

Abstract
Silylene transfer to α,β,γ,δ-unsaturated carbonyl compounds produced oxasilacyclopentenes that underwent thermal additions to aldehydes to produce trans-dioxasilacyclononenes as single stereoisomers. This reaction, which converts the δ-position the unsaturated carbonyl compound into a nucleophilic center, represents an inversion of polarity from the normal pattern of reactivity. The stereospecificity of the reaction suggests that the addition to aldehydes occurred through a closed, chair-like six-membered transition state. This reaction can be used to prepare enantiomerically pure materials by the use of chiral auxiliaries to control the formation of the oxasilacyclopentenes. Functionalization of the resulting trans-cycloalkene occurred with complete stereoselectively.
The different positions of unsaturated carbonyl compounds exhibit predictable patterns of reactivity. While the α- and γ-positions are donor sites, the β- and δ-positions are acceptors.1 For example, the aldol reaction, in which the α-position is nucleophilic, is a common transformation,2 and the vinylogous aldol reaction uses the γ-position as a nucleophile.3 Conjugate addition reactions capitalize on the electrophilicity of the β- and δ-positions.4 The polarity of these positions can be reversed in some cases. For example, formal homoaldol reactions employ the β-position as a nucleophilic site.5 Umpolung reactivity where the δ-position is nucleophilic, on the other hand, is uncommon.6
In this communication, we present a method for addition of aldehydes to dienoates at the δ-carbon. Silylene transfer to a dienoate forms a vinyl oxasilacyclopentene in which the δ-carbon becomes the nucleophilic site. These intermediates undergo nucleophilic addition to aldehydes, forming trans-dioxasilacyclononenes stereoselectively and stereospecifically.
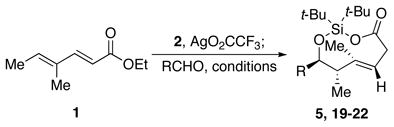
The one-flask conversion of dienoate 1 and benzaldehyde to the protected adduct 5 illustrates this transformation. Silver-catalyzed silylene transfer7 to dienoate 1 afforded vinyl oxasilacyclopentene 3 cleanly. Heating strained8 vinyl oxasilacyclopentene 3 with benzaldehyde produced the dienol ether 4 as a single diastereomer. Filtration through silica gel hydrolyzed the silyl ketene acetal to provide the corresponding trans-dioxasilacyclononene 5 as one diastereomer.9
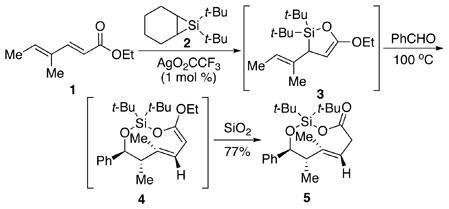 |
(1) |
The anti configuration of the addition product 4 is likely established through a Zimmerman-Traxler-like10 transition state in which the aldehyde is activated by coordination to the silicon center (A, Figure 1). Although E-allylic silanes typically react with aldehydes in the presence of Lewis acids through open transition states to give syn products,11,12 allylic silanes can react through closed transition states if the silicon atom is particularly Lewis acidic.8,13 Three facts support the closed transition state for the formation of adduct 4: (1) an external Lewis acid was not required to activate the addition of silane 3 to an aldehyde; (2) the E-allylic silane gave the anti product, not the syn product; and (3) no Mukaiyama14 α-aldol products were formed by reaction of the more nucleophilic silyl ketene acetal moiety.15
Figure 1.

The stereospecificity of the addition reaction also indicates that it proceeds through a closed transition state.16 The product obtained from the Z-dienoate 6 was the syn isomer of the trans-dioxasilacyclononene (7, eq 2).9 The relative configuration of trans-dioxasilacyclononene 7 is also consistent with its formation through a closed, chair-like transition state. 11,12
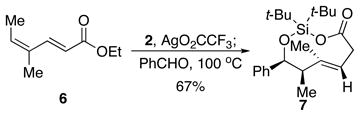 |
(2) |
The trans-cyclononene products can be formed with control of absolute stereochemistry. The chiral auxiliary of dienimide 8 controlled the silylene transfer reaction, resulting in stereoselective formation of intermediate 9 (Scheme 1). Heating this silane with benzaldehyde promoted diastereoselective formation of trans-dioxasilacyclononadiene 10.17 Treatment of diene 10 with acid under biphasic conditions removed the chiral auxiliary, providing enantioenriched trans-dioxasilacyclononene (−)-5.
Scheme 1.

Asymmetric Formation of trans-Dioxasilacyclononene
The addition of aldehydes at the δ-position of dienoates is general for a number of unsaturated esters. Reaction times, however, depend upon the nucleophilicity of the allylic silane formed after silylene transfer. Substrates that possessed a methyl substituent at the γ-position (Table 1, entries 1, 2, and 4) produced methallyl silanes that underwent faster addition relative to substrates that only had a hydrogen atom at that position.15 The longer reaction times of the substrates that only had a hydrogen atom at the γ-position led to more decomposition products and lower yields.
Table 1.
Dienoate Scope in Formation of trans-Dioxasilacyclononenes
| entry | Dienoate | R1 | R2 | R3 | Conditions | Product | Yield |
|---|---|---|---|---|---|---|---|
| 1 | 1 | H | Me | Me | 100 °C, 18 h | 5 | 77% |
| 2 | 6 | Me | H | Me | 100 °C, 18 h | 7 | 67% |
| 3 | 11 | H | Me | H | 100 °C, 5 d | 15 | 59% |
| 4 | 12 | H | H | Me | 100 °C, 2 d | 16 | 53% |
| 5 | 13 | Me | Me | H | 100 °C, 10 d | 17 | 38% |
| 6 | 14 | H | H | H | 100 °C, 5 d | 18 | 37% |
Products were formed as one isomer as detected by 1H NMR spectroscopy and GCMS. Yields reported are isolated yields.
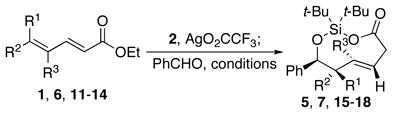 |
(3) |
A number of aldehydes participated in the addition reaction. Reactions of sterically hindered aldehydes required longer reaction times (Table 2, entries 3 and 4). A Lewis acid catalyzed the allylation, leading to faster reactions, even at room temperature (entry 4). The relative stereochemistry of the products formed by the Lewis acid-catalyzed process was again consistent with a closed, chair-like transition state. The Lewis acid likely coordinated to the oxygen atom of the O–Si bond of the vinyl oxasilacyclopentene 3, increasing the electrophilicity of the silicon center.18
Table 2.
Aldehyde Scope
| entry | RCHO | Conditions, yield | Product | |
|---|---|---|---|---|
| Thermal | SnBr4 (10 mol %), rt | |||
| 1 | PhCHO | 100 °C, 18 h, 77% | 3 h, 37% | 5 |
| 2 | n-BuCHO | 100 °C, 18 h, 72% | 3 h, 37% | 19 |
| 3 | i-PrCHO | 130 °C, 3 d, 73% | 18 h, 40% | 20 |
| 4 | 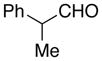 |
130 °C, 5 d, 72% | 18 h, 94% | 21 |
| 5 | 100 °C, 18 h, 80% | decomposition | 22 | |
Products were formed as one isomer as detected by 1H NMR spectroscopy and GCMS. Yields reported are isolated yields.
 |
(4) |
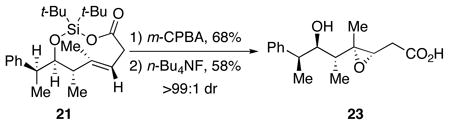
Stereochemically homogeneous products can be made by kinetic resolution. Addition to chiral aldehyde (Table 2, entry 4) produced the adduct as a single diastereomer.17 The relative stereochemistry of trans-dioxasilacyclononene 21 is consistent with a closed, chair-like transition state where the vinyl oxasilacyclopentene approached the chiral aldehyde on a Felkin trajectory.19 This result suggests that the use of chiral, non-racemic aldehydes would give enantioenriched products.
Because substituted trans-cyclononenes adopt specific conformations and are slow to isomerize,20 functionalization of the carbon–carbon double bond only occurred on the outside face. Treatment of trans-dioxasilacyclononene 21 with m-CPBA followed by deprotection afforded epoxide 23 as a single diastereomer. This selectivity is noteworthy because epoxidations of acyclic alkenes with m-CPBA in which the faces of the alkene are only differentiated by A1,2 strain are generally not diastereoselective.21 In addition, hydroxyl- directed epoxidation of free alcohols with structures analogous to cyclononene 21 would give epoxides with the opposite relative configuration compared to epoxide 23.21
 |
(5) |
In summary, silylene transfer to dienoates afforded intermediates that function as δ-enolate equivalents that react with aldehydes to form addition products stereoselectively and stereospecifically. Enantiopure products can be synthesized by employing a chiral auxiliary to control silylene transfer. Further functionalization of the trans-cycloalkene occurred diastereoselectively. This methodology has potential application for the synthesis of polypropionate natural products and related structures.22
Supplementary Material
Acknowledgments
This project was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM-54909). C. C. V. thanks the NIGMS for a predoctoral fellowship (F31GM080157). K. A. W. thanks Amgen and Lilly for awards to support research. We thank Dr. Phil Dennison (UCI) for assistance with NMR spectroscopy, Dr. Joseph W. Ziller (UCI) for X-ray crystallography, and Dr. John Greaves and Ms. Shirin Sorooshian (UCI) for mass spectrometry.
Footnotes
Supporting Information Available: Experimental procedures; spectroscopic, analytical, and X-ray data for the products (PDF,CIF). This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Seebach D. Angew Chem, Int Ed Engl. 1979;18:239–258. [Google Scholar]
- 2.Evans DA, Nelson JV, Taber TR. Top Stereochem. 1982;13:1–115. [Google Scholar]
- 3.Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem Rev. 200;100:1929–1972. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]
- 4.For a recent review of 1,4-additions, see: Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M. Chem Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515.For references of 1,6-additions to enoates, see: Fukuhara K, Urabe H. Tetrahedron Lett. 2005;46:603–606.and references therein
- 5.Nair V, Vellalath S, Babu BP. Chem Soc Rev. 2008;37:2691–2698. doi: 10.1039/b719083m. [DOI] [PubMed] [Google Scholar]
- 6.The dissolving metal reduction of a dienone resulted in protonation at the δ-position: Grimm K, Venkataramani PS, Reusch W. J Am Chem Soc. 1971;93:270–271.Crotylsilanes bearing carbonyl groups also give products that resemble electrophilic addition at the δ-position: Jain NF, Takenaka N, Panek JS. J Am Chem Soc. 1996;118:12475–12476.
- 7.Calad SA, Woerpel KA. J Am Chem Soc. 2005;127:2046–2047. doi: 10.1021/ja042893r. [DOI] [PubMed] [Google Scholar]
- 8.Kinnaird JWA, Ng PY, Kubota K, Wang X, Leighton JL. J Am Chem Soc. 2002;124:7920–7921. doi: 10.1021/ja0264908. [DOI] [PubMed] [Google Scholar]
- 9.The relative configuration of cyclononenes 5 and 7 was determined by nOe correlations.
- 10.Zimmerman HE, Traxler MD. J Am Chem Soc. 1957;79:1920–1923. [Google Scholar]
- 11.For reviews see: Masse CE, Panek JS. Chem Rev. 1995;95:1293–1316.Denmark SE, Almstead NG. J Mex Chem Soc. 2009;53:174–192.
- 12.Denmark SE, Almstead NG. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; Weinheim: 2000. pp. 299–402. [Google Scholar]
- 13.For different methods for activation of silicon centers, see: Matsumoto K, Oshima K, Utimoto K. J Org Chem. 1994;59:7152–7155.Prévost M, Woerpel KA. J Am Chem Soc. 2009;131:14182–14183. doi: 10.1021/ja906204a.Kobayashi S, Nishio K. J Org Chem. 1994;59:6620–6628.Sakurai H. Synlett. 1989:1–8.Shibato A, Itagaki Y, Tayama E, Hokke Y, Asao N, Maruoka K. Tetrahedron. 2000;56:5373–5382.Fürstner A, Voigtländer D. Synthesis. 2000;7:959–969.
- 14.Mukaiyama T, Banno K, Narasaka K. J Am Chem Soc. 1974;96:7503–7509. [Google Scholar]
- 15.Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66–77. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 16.Mukaiyama T. In: Organic Reactions. Dauben WG, editor. Vol. 28. Wiley-VCH: Weinheim; 1982. pp. 203–331. [Google Scholar]
- 17.The relative configuration of cyclononenes 10 and 21 was determined by X-ray crystallography.
- 18.Allylborations can also be accelerated by Lewis acids: Kennedy JWJ, Hall DG. J Am Chem Soc. 2002;124:11586–11587. doi: 10.1021/ja027453j.Ishiyama T, Ahiko T-a, Miyaura N. J Am Chem Soc. 2002;124:12414–12415. doi: 10.1021/ja0210345.
- 19.Roush WR. J Org Chem. 1991;56:4151–4160. [Google Scholar]
- 20.Deiters A, Mück-Lichtenfeld C, Fröhlich R, Hoppe D. Org Lett. 2000;2:2415–2418. doi: 10.1021/ol005998h. [DOI] [PubMed] [Google Scholar]
- 21.Carreira EM, Kvaerno L. Classics in Stereoselective Synthesis. Wiley-VCH; Weinheim: 2009. [Google Scholar]
- 22.(a) Su Q, Beeler AB, Lobkovsky E, John A, Porco J, Panek JS. Org Lett. 2003;5:2149–2152. doi: 10.1021/ol034608z. [DOI] [PubMed] [Google Scholar]; (b) Fleury E, Lannou MI, Bistri O, Sautel F, Massiot G, Pancrazi A, Ardisson J. J Org Chem. 2009;74:7034–7045. doi: 10.1021/jo9012833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.