

Abstract
Apurinic/apyrimidinic endonuclease (APE1) is an essential base excision repair protein that also functions as a reduction/oxidation (redox) factor in mammals. Through a thiol-based mechanism, APE1 reduces a number of important transcription factors including AP-1, p53, NF-κB, and HIF-1α. What is known about the mechanism to date is that the buried Cys residues 65 and 93 are critical for APE1’s redox activity. To further detail the redox mechanism, we developed a chemical footprinting/mass spectrometric assay using N-ethylmaleimide (NEM), an irreversible Cys modifier, to characterize the interaction of the redox inhibitor, E3330, with APE1. When incubated with E3330, two NEM-modified products were observed, one with 2 and a second with 7 added NEMs; this latter product corresponds to a fully modified APE1. In a similar control reaction without E3330, only the +2NEM product was observed in which the two solvent accessible Cys residues, C99 and C138, were modified by NEM. Through hydrogen-deuterium amide exchange with analysis by mass spectrometry, we found that the +7NEM modified species incorporates approximately 40 more deuterium atoms than the native protein, which exchanges nearly identically as the +2NEM product, suggesting that APE1 can be trapped in a partially unfolded state. E3330 was also found to increase disulfide bond formation involving redox critical Cys residues in APE1 as assessed by LC-MS/MS, suggesting a basis for its inhibitory effects on APE1’s redox activity. Collectively, our results suggest that APE1 adopts a partially unfolded state, which we propose is the redox active form of the enzyme.
Regulation of cellular processes by reduction-oxidation (redox) is critical for cell survival(1). Oxidation of proteins can result in loss of function owing to the formation of disulfide bonds, some of which are reduced by general redox factors such as thioredoxin and glutaredoxin (2, 3). Both thioredoxin and glutaredoxin include a C-X-X-C motif in which one Cys serves as the nucleophile forming a mixed disulfide bond with the substrate protein, and the second Cys resolves the mixed disulfide bond forming a disulfide bond in the redox factor thereby leaving the substrate protein in a reduced state (4). Thioredoxin is itself reduced by thioredoxin reductase whereas glutaredoxin is reduced by glutathione, which is then reduced by glutathione reductase (2, 3, 5).
A number of transcription factors are subject to redox regulation, the first known example being AP-1 (c-Jun/c-Fos) (6, 7). The nuclear factor responsible for reducing c-Jun/c-Fos was identified as Ref-1, also known as apurinic/apyrmidinic endonuclease (APE1), an essential base excision repair enzyme (8). As redox regulation of c-Jun was known to involve a Cys residue within its DNA-binding domain, reduction of c-Jun was proposed to involve a thiol-exchange mechanism. Of the seven Cys residues in APE1, substitution of a single Cys residue, namely Cys 65, with Ala results in loss of redox activity (9). The critical role of Cys 65 was further confirmed through mutational analysis of the redox inactive zebrafish APE (zAPE), which has a Thr residue (Thr 58) in the position equivalent to Cys 65. The Thr 58 to Cys substituted zAPE gains redox function through this single amino acid substitution (10). A second Cys residue implicated in the redox activity through mutational analysis is Cys 93 (9, 10). Not only does APE1 lack the C-X-X-C motif found in thioredoxin and glutaredoxin, but also the critical redox residues Cys 65 and Cys 93 are buried residues. Further, none of the Cys residues are positioned appropriately to form disulfide bonds as is revealed by the crystal structures reported for APE1(3). Thus, the mechanism by which APE1 acts as a redox factor poses an interesting problem to solve.
Toward our goal of elucidating the mechanism by which APE1 reduces transcription factors including AP-1, NF-κB, HIF-1α, p53, and others (3, 8, 11-13), we took advantage of the redox inhibitor, (E)-3-(2-(5,6-dimethoxy-3-methyl-1,4-benzoquinonyl))-2-nonyl propenoic acid (E3330), which was reported to interact directly with APE1 (14) and to inhibit its redox activity but not its DNA repair endonuclease activity (15). Here, we report an investigation of the nature of the interaction of APE1 and E3330 by using MS analysis of the amide hydrogen/deuterium exchange (HDX) (16-19) and of N-ethylmaleimide (NEM) labeling (20-22). Our results reveal a novel mode of interaction between E3330 and APE1 and provide new insights on the redox mechanism of APE1.
Experimental Procedures
Preparation of APE1 enzymes
Δ40APE1, an N-terminal truncation of APE1 including residues 40-318 was subcloned into pET28 using BamH1 and XhoI restriction sites with an N-terminal hexa-His affinity tag. The Δ40APE1 vector was transformed into Rosetta (DE3) E. coli (Novagen, Inc.), and cultures were grown at 37 °C in 20 μg/mL kanamycin and 34 μg/mL choramphenicol until OD at 600 nm reached 0.6 and then induced by using 1mM IPTG for 3 hours at 37 °C. The protein was purified as previously described by using Ni-NTA affinity purification followed by S-sepharose ion exchange chromatography (10). The hexa-His tag was then removed by digestion with thrombin, and the protein purified by using S-sepharose ion exchange chromatography. Site-directed mutagenesis using the Stratagene Quikchange kit was used to introduce C65A, C99A, C138A, and C99A/C138A substitutions in Δ40APE1 and confirmed by DNA sequencing analysis. Substituted Δ40APE1 proteins were expressed and purified as described for Δ40APE1.
Full-length APE1was subcloned into an N-terminal hexa-His- SUMO-fusion (Invitrogen) vector. The fusion construct was transformed into Rosetta (DE3) E. coli (Novagen, Inc.), grown in 3 L of LB media with 20 μg/mL kanamycin and 34 μg/mL chloramphenicol until the OD at 600 nm reached 0.6, and then induced overnight with 1 mM IPTG at 15 °C. The cultures were harvested by centrifugation at 4000 x g for 30 min, and the pellets were stored at −80 °C. The cell pellets were each resuspended in 20 mL of 50 mM sodium phosphate buffer pH 7.8, 0.3 M NaCl, 10 mM imidazole, and then lysed by using a French press (SLM-AMINCO, Spectronic Instruments, Rochester, USA) at 1000 psi. The suspension was centrifuged at 35,000 rpm for 35 min, and the supernatant was then loaded on a Ni-NTA column at 4 °C. The column was washed with 20 column volumes of 50 mM sodium phosphate buffer pH 7.8, 0.3 M NaCl, 20 mM imidazole protein and then incubated overnight with the SUMO-specific protease Ulp1, added at a molar ratio of ~1:1000 (Ulp1:APE1). Full-length APE1 was then eluted from the column in the same buffer and further purified using an S-Sepharose column run in 50 mM MES pH 6.5, 1 mM DTT, and a linear NaCl gradient (0.05-1 M). The peak fractions were then combined, concentrated, and subjected to gel filtration chromatographic separation using a Superdex 75 (Amersham Pharmacia) in 50 mM Tris pH 8.0, 0.1 M NaCl. Fractions containing full-length APE1 were then concentrated using Amicon ultra centrifugal concentrators and stored at −80 °C.
NanoESI (nESI) MS
Δ40APE1 was incubated in 1 M ammonium acetate (pH 7.5) with or without E3330 at room temperature (RT) for 4 h. E3330 was synthesized by the Vahlteich Medicinal Chemistry Core, University of Michigan, Dept. of Medicinal Chemistry, Ann Arbor, MI. The compound was dissolved in DMSO and stored at −20° C as a 100 mM stock solution. The stock solution was diluted prior to addition to protein samples resulting in a final DMSO concentration of 5%. Control samples also included 5% DMSO. Protein samples were analyzed by nESI-MS in the positive-ion mode on a Bruker MaXis UHR-TOF (ultra-high resolution time-of-flight) (Bruker Daltonics Inc., Billerica, MA) at a flow rate of 25 nL/min. nESI conditions were adjusted to observe the tetrameric form of lactate dehydrogenase in the mass spectrum as a control for the formation of weak complexes. The capillary voltage was set at −(1000-1200 V). Dry gas and temperature were at 5.0 L/min and 50 °C, respectively. The instrument was externally calibrated by using “Tuning Mix” (Agilent Technologies, Santa Clara, CA). The spray tips were made in-house by pulling a 150 μm i.d. × 365 μm o.d. fused silica capillary with a P-2000 Laser Puller (Sutter Instrument Co., Novato, CA). A four-step program was used with the parameter setup as follows with all other values set to zero: Heat = 290, velocity = 40, delay = 200; Heat = 280, velocity = 30, delay = 200; Heat = 270, velocity = 25, delay = 200; Heat = 260, velocity = 20, delay = 200. Tips were cut accordingly to allow a good spray under the experimental conditions. For each sample, a new tip was used to avoid cross contamination.
NEM Chemical Footprinting and ESI-MS
For NEM labeling, 10-20 μL of Δ40APE1/NEM (Δ40APE1:NEM = 1:5, mol/mol) and Δ40APE1/NEM/E3330 (Δ40APE1:NEM:E3330 = 1:5:5, mol/mol/mol) Δ40APE1 samples were incubated in 10 mM HEPES buffer (pH 7.5) at room temperature; the protein concentration was 100 μm. Both control and E3330 treated samples contained 5% DMSO. At a certain time, a 1 μL aliquot was removed and quenched with 1 μL of 20 mM DTT. Samples were then diluted with water to 1 μm followed by 5 μL injection for MS analysis. Mass spectra were collected on the Bruker MaXis UHR-TOF or Waters Micromass Q-TOF instrument (Waters-Micromass, Manchester, UK). The parameters for MaXis mass spectrometer were as follows: capillary voltage was −3600 V, nebulizer pressure was 0.4 bar, drying gas was 1.0 L/min, and drying temperature was 180°C. The instrument was calibrated using Tuning Mix (Agilent Technologies; Santa Clara, CA) as the external mass calibrant. The parameters for the Waters Q-TOF instrument were as follows: Z-Spray source was operated at 2.8 kV, the cone voltage was 150 V, and RF lens was 50. The source temperature and desolvation temperatures were 80 ° and 180 °C, respectively. The collision energy was 10 eV, and the MCP detector was 2,200 V. Protein samples were loaded on an Opti-Guard C18 column (10 mm × 1 mm i.d., Cobert Associates, St. Louis, MO) for desalting and then eluted to the mass spectrometer by using 50% (v/v) acetonitrile with 0.1% formic acid (FA) at 10 μL/min. Spectral deconvolution was performed using MaxEnt.
Data Processing of NEM-Labeling
A number of the equation parameters were extracted from the kinetic data by nonlinear least squares fitting of theoretical signals, computed from the parameter dependent system state trajectories, to experiment data. The system state was a vector that has the solution chemical species concentrations as the vector coordinates. In each trial of the search, the postulated parameters together with the system state of concentrations permitted the calculation of the time rate of change of the state by computing the fluxes into and out of each species as described by the system equations. This process implemented a vector first-order ordinary differential equation, which was solved by numerical integration for the time interval of reaction initiation to the longest reaction time to give the state time trajectory in each fitting trial. For comparison with the experiment data, the theoretical signal for each APE1 species was computed as a fraction of all APE1 species concentrations that were first weighted by a relative sensitivity factor that varied linearly with slope gN with the number of NEMs attached starting with one for Δ40APE1 by itself. The calculations were carried out in the computer application Mathcad 14.0 M010 (Parametric Technology Corporation, Needham, MA). The numerical integration of the differential equation was carried out by the adaptive fourth-order Runge-Kutta function “Rkadapt”.
HDX and Electrospray Ionization Mass Spectrometry (ESI-MS) of the NEM adducts of Δ40APE1
A 30 μL solution of Δ40APE1/E3330/NEM was incubated with 10 mM HEPES (pH 7.5) for 22 h at RT ([protein] = 100 μm; [E3330] = [NEM] = 500 μm). To quench the NEM labeling reaction, 0.5 μL of 1 M DTT was added to the above solution. An aliquot of the 2.8 μL DTT quenched solution was diluted to a final volume of 40 μL of 93% D2O medium with 10 mM HEPES (pH 7.5) and 150 mM KCl and incubated for various times to obtain the HDX kinetics at 25 °C. The HDX reaction was quenched by adding 1 μL of 1 M cold HCl. ESI-MS and data processing were the same as described above.
LC-MS/MS Experiments
Analysis of disulfide bonds
A 200 μL solution of Δ40APE1 (10 μm) and E3330 (50 μm) was incubated in 10 mM HEPES (pH = 7.5) at 37 °C for 1 h. Typically, a 100-fold molar excess of NEM was added to the sample immediately after the incubation to prevent disulfide-bond scrambling during the course of digestion. The Δ40APE1sample was diluted with water to a final concentration of 1 μm and then digested using a protein:trypsin ratio of 50:1 at 37 °C for 4 h. Native digestion conditions were similar to those successfully used for other problems involving purified protein samples(23, 24). The solution was then analyzed by LC-MS/MS whereby 5 μL of digestion solution was consumed for each experiment. Measurements were done for three independent experimental replicates. Reversed-phase capillary LC separations were performed with an Eksigent NanoLC-1D pump (Eksigent Technologies, Inc. Livermore, CA). The reversed-phase capillary column (0.075 mm × 150 mm) was packed in-house by using a PicoFrit™ tip (New Objective, Inc., Woburn, MA) with C18 particles (Magic, 5 μm, 120 Å, Michrom Bioresources, Inc., Auburn, CA). The mobile phase consisted of water, with 0.1% formic acid (solvent A) and acetonitrile with 0.1% formic acid (solvent B). Immediately after sample loading, the mobile phase was held at 98% A for 12 min. A linear gradient was used with 2% to 60% solvent B over 60 min, then to 80% solvent B over 10 min at 260 nL/min followed by a 12 min re-equilibration step by 100% solvent A. The flow was directed by PicoView Nanospray Source (PV550, New Objective, Inc., Woburn, MA) to the LTQ Orbitrap (Thermo Fisher Scientific, Inc., San Jose, CA). The spray voltage was 1.8-2.2 kV, and the capillary voltage was 27 V. The LTQ Orbitrap was operated in standard data-dependent MS/MS acquisition mode controlled by its Xcalibur 2.0.7 software, in which a full mass spectral scan was followed by six product-ion (MS/MS) scans. The mass spectra of the peptides were acquired at high mass resolving power (60,000 for ions of m/z 400) with the FT analyzer over the range of m/z 350-2000. The six most abundant precursor ions were dynamically selected in the order of highest to lowest signal intensity (minimal intensity of 1000 counts) and subjected to collision-induced dissociation (CID). Precursor activation was performed with an isolation width of 2 Da and activation time 30 ms. The normalized collision energy was 35% of the maximum available. The automatic gain control target value was regulated at 1 × 106 for the FT analyzer and 3 × 104 for the ion trap with a maximum injection time of 1000 ms for the FT analyzer and 200 ms for the ion trap. The instrument was externally calibrated by using a standard calibration mixture of caffeine, the peptide MRFA, and Ultramark 1621 (Thermo Fisher Scientific, Inc., San Jose, CA). To identify covalent modifications LC-MS/MS data were searched with Mascot 2.2 (Matrix Science, London, UK) against the NCBI database or MassMatix, an in-house search engine developed by Xu et al.(25-27). Parameters used in Mascot were: enzyme, trypsin; maximum missed cleavage, 3; peptide mass tolerance, 10 ppm with one C 13 peak; peptide charge, +1 to +3; product mass tolerance, 0.6 Da; instrument type, default (searching for all types of b and y ions). To locate disulfide bonds, LC-MS/MS data were searched with MassMatrix with the following parameter settings: enzyme, trypsin; maximum missed cleavage, 3; variable number of modifications by NEM on cysteines; precursor ion tolerance, 10 ppm; product ion tolerance, 0.8 Da; max # PTM per peptide, 2; minimum peptide length, 4 amino acids; maximum peptide length, 40 amino acids; min pp score, 5.0; min pptag score 1.3; max # match per peptide, 3; max # combination per match, 3; fragmentation method, CID; C13 isotope ions, 1; crosslink, disulfide; crosslink mode, exploratory; crosslink sites cleavability, not applicatble; max # crosslinks per peptide, 2.
Analysis of E3330 or NEM adducts
Samples of Δ40APE1 (100 μM) and E3330 (500 μM) were incubated at room temperature for varying lengths of time and analyzed for formation of covalent adducts with E3330 by LC-MS/MS as described above. NEM-modified samples, generated in the chemical footprinting experiment described above, were analyzed using the same protocol as described above for the disulfide bond analysis. Peptide coverage for all LC-MS/MS experiments was greater than 80%.
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed as described (10) with the following modifications. 0.3 mM purified APE1 proteins ( full-length APE1, Δ40APE1 and NEM modified full-length APE1 and Δ40APE1) were reduced with 1.0 mM DTT for 10 min and diluted to yield a final concentration of 0.06 mM with 0.2 mM DTT in PBS. 2 μL of each reduced APE1 protein (0.006 mM) was added to EMSA reaction buffer (10 mM Tris [pH 7.5], 50 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 5% [vol/vol] glycerol) with 2 μL of 0.007 mM protein mixture (1:1) of purified truncated c-Jun and c-Fos proteins containing the DNA-binding domain and the leucine zipper region (oxidized with 0.01 mM diamide for 10 minutes) in total volume 18 μL. Samples were incubated for 30 min at room temperature, and then the EMSA assay was performed as previously described (10).
Results
Interaction of E3330 with Δ40APE1
As a first step toward elucidating the mechanism by which APE1 reduces transcription factors, we considered that a study of the interaction of E3330, which inhibits the redox activity of APE1, may provide insight. We elected to perform our initial studies using Δ40APE1, a construct lacking the N-terminal 40 amino acids, which is easily purified from E. coli to greater than 95% homogeneity, behaves well in ESI mass spectrometry experiments, is fully functional as an endonuclease in vitro (28), and retains near wild-type redox activity (10). To examine the nature of the interaction, we subjected Δ40APE1 incubated with E3330 to both native (29-31) and denaturing ESI-MS analysis. As shown in Figures 1A and B, a weak interaction was observed in the native nESI-MS experiment. Furthermore, the peaks representing the charge-state distribution of the protein and of the protein/E3330 complexes were shifted to higher m/z. This shift could be caused by a small change in protein conformation induced by a weak interaction with E3330, making protonation more difficult and leading to lower charging.
Figure 1.


NanoESI mass spectra of Δ40APE1 without (A) and with E3330 (B). Samples were incubated in 1 M ammonium acetate at pH 7.5 for 4 h at RT and then directly analyzed by nESI-MS ([protein] = 100 μm; [E3330] = 500 μm). Mass spectra were collected on a Bruker MaXis UHR-TOF instrument. (C) ESImass spectrum of Δ40APE1 and E3330 with 50% ACN with 0.1% FA at 10 μL/min. Sample was incubated in 100 mM ammonium bicarbonate at pH 7.5 for 4 h at RT and before MS analysis: [protein] = 100 μm; [E3330] = 500 μm. The symbol * denotes peaks for the water and salt adducts. Data were collected on a Waters Micromass Q-TOF instrument, and deconvolution was done with MaxEnt1 algorithm provided with that system. Instrument parameters were the same as described in the HDX experiments except theflow rate was 10 μL/min, capillary voltage was 2.5 kV, and the collision energy was 5 eV.
Under denaturing ESI conditions, E3330 was observed to form adducts only under certain conditions. As shown in Figure 1C, small peaks were seen consistent with the association of E3330 with Δ40APE1 (Figure 1C) as indicated by a mass shift of 278 Da. In this case, it is not possible to determine directly the nature of the complex as both covalent and non-covalent adducts are expected to have the same mass difference. To determine whether E3330 forms covalent adducts with Δ40APE1, an LC-MS/MS analysis was performed. No E3330-modified peptides could be detected in this analysis indicating that the ESI-MS experiments under both native and denaturing conditions are consistent with the formation of a reversible adduct of E3330 and Δ40APE1.
Chemical footprinting analysis of the effects of E3330 on Δ40APE1
To elucidate further the nature of the interaction of E3330 with Δ40APE1, we developed a MS-based chemical footprinting assay using N-methyl maleimide (NEM) to irreversibly modify Cys residues in the protein. NEM specifically reacts with accessible cysteines via a Michael addition and is widely used in protein footprinting (21, 32-36). An important advantage of NEM-labeling is the ability to probe solvent-accessible Cys residues along with those that are exposed due to protein dynamics as demonstrated by Kaltahov and coworkers in their study of interferon β-1a (37). Both global ESI-MS and LC-MS/MS analyses were performed on Δ40APE1 samples. To establish the expected number of modifications by NEM, fully denatured full-length APE1 and Δ40APE1 samples were treated with NEM and subjected to ESI-MS analysis as described in the Supporting Information. A maximum of 7 modifications were observed (Figure S1), consistent with the 7 Cys residues present in APE1.
In the absence of E3330, NEM specifically modifies Δ40APE1 to give a +2NEM modified species as indicated by a shift in mass of 250 Da (Figure 2A). This product forms within 30 s of the addition of NEM and results in nearly 100% modification of Δ40APE1 within 10 min at room temperature as indicated by the time course of the reaction in Figure 3A, notably by the disappearance of the peaks corresponding to unmodified Δ40APE1 in the mass spectrum. Of the seven Cys residues within APE1, only Cys 99 and Cys 138, the two solvent-accessible Cys residues (Figure 2A) were modified by NEM as determined by LC-MS/MS of tryptic peptides produced from this NEM-labeled product.
Figure 2.

ESI mass spectra of Δ40APE1 after incubation without (A) and with E3330 (B) in the presence of NEM for 30 min (left panel) and 6 h (right panel). Samples were incubated in 10 mM HEPES with 150 mM KCl at pH 7.5([protein] = 100 μm; [E3330] = [NEM] = 500 μm). The symbol * denotes peaks for the water adducts. Mass spectra were collected on a Waters Micromass Q-TOF instrument and deconvolution was done with MaxEnt1 algorithm provided with that system.
Figure 3.

Kinetics of Δ40APE1 reaction with NEM (A) or NEM and E3330 (B). Both samples were prepared in 10 mM HEPES at pH 7.5 at RT ([protein] = 100 μm; [E3330] = [NEM] = 500 μm). Aliquots were quenched with DTT at various times. Mass spectra were collected on a Bruker MaXis UHR-TOF instrument, and deconvolution was done with MaxEnt1 algorithm provided with that system. The sums of intensities of different NEM adducts of Δ40APE1 were normalized to 1. Data were fitted with Mathcad using a relatively straight forward one-parameter fit of the + 2 NEM adduct kinetic curves and a three parameter fit varying kslowNEM, kON, and gN was used to fit the + 7 NEM adduct kinetic curves.
The data in Figure 3A can be fit with a kinetic model (see Experimental Procedures) that uses as free variables the rate constants for the reaction with NEM and the ionization efficiencies of the various NEM adducts. The best fit (shown in Figure 3A) afforded a rate constant for the reaction of NEM with a solvent-accessible Cys of 1020 min−1M−1. This value is important for considerations of the subsequent reactions of the proteins with NEM in the presence of E3330; it can be viewed as a measure of the intrinsic reactivity of a solvent-accessible Cys with NEM.
When Δ40APE1 was incubated with NEM and E3330, another major product corresponding to the addition of 7 NEMs was observed. The +2NEM product forms rapidly, whether E3330 is present or absent, whereas the +7NEM product arises slowly at room temperature (Figure 3B) only when E3330 is present. The only other product observed was a small amount of +3NEM product in reactions of Δ40APE1 with NEM or NEM and E3330 (Figure 2); LC-MS/MS analysis indicates that this product comes from the NEM reaction with other, non-Cys, solvent-accessible sites (e.g., Lys, His, and the – NH2 at the terminus). Although the general pKa values of Lys, His, and the terminal NH2 are higher than that of Cys, the immediate environment around these residues may allow for a small extent of the Michael addition. As for the reaction of Δ40APE1 and NEM, we also observed a small amount of the +3NEM product in the reaction in which E3330 was included.
To determine whether the + 7NEM adduct was the result of E3330 denaturing the protein, we incubated Δ40APE1 with E3330 for 24 h at room temperature and then added NEM. After the NEM reaction was allowed to occur for 0.5 h, we observed only the +2NEM product but not the +7NEM product. If E3330 were denaturing Δ40APE1, we would have expected to modify all seven Cys residues with NEM.
Modeling the kinetics of the reaction of NEM with Δ40APE1 in the presence of E3330 brings insight on the mechanism of Ape1’s interaction with E3330. We focus on the slow disappearance of the +2NEM species while the +7NEM product increased (Figure 3B). Although there are seven Cys residues in the protein, we chose to use an initial ratio of 5:1 for [NEM]:[ Δ40APE1] to limit non-specific reactions of NEM with APE1 at other nucleophilic sites than Cys. Once one or more of the five Cys residues remaining in the +2NEM-modified Δ40APE1 become exposed, the protein is converted rapidly to a +7NEM product until the [NEM] approaches zero. LC-MS/MS analysis confirmed that all seven Cys residues in the +7NEM species are modified. The +7NEM products become detectable in approximately 3 h, accumulating over a period of 24 h until the reagent NEM is depleted. Over the same time period and temperature, no +7NEM modified species is formed in the reaction of Δ40APE1 with NEM in the absence of E3330.
The +3NEM species does not change significantly during the time course of the reaction, suggesting that it is not an intermediate in the formation of the +7NEM product (Figure 3B). Furthermore, the appearance and abundance of this product are not affected by the presence of E3330. This +3NEM species can ultimately give +8NEM via reactions that are parallel to the reactions of +2NEM to give +7NEM.
The kinetics of the reaction of Δ40APE1 with NEM in the presence of E3330 (Figure 3B) are consistent with the mechanism shown in Scheme 1. We infer from this and other experiments described below that APE1 can adopt two different conformations, a native fully folded state (F) and a locally or partially unfolded state (LU) as shown in equation (1). Following the initial and rapid reactions of Δ40APE1 with two NEMs, equation (2) in Scheme 1, is a much slower reaction leading ultimately to modification the five remaining Cys residues of Δ40APE1. This reaction is not pseudo first order but second order as both the [Δ40APE1] and [NEM] are changing. The kinetic fit can be explained by the model shown in (3) of Scheme 1.
Scheme 1.

Equations are shown for an equilibrium of folded (F) and locally unfolded (LU) conformations of Δ40APE1 in (1), Δ40APE1’s initial fast reaction of solvent accessible Cys residues with NEM to give a +2NEM species (2), and the reaction of Δ40APE1 with NEM in the presence of E3330 leading to a +7NEM modified Δ40APE1.
In this model, the rate-determining step is the interaction of E3330 with Δ40APE1 resulting in the formation of a complex of E3330 with locally unfolded (LU) Δ40APE1. Although this reaction is potentially an equilibrium reaction, equilibrium cannot become established because LU is rapidly depleted by the reactions with NEM before it can revert. Reaction of a formerly buried Cys residue with NEM serves to trap APE1 in this partially unfolded state. The interaction with E3330 can be viewed a “wedging reaction” (i.e., one that holds open the unfolded protein until a newly exposed Cys is “trapped” by a relatively rapid reaction with NEM. The best fit to the kinetic data give a kon of 1.1 min−1M−1, koff = 0.006 min−1. The rate constant (kNEM) for reaction of newly exposed Cys residues is 1020 min−1M−1, which is that for reaction of a solvent-accessible Cys. This rate constant was multiplied by 5 for the reaction of the 3rd NEM because five Cys residues are available. This factor of 5 has little effect on the fit, however, because the reaction rate constant of solvent-accessible Cys is nearly 1000 times that of the interaction with E3330.
A second model consistent with the kinetic data is one in which E3330 interacts weakly with folded Δ40APE1 and is then converted to a locally unfolded Δ40APE1-E3330 complex with the remainder of the proposed reaction scheme being the same. Although fitting of the kinetic data cannot distinguish this model from that shown in Scheme 1, the kinetic fit, taken together with the data from all of our experiments, indicates that the most probable interaction of E3330 is with locally unfolded Δ40APE1. Both kinetic models require that the rate of reaction increase with increases in the concentration of E3330, which was observed (data not shown).
Temperature dependence of the E3330 induced NEM-modification of Δ40APE1
If formation of the +7NEM modified Δ40APE1 at room temperature occurred over a time period of several hours, consistent with partial unfolding of the protein, we would expect the reaction to occur more rapidly at higher temperature. Furthermore, if E3330 stabilizes one or more locally unfolded states of the protein, allowing labeling of all seven Cys residues, we might expect to populate this conformation at higher temperatures even in the absence of E3330. Indeed, Δ40APE1, when treated with NEM at 37 °C, formed the expected +2NEM product along with a small percentage of +7NEM product confirming that the protein does adopt this conformation even in the absence of E3330 (Figure 4). In addition, the modification of Δ40APE1 by NEM in the presence of E3330 should occur more rapidly at a higher temperature. At 37 °C, the major product is the +7NEM modified species for a reaction time of 30 min (Figure 4). These results also support partial unfolding of Δ40APE1 to expose the one or more buried Cys residues, which then react with NEM. This is more evidence that folded Δ40APE1 exists in an equilibrium with locally unfolded Δ40APE1 as shown in (1) of Scheme 1 with a distribution favoring the folded protein in the absence of perturbation.
Figure 4.

ESI mass spectra of ApeΔ40 without and with incubation with E3330 in the presence of NEM for 0.5 h at 37 °C. Sample was incubated in 10 mM HEPES at pH 7.5([protein] = 10 μm; [E3330] = [NEM] = 50 μm). Mass spectrum was collected on a Bruker MaXis UHR-TOF instrument and deconvolution was done with MaxEnt1 algorithm.
Role of Cys residues in the reaction of NEM/E3330 with Δ40APE1
Cys 65 is known to play a critical role in the redox activity of Δ40APE1. Thus, it was of interest to determine whether Δ40APE1 must have Cys at site 65 to adopt a conformation that reacts with NEM, leading ultimately to fully modified APE1. When C65A APE1 was reacted with NEM in the presence of E3330, a +6 NEM species was observed, consistent with labeling of all Cys residues in the protein (Figure S2). This result indicates that labeling of the buried Cys residues in Δ40APE1 does not depend on the presence of Cys 65.
Another concern was whether the initial modification of Δ40APE1 forming the +2NEM species labeled at Cys 99 and Cys 138 in some way facilitated or was required for NEM reaction with the remaining Cys residues. Therefore, we reacted the C99A/C138A substituted protein with NEM in the presence of E3330. The outcome of this reaction was slow formation of a +5NEM labeled species in the same time frame as observed for the unsubstituted protein (Figure S2). For this +5NEM labeled species, all buried Cys residues of Δ40APE1 were modified with NEM, as determined by LC-MS/MS analysis of a tryptic digest. Therefore, initial modification of Cys 99 and Cys 138 is not required for the subsequent labeling of the buried Cys residues.
Characterization of the NEM-modified Δ40APE1 species by HDX
To assess the conformational state of the +2NEM and the +7NEM Δ40APE1 species, we measured the exchange of hydrogen for deuterium atoms within the peptide amides as a function of time. Amides involved in hydrogen-bonding interactions within the protein should exchange only slowly under the conditions used in this experiment. For the +2NEM modified Δ40APE1, the kinetics of deuterium uptake were identical to that of the unmodified Δ40APE1 protein; 144 hydrogen atoms underwent exchange to deuterium (Figure 5 and Figure S3). For the +7NEM modified Δ40APE1, the deuterium uptake was significantly higher; a total of 188 deuterium atoms exchanged (Figure 5). Using a kinetic model (17), we fit the data by “binning” the number of amides that exchanged in 4 h into fast, intermediate, and slow. We limited the model to that time because the exchange had become “very slow” after 4 h, as indicated by the nearly flat kinetic curves at that time. For the +2NEM adduct, the number of fast, intermediate, and slow exchanging amide H’s are 81± 1, 20 ± 4, 43 ± 2, respectively; for the + 3NEM adduct, the number of amides are 81 ± 2, 19 ± 2, 41 ± 1, whereas for the + 7NEM adduct, the numbers are 136 ± 3, 14 ± 1, 38 ± 4. Clearly, the number of fast exchanging amide H’s has increased substantially for the +7NEM adduct, consistent with it being a partially unfolded conformer that becomes “locked-in” or “trapped” as a result of the reaction with NEM. The +7NEM adduct has ~40 more exchanging amide H’s than Δ40APE1, +2NEM adduct, or the + 3NEM adduct. The protein contains a total of 278 amide hydrogen atoms, of which 51% are exchanged in the wild-type protein and 67% in the +7NEM adduct. Therefore, the +7NEM modified Δ40APE1 represents a partially but not completely unfolded state of the enzyme; it is clearly distinct from wild-type or the +2NEM modified protein.
Figure 5.
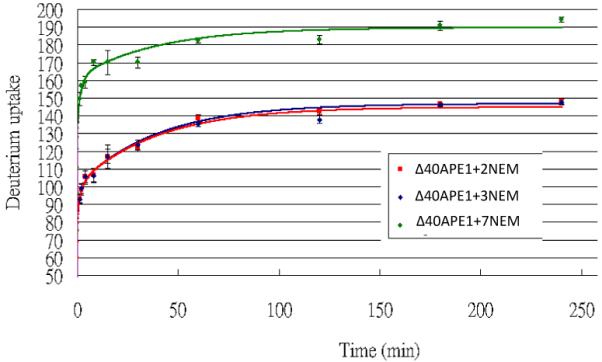
HDX kinetics results of “Δ40APE1 + 2 NEM”, “Δ40APE1 + 3 NEM” and “Δ40APE1 + 7 NEM”. Δ40APE1/E3330/NEM sample was incubated in 10 mM HEPES with 150mM KCl (pH 7.5) for 22 h at RT ([protein] = 100 μm; [E3330] = [NEM] = 500 μm). Reaction was quenched by adding DTT. HDX was conducted in a 93% D2O medium with 10 mM HEPES (pH 7.5) and 150 mM KCl at 25 °C and quenched by adding sufficient 1 M cold HCl to give a pH of 2.5. The curves were plotted with data from 3 independent experimental replicates.
The effect of NEM modification on APE1’s redox activity
Given that it is the +2NEM-modified Δ40APE1 that is converted to a +7NEM product, we sought to determine whether the +2 NEM species retains redox activity and, therefore, represents a biologically active form of the enzyme. Accordingly, NEM-treated Δ40APE1 and full-length APE1 were subjected to global ESI-MS analysis to confirm that the reactions produced a +2NEM modified species for each sample. The products were then analyzed for redox activity using an EMSA redox assay. In this assay, the redox activities of +2NEM-modified APE1 samples were compared to those of a redox-inactive control, C65A APE1 and of Δ40APE1 or full-length APE1. As shown in Figure 6, the +2NEM-labeled samples retained ~90% of full-length or Δ40APE1 activity. Therefore, the +2NEM modification of APE1 does not significantly reduce its redox activity. This result is consistent with results obtained for single Cys mutants of APE1 in which C99A and C138A APE1 samples retain near wild-type redox activity (9, 10). Modification of all 7 Cys residues in APE1 by NEM when E3330 is present would be expected to cause loss of redox activity.
Figure 6.


EMSA redox assay comparing the redox activity of +2 NEM modified full-length APE1 and Δ40APE1 to the unmodified enzymes. The redox inactive C65A enzyme is included as a control. APE1 samples either untreated (left lanes labeled “unreduced proteins”) or pretreated (right lanes labeled “reduced proteins with 0.02 mM DTT”) were used in the assay. APE1 proteins were reduced with 1 mM DTT as described in the Experimental Procedures and diluted yielding final concentrations in the assay of 0.006 mM APE1 and 0.02 mM DTT. Oxidized c-Jun/c-Fos (0.007 mM) was incubated for 30 min with reduced APE1 in EMSA reaction buffer. The EMSA assay was performed as described in the Experimental Procedures. (A) Gel and (B) quantitation of the gel. At least three independent experiments were done, and the results of a representative experiment are shown here.
Effect of E3330 on disulfide bond formation in Δ40APE1
The redox activity of APE1 requires Cys 65 and, therefore, is thought to involve a thiol-mediated disulfide exchange reaction. If Cys 65 serves as the nucleophilic thiol in the reduction of transcription factors by APE1, it must exist in a reduced state. Further, other participating Cys residues must be available to reduce the mixed disulfide formed between Cys 65 and a Cys residue within the transcription factor. Therefore, we asked whether E3330 affects the formation of disulfide bonds in Δ40APE1, particularly those that play an important role in the redox activity. When Δ40APE 1 was treated with E3330, the relative abundance of disulfide bonds in Δ40APE1 of C65-C93, C93-C99, C93-C138, and C65-C99 increased from 0.16, 0.04, 0.01, and 0.01 to 8.2, 5.8, 2.6, and 2.6%, respectively, as determined by LC-MS/MS analysis of tryptic fragments (results in Figure 7). Percentage values reflect the abundance of disulfide-bonded peptides as normalized to an internal peptide that was used for quantitation (38, 39) as described in the Experimental Procedures. Product-ion (MS2) spectra identifying the disulfide linkages (Figure S4) show that the relative abundance of each of the disulfide-bonded peptides increased significantly following treatment with E3330. Both Cys 65 and Cys 93 play an important role in the redox activity of APE1(9, 10), and all of the observed disulfide bonds involve either Cys 65 or Cys 93. Therefore, all of these disulfide bonds would be expected to impact the redox activity of APE1.
Figure 7.

Normalized percentages of disulfide linkages in the Δ40APE1 control and Δ40APE1/E3330 samples. Samples were prepared by incubating the protein without (control) and with E3330 in 10 mM HEPES at pH 7.5 for 1 h at 37 °C ([protein] = 10 μm; [E3330] = 50 μm). An excess of NEM (protein/NEM = 1/50, mol/mol) was immediately added to quench the disulfide crosslinking reaction and prevent disulfide scrambling during the subsequent digestion process. Data base searching was done with MassMatrix, an algorithm with the ability to identify disulfide-linked and crosslinked peptides (25-27). The amounts of peptides were estimated by dividing the peak areas in the LC chromatogram of the disulfide-linked peptides by that of a “standard” peptide “WDEAFR” in the sequence, which does not become modified during the sample preparation and LC-MS/MS analysis process. The assumption for this method is that there is no or little ionization discrimination between the “standard” peptide and the didulfide linked peptides. No C65-C138 linkage was observed in the control sample. Bars represent “mean ± standard derivation” taken from triplicate measurements.
Comparison of full-length APE1 with Δ40APE1
An important question is whether the N-terminal residues in the full-length APE1 affect the ability of the protein to unfold partially, providing access of NEM to buried Cys residues as is true for the Δ40APE1 protein. Following incubation of full-length APE1 with NEM/E3330 under conditions similar to those used for the Δ40APE1 experiments, we observed the same major products by ESI-MS, +2NEM and +7NEM adducts (Figure 8). Thus, the full-length APE1 behaves similarly to Δ40APE1, and the conclusions from MS study of Δ40APE1 are applicable to the full-length APE1, which also undergoes the partial unfolding event(s) allowing NEM to react with buried Cys residues within APE1.
Figure 8.

ESI mass spectrum of full-length APE1 (FL-APE1) with E3330 in the presence of NEM for 12 h. Sample was incubated in 10 mM HEPES at pH 7.5([protein] = 100 μm; [E3330] = [NEM] = 500 μm). Mass spectrum was collected on a Bruker MaXis UHR-TOF instrument and deconvolution was done with MaxEnt1 algorithm.
Discussion
Taken together, our data provide the first definitive evidence that APE1 can adopt a partially or locally unfolded conformation. Three lines of evidence confirm the partially unfolded conformation. First, the chemical footprinting experiments with NEM in the presence of E3330 show that all seven Cys residues react with NEM although only two of the seven Cys residues are solvent-accessible in the native state of the protein. Therefore, an unfolding event must occur to allow the remaining Cys residues to be modified by NEM. Second, the kinetics modeling is consistent with a rate-determining reaction of E3330 with a small population of locally unfolded protein that possesses one or more newly exposed Cys residues. The reaction is necessarily slow because the reactive concentration is small. The reaction stabilizes the partially unfolded protein, allowing that small fraction to react rapidly with NEM. Subsequent additional unfolding exposes more Cys residues for reaction with NEM until all seven are rapidly modified. Third, the HDX kinetics, as determined by mass spectrometry, demonstrate an additional uptake of approximately 40 deuterium atoms in the +7NEM adduct as compared to the +2NEM or wild-type protein.
An important feature of the locally unfolded conformation of APE1 is that Cys 65, the residue critical for the redox activity of APE1, becomes exposed as demonstrated by reactivity with NEM in the presence of E3330. However, E3330 is not necessary for APE1 to sample this locally unfolded state. This is demonstrated by reaction, in the absence of E3330, of buried Cys residues with NEM, at the physiologically relevant temperature of 37 °C. As expected, at an elevated temperature, the equilibrium between the folded and locally unfolded states of APE1 is shifted in favor of the unfolded species, allowing a small amount of the +7NEM product to be produced more rapidly (i.e. within 30 min). In the presence of E3330 at 37 °C, approximately 80% of the sample is fully modified by NEM within 30 min. As demonstrated in the chemical footprinting experiments, perturbation of the equilibrium between folded and locally unfolded states of APE1 occurs through interaction with E3330, a redox inhibitor of APE1. In its capacity as a redox factor, we suggest that interaction with oxidized transcription factors similarly perturbs this equilibrium governing the conformational states of APE1.
Our studies also provide a basis for understanding the mechanism by which E3330 inhibits the redox activity of APE1. Although our experiments do not support strong binding of E3330 as indicated by the previously reported Kd of 1.6 nM for E3330 binding to APE1(14), they do confirm a novel mode of interaction for E3330 with APE1. Based on our chemical footprinting and HDX experiments, we propose that E3330 interacts with the locally unfolded state of APE1, stabilizing it so that the normally buried Cys residues can react with NEM. In shifting the equilibrium toward the locally unfolded state of APE1, E3330 also facilitates disulfide bond formation in APE1. The mechanism by which E3330 increases disulfide bond formation may involve reversible activation of a Cys residue by E3330, making that Cys residue susceptible to nucleophilic attack by a reduced Cys residue. Of particular relevance is the fact that all of the disulfide bonds identified involve the critical redox residues Cys 65 and/or Cys 93 (9, 10) and that both of these Cys residues are buried in the folded conformation of APE1. This indicates that disulfide bond formation occurs in the locally unfolded form of the protein. Our current model is that E3330 acts by increasing disulfide bond formation involving Cys 65 and/or Cys 93, effectively decreasing the redox active population of APE1 molecules.
The results from interaction of APE1 with E3330 have implications on the general redox properties of this protein. It may be that the redox active form of APE1 is the locally unfolded unfolded form of the enzyme; the unfolding exposes the critical Cys 65 residue, thereby making it available for a thiol-mediated/disulfide exchange reaction with a transcription factor.
Supplementary Material
Acknowledgements
We thank Richard Huang, Hao Zhang, Weidong Cui from Gross laboratory at Washington University in St. Louis for their help with this work. We also thank Dr. Tom Hurley and colleagues at Indiana University School of Medicine for helpful discussions and Dr. Karl Dria, Department of Chemistry and Chemical Biology, IUPUI for his assistance.
This work was supported by a grant from the National Institutes of Health, CA14571 to M.M.G., a grant from the National Institutes of Health, NCRR (2P41RR000954), to M.L.G., and a grant from the National Science Foundation to R.E.M. (NSF MRI DBI 0821661).
Abbreviations
- APE1
apurinic/apyrimidinic endonuclease
- ESI-MS
electrospray ionization mass spectrometry
- E3330
(E)-3-(2-(5,6-dimethoxy-3-methyl-1,4-benzoquinonyl))-2-nonyl propenoic acid
- nESI
nano ESI
- NEM
N-ethyl maleimide
- Cys
cysteine
- His
histidine
- Lys
lysine
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supporting information includes three additional figures with NEM-footprinting mass spectrometry data for two substituted APE1 proteins, HDX data for APE1, and LC-MS/MS data for the peptides including disulfide bonds for APE1 treated with E3330. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 3.Luo M, He H, Kelley MR, Georgiadis MM. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 5.Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780:1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Abate C, Patel L, Rauscher FJ, 3rd, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 7.Abate C, Luk D, Curran T. A ubiquitous nuclear protein stimulates the DNA-binding activity of fos and jun indirectly. Cell Growth Differ. 1990;1:455–462. [PubMed] [Google Scholar]
- 8.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol. 1993;13:5370–5376. doi: 10.1128/mcb.13.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Georgiadis M, Luo M, Gaur R, Delaplane S, Li X, Kelley M. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jayaraman L, Murthy KG, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 12.Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem. 1999;274:27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 13.Lando D, Pongratz I, Poellinger L, Whitelaw ML. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1alpha and the HIF-like factor. J Biol Chem. 2000;275:4618–4627. doi: 10.1074/jbc.275.7.4618. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu N, Sugimoto K, Tang J, Nishi T, Sato I, Hiramoto M, Aizawa S, Hatakeyama M, Ohba R, Hatori H, Yoshikawa T, Suzuki F, Oomori A, Tanaka H, Kawaguchi H, Watanabe H, Handa H. High-performance affinity beads for identifying drug receptors. Nat Biotechnol. 2000;18:877–881. doi: 10.1038/78496. [DOI] [PubMed] [Google Scholar]
- 15.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, II, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small molecule inhibition of Ape1’s redox function. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katta V, Chait BT. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1991;5:214–217. doi: 10.1002/rcm.1290050415. [DOI] [PubMed] [Google Scholar]
- 17.Zhu MM, Rempel DL, Zhao J, Giblin DE, Gross ML. Probing Ca2+-induced conformational changes in porcine calmodulin by H/D exchange and ESI-MS: effect of cations and ionic strength. Biochemistry. 2003;42:15388–15397. doi: 10.1021/bi035188o. [DOI] [PubMed] [Google Scholar]
- 18.Garcia RA, Pantazatos D, Villarreal FJ. Hydrogen/deuterium exchange mass spectrometry for investigating protein-ligand interactions. Assay Drug Dev. Technol. 2004;2:81–91. doi: 10.1089/154065804322966342. [DOI] [PubMed] [Google Scholar]
- 19.Weis DD, Kaveti S, Wu Y, Engen JR. Probing protein interactions using hydrogen-deuterium exchange mass spectrometry. In: Downard KM, editor. Mass Spectrometry of Protein Interactions. John Wiley & Sons, Inc.; Hoboken, NJ.: 2007. pp. 45–61. [Google Scholar]
- 20.Kim YJ, Pannell LK, Sackett DL. Mass spectrometric measurement of differential reactivity of cysteine to localize protein-ligand binding sites. Application to tubulin-binding drugs. Anal. Biochem. 2004;332:376–383. doi: 10.1016/j.ab.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 21.Schilling B, Yoo CB, Collins CJ, Gibson BW. Determining cysteine oxidation status using differential alkylation. International Journal of Mass Spectrometry. 2004;236:117–127. [Google Scholar]
- 22.Kurono S, Kurono T, Komori N, Niwayama S, Matsumoto H. Quantitative proteome analysis using D-labeled N-ethylmaleimide and 13C-labeled iodoacetanilide by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Bioorg. Med. Chem. 2006;14:8197–8209. doi: 10.1016/j.bmc.2006.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gau BC, Chen H, Zhang Y, Gross ML. Sulfate radical anion as a new reagent for fast photochemical oxidation of proteins. Anal Chem. 2010;82:7821–7827. doi: 10.1021/ac101760y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hambly DM, Gross ML. Laser flash photochemical oxidation to locate heme beinding and conformational changes in myglobin. International Journal of Mass Spectrometry. 2007;259:124–129. [Google Scholar]
- 25.Xu H, Freitas MA. A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, Yang L, Freitas MA. A robust linear regression based algorithm for automated evaluation of peptide identifications from shotgun proteomics by use of reversed-phase liquid chromatography retention time. BMC Bioinformatics. 2008;9:347. doi: 10.1186/1471-2105-9-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu H, Zhang L, Freitas MA. Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J Proteome Res. 2008;7:138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bapat A, Glass LS, Luo M, Fishel ML, Long EC, Georgiadis MM, Kelley MR. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther. 2010;334:988–998. doi: 10.1124/jpet.110.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernandez H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 30.Sandercock AM, Robinson CV. Electrospray Ionization Mass Spectrometry and the Study of Protein Complexes. In: Schuck P, editor. Protein Interactions. Springer US; 2007. pp. 447–468. [Google Scholar]
- 31.Sharon M, Robinson CV. The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem. 2007;76:167–193. doi: 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- 32.Titani Y, Tsuruta Y. Some chemical and biological characteristics of showdomycin. J Antibiot (Tokyo) 1974;27:956–962. doi: 10.7164/antibiotics.27.956. [DOI] [PubMed] [Google Scholar]
- 33.Kim YJ, Pannell LK, Sackett DL. Mass spectrometric measurement of differential reactivity of cysteine to localize protein-ligand binding sites. Application to tubulin-binding drugs. Anal Biochem. 2004;332:376–383. doi: 10.1016/j.ab.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 34.Rishavy MA, Pudota BN, Hallgren KW, Qian W, Yakubenko AV, Song JH, Runge KW, Berkner KL. A new model for vitamin K-dependent carboxylation: the catalytic base that deprotonates vitamin K hydroquinone is not Cys but an activated amine. Proc Natl Acad Sci U S A. 2004;101:13732–13737. doi: 10.1073/pnas.0404989101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurono S, Kurono T, Komori N, Niwayama S, Matsumoto H. Quantitative proteome analysis using D-labeled N-ethylmaleimide and 13C-labeled iodoacetanilide by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Bioorg Med Chem. 2006;14:8197–8209. doi: 10.1016/j.bmc.2006.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan L, Kaback HR. Site-directed alkylation of cysteine to test solvent accessibility of membrane proteins. Nat Protoc. 2007;2:2012–2017. doi: 10.1038/nprot.2007.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bobst CE, Abzalimov RR, Houde D, Kloczewiak M, Mhatre R, Berkowitz SA, Kaltashov IA. Detection and characterization of altered conformations of protein pharmaceuticals using complementary mass spectrometry-based approaches. Anal Chem. 2008;80:7473–7481. doi: 10.1021/ac801214x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiener MC, Sachs JR, Deyanova EG, Yates NA. Differential mass spectrometry: a label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal Chem. 2004;76:6085–6096. doi: 10.1021/ac0493875. [DOI] [PubMed] [Google Scholar]
- 39.Podwojski K, Eisenacher M, Kohl M, Turewicz M, Meyer HE, Rahnenfuhrer J, Stephan C. Peek a peak: a glance at statistics for quantitative label-free proteomics. Expert Rev Proteomics. 2010;7:249–261. doi: 10.1586/epr.09.107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.