

Abstract
We suggest a possible mechanism of how 8-aminoquinolines (8-AQ’s) causes hemotoxicity by oxidizing hemoglobin to methemoglobin. In our DFT calculations, we found that 5-hydroxyprimaquine is able to donate an electron to O2 to facilitate its conversion to H2O2. Meanwhile, Fe(II) is oxidized to Fe(III) and methemoglobin is formed. In this mechanism, the 8-AQ drug plays a similar role as that of H4B in nitric oxide synthase. Furthermore, our study offers an approach to inform the design of less toxic antimalarial drugs.
There is a great need for new antimalarial drugs that can radically cure the liver stage of malaria.1,2 At present the only FDA-approved drug which can kill liver hypnozoites is primaquine3 (1) (Figure 1), a member of the 8-aminoquinoline (8-AQ) family. In vivo, primaquine is believed to form a number of metabolites, such as carboxyprimaquine4 (2), 5-hydroxyprimaquine5 (5-HPQ, 3), and 6-methoxy-8-aminoquinoline6 (4) (Figure 1). A major concern regarding primaquine and its metabolites is that they can cause life-threatening hemolysis in G6PD-deficient patients.7 The hemolysis may be related to the propensity of 8-AQ metabolites to oxidize hemoglobin to methemoglobin, an Fe(III) protein which is unable to carry oxygen.8 The reaction also results in the formation of reactive oxygen species (ROS), such as hydrogen peroxide. Since primaquine was first synthesized in 1946,9 numerous efforts have been made to reduce the toxicity of 8-AQ drugs.10 Unfortunately, our knowledge of such toxicity has been limited by a lack of understanding of its chemical mechanism.4,11
Figure 1.
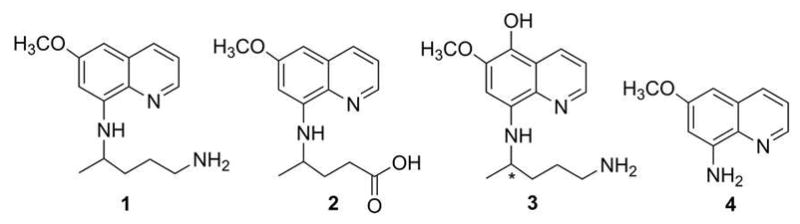
A schematic illustration of primaquine and metabolites.
In this communication, we suggest a possible mechanism for the methemoglobinemia of 8-AQ drugs. Considering the process of converting O2 to H2O2, it is apparent that in addition to two protons, two electrons must be supplied to the π* orbital of O2. The question is: where do the two electrons come from? Obviously, iron is able to provide one electron with Fe(II) being oxidized to Fe(III), which results in the conversion of hemoglobin to methemoglobin. The source of the second electron, however, is not clear. In this work we hypothesize that an 8-AQ metabolite may provide this second electron, itself being converted into a radical cation. Interestingly, if this were true, the 8-AQ metabolite then plays a similar role to that of tetrahydrobiopterin (H4B) in nitric oxide synthase (NOS). NOS is an iron-heme containing enzyme that catalyzes the formation of nitric oxide from L-arginine. It has been found that NOS needs the cofactor H4B during catalysis to transfer an electron to the Fe–O2 moiety.12 In particular, Shaik and coworkers13 studied the active site protonation states of NOS using QM/MM methods and found that when O2 is doubly protonated to form the Fe–H2O2 complex, H4B is converted to a radical cation and transfers an electron to aid the formation of H2O2. Similarly, when complexed with hemoglobin, an 8-AQ or an 8-AQ metabolite could assist the formation of H2O2 analogously as does H4B in NOS.
To test this hypothesis, we performed a density functional theory (DFT) study on unprotonated, singly protonated and doubly protonated hemoglobin–8-AQ complexes. 5-HPQ (3) was chosen for this study because it is known to cause methemoglobinemia directly and is able to form H2O2.14 By contrast, primaquine itself appears to require metabolic activation to elicit methemoglobin formation.10 The terminal amine of the 5-HPQ’s 8-amino alkyl chain was assumed to be protonated to match physiological pH and the asymmetric carbon alpha to the 8-amine (see Figure 1) was chosen in the S form. The initial 5-HPQ–hemoglobin complex was obtained by docking 5-HPQ into hemoglobin using the Glide program.15 The crystal structure (PDB: 2D5X),16 in which a ligand 2-[4-(3,5-dichlorophenylureido) phenoxy]-2-methylpropionic acid makes a hydrogen bond with the propionate group of heme, was used for the docking. Detailed docking procedures are provided in the Supporting Information. The docked structure showed an interaction of the terminal 8-amino –NH3+ group of 5-HPQ with the carboxylic side chain of heme (Figure 2a), most likely due to the formation of a strong electrostatic interaction. On the basis of this result, a chemical model to be used for DFT calculations was derived (Figure 2b). All DFT calculations were performed using the spin unrestricted approach. Geometries were optimized using the Jaguar program17 at the B3LYP18 level with the LACVP* basis set. Relative energies were obtained at those geometries using Jaguar’s Poisson-Boltzmann self-consistent reaction field method with a dielectric constant (ε) of 4.0 at the B3LYP/LACV3P** level of theory. The above computational approach to study metalloenzymes has recently been applied to similar systems and reviewed in depth.19
Figure 2.

(a) The interaction of 5-HPQ with hemoglobin from docking into 2D5X. For clarity, only 5-HPQ and heme are shown. The sidechain terminal amine was found to be in close proximity to the heme carboxyl moiety, whereas the alkyl group at the 8-position and the quinoline ring are more than 3 Å away from the heme and form hydrophobic interactions with Leu86 and Leu83. (b) The chemical model used for DFT calculations.
Figure 3 shows the optimized structures of unprotonated, singly protonated, and doubly protonated O2–hemoglobin–5-HPQ complexes. Upon optimization, the terminal –NH3+ group of 5-HPQ in all the protonation states transfers a proton to the carboxylic group of heme, forming a –H2N···HOOC– hydrogen bond. For the unprotonated complex, the lowest energy structure is in the singlet spin state (Table 1). In this spin state, an electron is transferred from iron to one of the π* orbitals of O2 to form FeIII -O2•−. The O2 moiety shows superoxide character (electron configuration σx2 πx2 πy2 πx*2 πy*1 σx*0) with the O–O bond length at 1.27 Å. Thus, the O2 is in the one-electron reduced state. This electronic assignment is in agreement with computational studies on similar systems.20–24 It should be noted that in all the possible spin states, the spin density on 5-HPQ is 0.00, suggesting that no electron is transferred from 5-HPQ to O2.
Figure 3.
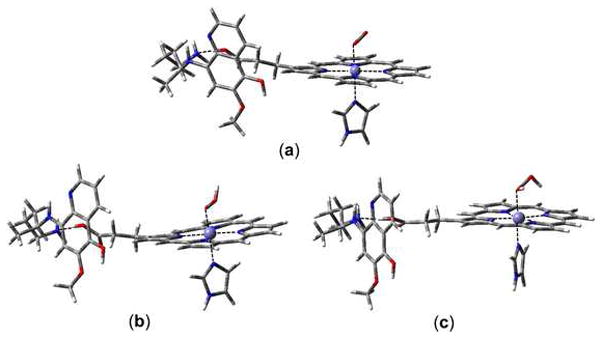
The optimized structures of (a) unprotonated, (b) singly protonated and (c) doubly protonated O2 hemoglobin–5-HPQ complexes in the lowest energy spin states.
Table 1.
Relative Energies (kJ mol−1) and Spin Densities for the Unprotonated (O2), Singly Protonated (OOH) and Doubly Protonated (H2O2) Species in all the Possible Spin Multiplicities.
| Structure | M | ΔE | Spin Density |
||||
|---|---|---|---|---|---|---|---|
| Fe | Oinner | Oouter | heme | 5-HPQ | |||
| O2 | 1 | 0.0 | 1.37 | −0.56 | −0.73 | −0.08 | 0.00 |
| 3 | 16.3 | 1.10 | 0.33 | 0.62 | −0.04 | 0.00 | |
| 5 | 32.3 | 2.14 | 0.94 | 0.97 | −0.08 | 0.00 | |
| 7 | 17.5 | 3.80 | 1.00 | 0.99 | 0.17 | 0.00 | |
| OOH | 1 | 0.3 | 0.84 | 0.19 | 0.02 | −0.42 | −0.63 |
| 3 | 0.0 | 0.85 | 0.19 | 0.03 | 0.32 | 0.64 | |
| 5 | 62.1 | 3.06 | −0.16 | −0.07 | 0.62 | 0.59 | |
| 7 | 31.4 | 4.13 | 0.40 | 0.09 | 0.75 | 0.61 | |
| HOOH | 1 | 11.8 | 1.05 | 0.00 | 0.00 | −0.03 | −1.01 |
| 3 | 0.0 | 1.06 | 0.00 | 0.00 | −0.04 | 1.01 | |
| 5 | 38.7 | 3.03 | 0.02 | 0.00 | −0.12 | 1.01 | |
| 7 | 10.2 | 4.26 | 0.04 | 0.00 | 0.59 | 1.01 | |
In the singly protonated complex, the lowest energy structure is in the triplet spin state. The singlet spin state, however, lies only marginally higher in energy by 0.3 kJ mol−1. In the NOS system complexed with OOH, the singlet and triplet states were similarly found to be very close in energy.13 The affinity of the unprotonated complex to accept one proton was calculated to be 1133.5 kJ mol−1 using the free energies25 of the unprotonated and singly protonated complexes in the ground state. In the triplet state, iron is still in the Fe(III) state. The O–O bond is further lengthened to 1.44 Å, indicating that it is essentially a single bond. Thus, an electron has been promoted to the other π* orbital of O2•− to convert it to O22− with electron configuration σx2πx2 πy 2πx*2 πy *2σx*0. Hence, the O2 is now in the two-electron reduced state. From the calculated spin distribution (Table 1), it can be concluded that this electron is partially supplied by each of heme and 5-HPQ, with 5-HPQ contributing ~0.64 electrons. In the triplet, quintet and septet states, the unpaired electron shared between heme and 5-HPQ aligns ferromagnetically with that in iron, while in the single state, they align antiferromagnetically.
In the doubly protonated complex, the lowest energy structure is also in the triplet state. The affinity of the singly protonated complex to accept an additional proton was calculated to be 1126.6 kJ mol−1 using the free energies25 of the singly protonated and doubly protonated complexes in the ground state. The proton affinity of H2O was calculated to be 997.2 kJ mol−1 at the same level. The affinities of O2 to accept protons are larger than that of H2O, suggesting that the hemoglobin ···5-HPQ bound O2 is more basic than H2O and should be able to be protonated by H3O+. In the triplet spin state of the doubly protonated complex, iron remains in the Fe(III) state. The O–O bond lengthens slightly further to 1.45 Å and the spin densities on both oxygen atoms are 0.00. Thus, as for the singly protonated complex, O–O is closed-shell and single bonded with electron configuration σx2πx2πy2πx*2 πy*2 σx*0. However, in addition to the electron donated by iron, the second electron transferred to the π* orbital of O2 is now purely provided by 5-HPQ, which is converted to a radical cation, as shown from its calculated spin density of 1.01.
In a separate set of gas phase calculations, we fixed the lengths of the two terminal N–H bonds to prevent the proton transfer to the carboxylic acid side chain of the heme. This restriction to the –H3N+···−OOC– interaction, however, did not affect the conclusions obtained (see Supporting Information). In addition, to take into account that the binding site is located at the protein surface, single point calculations were performed on selected structures with ε of 80.37 (representing water), but in this case also similar results were obtained (Supporting Information).
These results show that 5-HPQ indeed plays a similar role for hemoglobin as H4B does for NOS. It is, in fact, an even better electron donor than H4B. In Shaik et al.’s study13 on NOS, the process of electron transfer from H4B was only observed when O2 is doubly protonated. In contrast, when O2 is singly protonated in the hemoglobin–5-HPQ complex, 5-HPQ donates nearly two thirds of an electron to O2. This may be explained in part by the fact that 5-HPQ has a lower ionization energy than H4B. The gas phase calculation at the B3LYP/6-311+G(2df,p)//B3LYP/6-31G(d,p) level shows that H4B has an ionization energy of 593.9 kJ mol−1. However, that of 5-HPQ is 13.2 kJ mol−1 lower in energy at 580.7 kJ mol−1. Thus, 5-HPQ is more capable of donating an electron than H4B.
Furthermore, it should be noted that the binding pocket is located at the surface of hemoglobin. In addition to the –H2N···HOOC– hydrogen bond, the ligand interacts hydrophobically with the amino acid residues of the protein. The relatively non-specific hydrophobic interactions suggest that the proposed chemical mechanism may be general to the class of aromatic compounds that can form a hydrogen bond with the carboxylic arm of heme via an amine or a hydroxyl group. Indeed, certain small compounds such as aniline and its metabolites are also able to catalyze the formation of methemoglobin in vivo.26 These processes may follow a similar mechanism to that discussed in this communication.
In the present work, we suggest a possible explanation of the methemoglobinemia caused by 8-AQ drugs. Both iron and 8-AQ donate an electron to the π* orbital of O2. This thus facilitates the formation of H2O2. In the meantime, Fe(II) is converted to Fe(III). Therefore, methemoglobin, which cannot bind O2, is formed. Notably, in this mechanism 8-AQ plays a similar role as that of H4B in NOS. In principle, modification of the exocyclic substituents of 8-AQ’s will affect their electron donating ability, which suggests a rational plan for discovery of new, less toxic 8-AQ drugs.
Supplementary Material
Acknowledgments
We thank the US Department of Defense for financial support through award #W81XWH0720095; LAW and NPDN are partially supported by a U.S. Department of Agriculture, Agriculture Research Service, Cooperative Agreement # 58-6408-2-0009. Computer time and resources from the Department of Medicinal Chemistry and the Mississippi Center for Supercomputer Research are greatly appreciated. This investigation was conducted in part in a facility constructed with support from Research Facilities Improvements Program (C06 RR-14503-01) from the NIH National Center for Research Resources.
Footnotes
Supporting Information Available: The detailed docking procedures and the top 10 binding poses generated by the Glide SP method, the effects of proton transfer and more polarized environment, and optimized xyz coordinates of all structures considered in this study. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wells TNC, Burrows JN, Baird JK. Trends in Parasitol. 2010;26:145–151. doi: 10.1016/j.pt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 2.White NJ. Malaria J. 2008;7:6. doi: 10.1186/1475-2875-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird JK, Hoffman SL. Clin Infect Dis. 2004;39:1336–1345. doi: 10.1086/424663. [DOI] [PubMed] [Google Scholar]
- 4.Vale N, Moreira R, Gomes P. Eur J Med Chem. 2009;44:937–953. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Bowman ZS, Oatis JE, Whelan JL, Jollow DJ, McMillan DC. J Pharmacol Exp Ther. 2004;309:79–85. doi: 10.1124/jpet.103.062984. [DOI] [PubMed] [Google Scholar]
- 6.Bolchoz LJC, Gelasco AK, Jollow DJ, McMillan DC. J Pharmacol Exp Ther. 2002;303:1121–1129. doi: 10.1124/jpet.102.041459. [DOI] [PubMed] [Google Scholar]
- 7.Beutler E, Duparc S. Am J Trop Med Hyg. 2007;77:779–789. [PubMed] [Google Scholar]
- 8.Ganesan S, Tekwani BL, Sahu R, Tripathi LM, Walker LA. Toxicol Appl Pharmacol. 2009;241:14–22. doi: 10.1016/j.taap.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Elderfield RC, Gensler WJ, Head JD, Hageman HA, Kremer CB, Wright JB, Holley AD, Williamson B, Galbreath J, Wiederhold L, III, Frohardt R, Kupchan SM, Williamson TA, Birstein O. J Am Chem Soc. 1946;68:1524–1529. [Google Scholar]
- 10.(a) Carroll FI, Berrang B, Linn CP, Twine CE. J Med Chem. 1979;22:694–699. doi: 10.1021/jm00192a016. [DOI] [PubMed] [Google Scholar]; (b) Nanayakkara NPD, Ager AL, Jr, Bartlett MS, Yardley V, Croft SL, Khan IA, McChesney JD, Walker LA. Antimicrob Agents Chemother. 2008;52:2130–2137. doi: 10.1128/AAC.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pradines B, Mamfoumbi MM, Tall A, Sokhna C, Koeck JL, Fusai T, Mosnier J, Czarnecki E, Spiegel A, Trape JF, Kombila M, Rogier C. Antimicrob Agents Chemother. 2006;50:3225–3226. doi: 10.1128/AAC.00777-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jain M, Vangapandu S, Sachdeva S, Singh S, Singh PP, Jena GB, Tikoo K, Ramarao P, Kaul CL, Jain R. J Med Chem. 2003;47:285–287. doi: 10.1021/jm0304562. [DOI] [PubMed] [Google Scholar]
- 11.Kouznetsov VV, Gómez-Barrio A. Eur J Med Chem. 2009;44:3091–3113. doi: 10.1016/j.ejmech.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hurshman AR, Krebs C, Edmondson DE, Huynh BH, Marletta MA. Biochemistry. 1999;38:15689–15696. doi: 10.1021/bi992026c. [DOI] [PubMed] [Google Scholar]; (b) Bec N, Gorren AFC, Mayer B, Schmidt PP, Andersson KK, Lange RJ. Inorg Biochem. 2000;81:207–211. doi: 10.1016/s0162-0134(00)00104-5. [DOI] [PubMed] [Google Scholar]; (c) Hurshman AR, Marletta MA. Biochemistry. 2002;41:3439–3456. doi: 10.1021/bi012002h. [DOI] [PubMed] [Google Scholar]; (d) Wei CC, Wang ZQ, Wang Q, Meade AL, Hemann C, Hille R, Stuehr DJ. J Biol Chem. 2001;276:315–319. doi: 10.1074/jbc.M008441200. [DOI] [PubMed] [Google Scholar]; (e) Wei CC, Wang ZQ, Hemann C, Hille R, Stuehr DJ. J Biol Chem. 2003;278:46668–46673. doi: 10.1074/jbc.M307682200. [DOI] [PubMed] [Google Scholar]; (f) Cho KB, Carvajal MA, Shaik S. J Phys Chem B. 2008;113:336–346. doi: 10.1021/jp8073199. [DOI] [PubMed] [Google Scholar]
- 13.Cho KB, Derat E, Shaik S. J Am Chem Soc. 2007;129:3182–3188. doi: 10.1021/ja066662r. [DOI] [PubMed] [Google Scholar]
- 14.Fletcher KA, Barton PF, Kelly JA. Biochem Pharmacol. 1988;37:2683–2690. doi: 10.1016/0006-2952(88)90263-8. [DOI] [PubMed] [Google Scholar]
- 15.Glide. Schrodinger, LLC; New York, NY: 2009. [Google Scholar]
- 16.Yokoyama T, Neya S, Tsuneshige A, Yonetani T, Park S-Y, Tame JRH. J Mol Biol. 2006;356:790–801. doi: 10.1016/j.jmb.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 17.Jaguar. Schrodinger, LLC; New York, NY: 2009. [Google Scholar]
- 18.(a) Becke AD. J Chem Phys. 1993;98:1372–1377. [Google Scholar]; (b) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (c) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 19.(a) Noodleman L, Lovell T, Han WG, Li J, Himo F. Chem Rev. 2004;104:459–508. doi: 10.1021/cr020625a. [DOI] [PubMed] [Google Scholar]; (b) Siegbahn PEM, Borowski T. Acc Chem Res. 2006;39:729–738. doi: 10.1021/ar050123u. [DOI] [PubMed] [Google Scholar]; (c) Siegbahn PEM, Himo F. J Biol Inorg Chem. 2009;14:643–651. doi: 10.1007/s00775-009-0511-y. [DOI] [PubMed] [Google Scholar]; (d) Himo F. Theor Chem Acc. 2006;116:232–240. [Google Scholar]
- 20.Chen H, Ikeda-Saito M, Shaik S. J Am Chem Soc. 2008;130:14778–14790. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]
- 21.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 22.Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem Rev. 2009;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 23.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 24.Unno M, Chen H, Kusama S, Shaik S, Ikeda-Saito M. J Am Chem Soc. 2007;129:13394–13395. doi: 10.1021/ja076108x. [DOI] [PubMed] [Google Scholar]
- 25.Free energy corrections were obtained by performing frequency calculations on the ground states using the Gaussian 09 program.
- 26.(a) Harrison JH, Jollow D. J Mol Pharmacol. 1987;32:423–431. [PubMed] [Google Scholar]; (b) Singh H, Purnell ET. J Environ Pathol Toxicol Oncol. 2005;24:57–65. doi: 10.1615/jenvpathtoxoncol.v24.i1.60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.