyte-endothelial interactions in inflammation. A direct vital microscopic study. J. Leukoc. Biol. 1995;57:379–386. doi: 10.1002/jlb.57.3.379. [DOI] [PubMed] [Google Scholar]
- 17.Fingerle-Rowson G, Koch P, Bikoff R, Lin X, Metz CN, Dhabhar FS, Meinhardt A, Bucala R. Regulation of macrophage migration inhibitory factor expression by glucocortocoids in vivo. Am. J. Pathol. 2003;162:47–56. doi: 10.1016/S0002-9440(10)63797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Muller W, Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci U S A. 2003;100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flaster H, Bernhagen J, Calandra T, Bucala R. The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol. 2007;21:1267–1280. doi: 10.1210/me.2007-0065. [DOI] [PubMed] [Google Scholar]
- 20.Foote A, Brigante FM, Kipen Y, Santos L, Leech M, Morand EF. Macrophage migration inhibitory factor in systemic lupus erythematosus. J. Rheumatol. 2004;31:268–273. [PubMed] [Google Scholar]
- 21.Gregory JL, Leech MT, David J, Yang YH, Dacumos A, Hickey MJ. Reduced leukocyte-endothelial cell interactions in the inflamed microcirculation of macrophage migration inhibitory factor-deficient Mice. Arthritis Rheum. 2004;50:3023–3034. doi: 10.1002/art.20470. [DOI] [PubMed] [Google Scholar]
- 22.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, McColl SR, Hickey MJ. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J. Immunol. 2006;177:8072–8079. doi: 10.4049/jimmunol.177.11.8072. [DOI] [PubMed] [Google Scholar]
- 23.Heit B, Colarusso P, Kubes P. Fundamentally different roles for LFA-1, Mac-1 and α4 integrin in neutrophil chemotaxis. J. Cell Sci. 2005;118:5205–5220. doi: 10.1242/jcs.02632. [DOI] [PubMed] [Google Scholar]
- 24.Hermann C, von Aulock S, Dehus O, Keller M, Okigami H, Gantner F, Wendel A, Hartung T. Endogenous cortisol determines the circadian rhythm of lipopolysaccharide-- but not lipoteichoic acid--inducible cytokine release. Eur J Immunol. 2006;36:371–379. doi: 10.1002/eji.200535470. [DOI] [PubMed] [Google Scholar]
- 25.Hirasawa N, Watanabe M, Mue S, Watanabe K, Tsurufuji S, Ohuchi K. Induction of neutrophil infiltration by rat chemotactic cytokine (CINC) and its inhibition by dexamethasone in rats. Inflammation. 1992;16:187–196. doi: 10.1007/BF00918958. [DOI] [PubMed] [Google Scholar]
- 26.Hoi AY, Hickey MJ, Hall P, Yamana J, O'Sullivan KM, Santos LL, James WG, Kitching AR, Morand EF. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr Mice. J. Immunol. 2006;177:5687–5696. doi: 10.4049/jimmunol.177.8.5687. [DOI] [PubMed] [Google Scholar]
- 27.Huang XR, Chun Hui CW, Chen YX, Wong BC, Fung PC, Metz C, Cho CH, Hui WM, Bucala R, Lam SK, Lan HY. Macrophage migration inhibitory factor is an important mediator in the pathogenesis of gastric inflammation in rats. Gastroenterology. 2001;121:619–630. doi: 10.1053/gast.2001.27205. [DOI] [PubMed] [Google Scholar]
- 28.Kamiyama K, Matsuda N, Yamamoto S, Takano K, Takano Y, Yamazaki H, Kageyama S, Yokoo H, Nagata T, Hatakeyama N, Tsukada K, Hattori Y. Modulation of glucocorticoid receptor expression, inflammation, and cell apoptosis in septic guinea pig lungs using methylprednisolone. Am J Physiol Lung Cell Mol Physiol. 2008;295:L998–L1006. doi: 10.1152/ajplung.00459.2007. [DOI] [PubMed] [Google Scholar]
- 29.Kawahara RS, Deng ZW, Deuel TF. PDGF and the small inducible gene (SIG) family: roles in the inflammatory response. Adv. Exp. Med. Biol. 1991;305:79–87. doi: 10.1007/978-1-4684-6009-4_10. [DOI] [PubMed] [Google Scholar]
- 30.Lacey D, Sampey A, Mitchell RA, Bucala R, Santos L, Leech M, Morand EF. Control of fibroblast-Like synoviocyte proliferation by macrophage migration inhibitory factor. Arthritis Rheum. 2003;48:103–109. doi: 10.1002/art.10733. [DOI] [PubMed] [Google Scholar]
- 31.Lan HY, Yang N, Nikolic-Paterson DJ, Yu XQ, Mu W, Isbel NM, Metz CN, Bucala R, Atkins RC. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. 2000;57:499–509. doi: 10.1046/j.1523-1755.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- 32.Leech M, Huang XR, Morand EF, Holdsworth SR. Endogenous glucocorticoids modulate experimental anti-glomerular basement membrane glomerulonephritis. Clin. Exp. Immunol. 2000;119:161–168. doi: 10.1046/j.1365-2249.2000.01086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leech M, Hutchinson P, Holdsworth SR, Morand EF. Endogenous glucocorticoids modulate neutrophil migration and synovial P-selectin expression but not neutrophil phagocytic or oxidative function in experimental arthritis. Clin. Exp. Immunol. 1998;112:383–388. doi: 10.1046/j.1365-2249.1998.00601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leech M, Metz CN, Hall P, Hutchinson P, Gianis K, Smith M, Weedon H, Holdsworth SR, Bucala R, Morand EF. Macrophage migration inhibitory factor in rheumatoid arthritis: Evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999;42:1601–1608. doi: 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 35.Leech M, Metz CN, Santos L, Peng T, Holdsworth SR, Bucala R, Morand EF. Involvement of macrophage migration inhibitory factor in the evolution of rat adjuvant arthritis. Arthritis Rheum. 1998;41:910–917. doi: 10.1002/1529-0131(199805)41:5<910::AID-ART19>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Whiteman M, Moore PK. Dexamethasone inhibits lipopolysacharide-induced hydrogen sulfide biosynthesis in intact cells and in an animal model of endotoxic shock. J Cell Mol Med. 2008 Dec 16; doi: 10.1111/j.1582-4934.2008.00610.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin X, Sakuragi T, Metz CN, Ojamaa K, Skopicki HA, Wang P, Al-Abed Y, Miller EJ. Macrophage migration inhibitory factor within the alveolar spaces induces changes in the heart during late experimental sepsis. Shock. 2005;24:556–563. doi: 10.1097/01.shk.0000183238.70374.a8. [DOI] [PubMed] [Google Scholar]
- 38.Lister KJ, James WG, Hickey MJ. Immune complexes mediate rapid alterations in microvascular permeability: roles for neutrophils, complement, and platelets. Microcirculation. 2007;14:709–722. doi: 10.1080/10739680701404879. [DOI] [PubMed] [Google Scholar]
- 39.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander W, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Invest. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J. Immunol. 1997;158:5514–5517. [PubMed] [Google Scholar]
- 41.Mitchell RA, Metz C, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF) J. Biol. Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 42.Moore SL, Fewell JE. Mifepristone (RU38486) influences the core temperature response of term pregnant rats to intraperitoneal lipopolysaccharide. Exp Physiol. 2006;91:741–746. doi: 10.1113/expphysiol.2006.033688. [DOI] [PubMed] [Google Scholar]
- 43.Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006;5:399–410. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 44.Morand EF, Leech M, Weedon H, Metz C, Bucala R, Smith MD. Macrophage migration inhibitory factor in rheumatoid arthritis: clinical correlations. Rheumatology (Oxford) 2002;41:558–562. doi: 10.1093/rheumatology/41.5.558. [DOI] [PubMed] [Google Scholar]
- 45.Mori N, Horie Y, Gerritsen ME, Anderson DC, Granger DN. Anti-inflammatory drugs and endothelial cell adhesion molecule expression in murine vascular beds. Gut. 1999;44:186–195. doi: 10.1136/gut.44.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mukaida N, Zachariae CCO, Gusella GL, Matsushima K. Dexamethasone inhibits the induction of monocyte chemotactic-activating factor production by IL-1 or tumor necrosis factor. J. Immunol. 1991;146:1212–1215. [PubMed] [Google Scholar]
- 47.Neeck G, Kluter A, Dotzlaw H, Eggert M. Involvement of the glucocorticoid receptor in the pathogenesis of rheumatoid arthritis. Ann N Y Acad Sci. 2002;966:491–495. doi: 10.1111/j.1749-6632.2002.tb04252.x. [DOI] [PubMed] [Google Scholar]
- 48.Norman MU, Lister KJ, Yang YH, Issekutz A, Hickey MJ. TNF regulates leukocyte-endothelial cell interactions and microvascular dysfunction during immune complex-mediated inflammation. Br. J. Pharmacol. 2005;144:265–274. doi: 10.1038/sj.bjp.0706081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norman MU, Van De Velde N, Timoshanko JR, Issekutz A, Hickey MJ. Overlapping roles of endothelial selectins and VCAM-1 in immune complex-induced leukocyte recruitment in the cremasteric microvasculature. Am. J. Pathol. 2003;163:1491–1503. doi: 10.1016/S0002-9440(10)63506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Onodera S, Tanji H, Suzuki K, Kaneda K, Mizue YU, Sagawa A, Nishihira J. High expression of macrophage migration inhibitory factor in the synovial tissues of rheumatism Joints. Cytokine. 1998;11:163–167. doi: 10.1006/cyto.1998.0402. [DOI] [PubMed] [Google Scholar]
- 51.Pan J, Sukhova GK, Yang J, Wang B, Xie T, Fu H, Zhang Y, Satoskar AR, David J, Metz C, Bucala R, Fang K, Simon DI, Chapman HA, Libby P, Shi G. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109:3149–3153. doi: 10.1161/01.CIR.0000134704.84454.D2. [DOI] [PubMed] [Google Scholar]
- 52.Peeters BW, Ruigt GS, Craighead M, Kitchener P. Differential effects of the new glucocorticoid receptor antagonist ORG 34517 and RU486 (mifepristone) on glucocorticoid receptor nuclear translocation in the AtT20 cell line. Ann N Y Acad Sci. 2008;1148:536–541. doi: 10.1196/annals.1410.072. [DOI] [PubMed] [Google Scholar]
- 53.Powell ND, Papenfuss TL, McClain MA, Gienapp IE, Shawler TM, Satoskar AR, Whitacre CC. Cutting Edge: Macrophage migration inhibitory factor is necessary for progression of experimental autoimmune encephalomyelitis. J. Immunol. 2005;175:5611–5614. doi: 10.4049/jimmunol.175.9.5611. [DOI] [PubMed] [Google Scholar]
- 54.Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene transcription. J. Leukoc. Biol. 1999;66:876–888. doi: 10.1002/jlb.66.6.876. [DOI] [PubMed] [Google Scholar]
- 55.Roger T, Chanson A, Knaup-Reymond M, Calandra T. Macrophage migration inhibitory factor promotes innate immune responses by supressing glucocorticoid-induced expression of mitogen-activated protein kinase phosphatase-1. Eur. J. Immunol. 2005;35:3405–3413. doi: 10.1002/eji.200535413. [DOI] [PubMed] [Google Scholar]
- 56.Rovai LE, Herschman HR, Smith JB. The murine neutrophil-chemoattractant chemokines LIX, KC, and MIP-2 have distinct induction kinetics, tissue distributions, and tissue-specific sensitivities to glucocorticoid regulation in endotoxemia. J Leukoc Biol. 1998;64:494–502. doi: 10.1002/jlb.64.4.494. [DOI] [PubMed] [Google Scholar]
- 57.Santos L, Hall C, Metz CN, Bucala R, Morand EF. Role of macrophage migration inhibitory factor (MIF) in murine antigen-induced arthritis: interactions with glucocorticoids. Clin. Exp. Immunol. 2001;123:309–314. doi: 10.1046/j.1365-2249.2001.01423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santos L, Lacey D, Yang Y, Leech M, Morand EF. Activation of synovial cell p38 MAP kinase by macrophage migration inhibitory factor. J. Rheumatol. 2004;31:1038–1043. [PubMed] [Google Scholar]
- 59.Schramm R, Schaefer T, Menger MD, Thorlacius H. Acute mast cell-dependent neutrophil recruitment in the skin is mediated by KC and LFA-1: inhibitory mechanisms of dexamethasone. J Leukoc Biol. 2002;72:1122–1132. [PubMed] [Google Scholar]
- 60.Suzuki H, Zweifach BW, Forrest MJ, Schmid-Schonbein GW. Modification of leukocyte adhesion in spontaneously hypertensive rats by adrenal corticosteroids. J. Leukoc. Biol. 1995;57:20–26. [PubMed] [Google Scholar]
- 61.Tailor A, Tomlinson A, Salas A, Panes J, Granger DN, Flower RJ, Perretti M. Dexamethasone inhibition of leucocyte adhesion to rat mesenteric postcapillary venules: role of intercellular adhesion molecule 1 and KC. Gut. 1999;45:705–712. doi: 10.1136/gut.45.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tessier P, Cattaruzzi P, McColl SR. Inhibition of lymphocyte adhesion to cytokine-activated synovial fibroblasts by glucocorticoids involves the attenuation of vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 gene expression. Arthritis Rheum. 1996;39:226–234. doi: 10.1002/art.1780390208. [DOI] [PubMed] [Google Scholar]
- 63.Wang F, Gao F, Jing L. Is macrophage migration inhibitory factor (MIF) the "control point" of vascular hypo-responsiveness in septic shock? Med Hypotheses. 2005;65:1082–1087. doi: 10.1016/j.mehy.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 64.Weber C, Kraemer S, Dreschler M, Lue H, Koenen RR, Kapurniotu A, Zernecke A, Bernhagen J. Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16278–16283. doi: 10.1073/pnas.0804017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weber KS, Draude G, Erl W, de Martin R, Weber C. Monocyte arrest and transmigration on inflamed endothelium in shear flow is inhibited by adenovirus-mediated gene transfer of IκB-α. Blood. 1999;93:3685–3693. [PubMed] [Google Scholar]
- 66.Willis MS, Carlson DL, DiMaio JM, White MD, White DJ, Adams GA, Horton JW, Giroir BP. Macrophage migration inhibitory factor mediates late cardiac dysfunction after burn injury. Am. J. Physiol. 2004;288:H795–H804. doi: 10.1152/ajpheart.00189.2004. [DOI] [PubMed] [Google Scholar]
- 67.Zentay Z, Sharaf M, Qadir M, Drafta D, Davidson D. Mechanism for dexamethasone inhibition of neutrophil migration upon exposure to lipopolysaccharide in vitro: role of neutrophil interleukin-8 release. Pediatr. Res. 1999;46:406–410. doi: 10.1203/00006450-199910000-00008. [DOI] [PubMed] [Google Scholar]