

Abstract
The synthesis of diverse nitric oxide (NO)-releasing network polyesters is described. The melt phase condensation of polyols with a calculated excess of diacid followed by thermal curing generates crosslinked polyesters containing acid end groups. Varying the composition and curing temperatures of the polyesters resulted in materials with tunable thermal and degradation properties. Glass transition temperatures for the synthesized materials range from −25.5 °C to 3.2 °C, while complete degradation of these polyesters occurs within a minimum of nine weeks under physiological conditions (pH 7.4, 37 °C). Post-polymerization coupling of aminothiols to terminal carboxylic acids generate thiol-containing polyesters, with thermal and degradation characteristics similar to those of the parent polyesters. After nitrosation, these materials are capable of releasing up to 0.81 μmol NO cm−2 for up to 6 d. The utility of the polyesters as antibacterial biomaterials was indicated by an 80% reduction of Pseudomonas aeruginosa adhesion compared to unmodified controls.
Introduction
The formulation of diverse biodegradable polymers has become an increasingly important aspect in the advancement of biomedical materials.1 Absorption of degradable scaffolds by host tissues may overcome long-term biocompatibility problems associated with persistent implants, as well as eliminate the high costs and patient morbidity associated with follow-up surgeries.2–4 Additionally, these materials may facilitate the emergence of unique therapies (e.g., drug delivery) that exploit characteristic degradation patterns.1 The development of degradable polymers for use in vascular stents, sutures, tissue engineering scaffolds, and joint replacement applications has been widely reported.5–9 Despite their promise in addressing specific challenges, the implementation of such materials remains limited due to the prevalence of infection associated with implantable medical devices in general.10 Indeed, opportunistic microbes at surgical sites cause over two million hospital-acquired infections each year in the United States alone.2 The cost associated with treatment and extended hospitalization for patients with nosocomial infections is > $3 billion and expected to increase as the average age of our population rises.2 Effective treatments and reducing the incidence of such infections is of critical importance to health care.
Hospital-acquired infections occur due to microbe propagation at the surgical site and subsequent formation of a biofilm surrounding the implant. The biofilm matrix protects bacteria, making their eradication particularly challenging without explantation of the associated medical device.11 The polymicrobial nature and advanced drug resistance mechanisms of biofilms often require the administration of elevated doses of antimicrobial agents but with less than adequate results. Incomplete biofilm elimination often leads to the emergence of drug-resistant strains, further complicating treatments.11 Consequently, prevention of the initial bacterial adherence and colonization at the medical device surface is the most ideal strategy for reducing the risk and severity of surgical infections.12
The endogenously produced diatomic free radical nitric oxide (NO) has been implicated in a number of physiological roles including vasodilation,13 angiogenesis,14 wound healing,15 and the elimination of pathogens.16 The effectiveness of NO against numerous types of bacteria associated with hospital-acquired infections (e.g. Pseudomonas aeruginosa and Staphylococcus aureus) are well documented.17 Since NO is highly reactive, the formulation of scaffolds capable of releasing NO in a controlled manner are essential to its use as a therapeutic. As a result, numerous researchers have investigated methods aimed at the storage and controlled release of NO.18–20 S-nitrosothiols and N-diazeniumdiolates represent two of the most well-known NO donor molecules.20 While diazeniumdiolates release their NO payload in the presence of a H+ donor,19 S-nitrosothiols liberate their NO in the presence of heat, light, and/or trace metals.21
Due to its short half-life in physiological milieu (~3 s), NO must be generated at the site of interest to maximize its efficacy as a therapeutic and minimize potential side reactions and toxicity. Numerous NO-releasing coatings have been designed to minimize surface biofouling including the polymers doped with small molecule NO donors (i.e. S-nitrosoglutathione [GSNO]22 and the diazeniumdiolate form of proline [PROLI/NO]23), and those whereby the NO donor is covalently attached to a polymer. In both cases, NO release proved useful in reducing platelet and bacterial adhesion.24–27 However, the fact that these polymer coatings are not biodegradable limit their potential uses to short-term implants such as catheters or sensors.
The development of NO-releasing biodegradable polymers will further extend the utility of NO release in the biomedical arena. Herein, we report the synthesis of crosslinked polymeric materials capable of both controlled NO release and matrix degradation under physiological conditions. The prepolymer preparation by a melt phase reaction and subsequent thermal curing allows for formation of crosslinked products in a variety of architectures. This tunability may be used to tailor the materials for a desired application. Nitric oxide donor functionalization is accomplished via straightforward coupling reactions to terminal carboxylic acids present after thermal crosslinking. A range of degradation rates and NO-storage capabilities are achieved by employing interchangeable starting materials consisting of polyfunctional alcohols (glycerol and pentaerythritol), diacids (glutaric acid and adipic acid), and the NO-donor precursors (cysteamine and penicillamine). The antibacterial efficacy of these materials against Pseudomonas aeruginosa is examined using a static adhesion assay.
Experimental Section
Materials
Glutaric acid (GA), adipic acid (AA), pentaerythritol, cysteamine, penicillamine, and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were purchased from the Aldrich Chemical Company (Milwaukee, WI). Glycerol, N-hydroxysuccinimide (NHS), and laboratory grade salts and solvents were purchased from Fisher Scientific (Fair Lawn, NJ). Sodium nitrite was purchased from Acros Organics (Morris Plains, NJ). Diethylenetriamine-pentaacetic acid (DTPA) was purchased from Fluka (Buchs, Switzerland). Tryptic soy broth (TSB) and Minimum Essential Media (MEM) was purchased from Becton, Dickinson, and Company (Sparks, MD). Nitric oxide calibration gas (25.85 ppm; balance nitrogen) was purchased from National Welders Supply Co. (Durham, NC). Pseudomonas aeruginosa (ATCC #19143) was obtained from American Type Culture Collection (Manassas, VA). Water was purified to 18.2 MΩ·cm using a Millipore Milli-Q Gradient A-10 purification system (Bedford, MA).
Characterization
1H and 13C NMR was performed in methyl sulfoxide-d6 on a Bruker 400 MHz AVANCE Nuclear Magnetic Resonance spectrometer. Gel-permeation chromatography was carried out using a Waters GPC system with a Wyatt Optilab DSP interferometric refractometer and a Wyatt Dawn EOS detector with polystyrene standards. Static water contact angle measurements were acquired using a CAM 200 optical angle goniometer. Measurements obtained were the average of five frames taken over a period of 5 s immediately after administering a drop. Thermogravimetric analysis was performed under a nitrogen atmosphere on a Perkin-Elmer Pyris 1 TGA with a heating rate of 10 °C/min. Transition temperatures were measured using a Seiko 220C differential scanning calorimeter with heating and cooling rates of 10 °C/min in a nitrogen atmosphere. Nitric oxide release was measured using a Sievers 280i Nitric Oxide Analyzer (Boulder, CO) operating at a flow rate of 200 mL min−1. A two-point calibration line was generated using 25.85 ppm NO gas (balance nitrogen) and air passed through a Sievers NO zero filter.
Polyester Synthesis
Prepolymer melts were formed by heating the desired diacid precursor just above its melting temperature for 2 h in either a 1.60:1.00 molar ratio with glycerol, or a 2.15:1.00 molar ratio with pentaerythritol. Hybrid polyesters were prepared similarly using diacid:glycerol:pentaerythritol molar ratios of 1.00:0.31:0.23. After 2 h, the melt was carefully poured into a mold and the prepolymer cured for 24 h at elevated temperatures determined to be the minimum temperature required for complete curing (75, 120 or 160 °C). After curing, the solid polyester was removed from the mold and cut to produce 1 cm × 1 cm squares of roughly 0.2 cm thickness.
Polyester Functionalization
By controlling the stoichiometry of the prepolymer synthesis, free carboxylic acid functionalities remain after curing, permitting conjugation of NO donors using straightforward NHS/EDC coupling techniques. Briefly, a polymer square was added to a glass vial containing 10 mL of water. Exactly 1 mL of 1.56 M NHS (aq). was then added to the solution. After chilling in an ice bath for 5 min, 1 mL of 1.56 M EDC (aq) was added, and the vial returned to the ice bath for 10 min. After allowing the polymer to soak at room temperature for 24 h, the square was removed from the NHS/EDC solution, rinsed with chilled water and added to another vial containing 10 mL of aqueous 0.25 M solutions of either cysteamine or penicillamine. To prevent premature degradation of the polymer squares, the pH of the solutions was neutralized only immediately prior to study. After soaking for an additional 24 h at room temperature, the polymer squares were removed from solution, rinsed thoroughly with chilled water and dried in vacuo. The polymer (thiols) was nitrosated by submerging the squares in 8 mL of 10 mg/mL NaNO2 (aq) and adding 8 mL of 0.5 M HCl at 0 °C (ice bath) for 30 min in the dark. The polymer squares were then removed from solution, rinsed with a chilled solution of 100 μM DTPA (aq) and dried in vacuo. Nitrosated squares were stored at −20 °C until analysis.
In Vitro Degradation
Rates of polymer degradation were assessed by placing polymer squares weighing ~250 mg in 10 mL of PBS at pH 7.4. After incubation at 37 °C, samples were removed, rinsed with water, and dried in vacuo overnight. The percent mass lost during solution immersion was recorded for each sample with the % degradation expressed using the average of three samples for each time point. Polymer degradation was monitored weekly for the initial polymeric materials (PE1, PE2, PE3, PE4) and at 1, 5, and 10 week intervals for the functionalized squares. To prevent saturation, incubation solutions were changed weekly.
NO Release Analysis
Nitric oxide release was measured using a chemiluminescence nitric oxide analyzer. Briefly, a nitrosated polyester square was added to a flask containing 30 mL of deoxygenated phosphate buffered saline (PBS) containing 500 μM DTPA as a copper chelator at pH 7.4 and 37 °C. Nitrogen was flowed through the solution at a flow rate of 70 mL/min, carrying any liberated NO to the analyzer. Additional nitrogen flow was supplied to the flask to match the collection rate of the instrument at 200 mL/min. The analyte solution was shielded from ambient light to ensure that all NO release was thermally triggered, unless noted otherwise.
Bacterial Viability
P. aeruginosa cultures were grown in TSB to concentrations of 108 colony forming units (CFU) mL−1. Polymer squares were incubated individually in 15 mL conical vials containing 4 mL of the bacterial suspension in PBS for 3 h at 37 °C with gentle agitation. The squares were removed from the bacterial suspension, rinsed lightly with sterile water, and dried under stream of nitrogen. The extent of bacterial adhesion was measured by phase contrast optical microscopy using a Zeiss Axiovert 200 inverted microscope (Chester, VA) coupled with a Zeiss Axiocam digital camera (Chester, VA). Cell adhesion was quantified by digitally processing the images and applying a threshold value to differentiate the background from adhered cells. Materials with rapid degradation rates (PE1 and PE2) were not analyzed in this manner due to the onset of surface roughness contributing to localized darkness in the phase contrast optical micrographs and decreased optical clarity due to water uptake. As such, only polyesters that remained optically transparent after soaking were selected for comparison (PE3, PE4, and PE6).
In vitro toxicity of polyesters
Polyester samples (250 mg) were soaked in PBS for 24 h at 37 °C to remove any soluble or degraded materials from the polymers. L929 mouse fibroblasts were grown in MEM supplemented with 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin and incubated in 5% CO2 under humidified conditions at 37 °C. Following 80% confluency, the cells were trypsinized and seeded onto tissue-culture treated polystyrene 96-well plates at a density of 3 × 105 cells/ml. Cells were incubated with the polyester soak solutions for a period of 24 h. Following the incubation period, the supernatant was aspirated and cells were washed with sterile PBS three times. Fresh media (100 μl) was added to the cells along with 20 μl of the MTS assay reagent (CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay, Promega, Madison, WI). The mitogenic MTS assay for cell viability relies upon the reduction of a yellow MTS compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) as a result of mitochondrial respiration. The reduction to a purple formazan derivative occurs only in viable cells. The absorbance of this colored solution was quantified at 490 nm using a plate reader (Thermoscientific Multiskan® EX, Waltham, MA). Untreated cells were used as control and results were expressed as % viability with respected to untreated controls. Average percent cell viabilities were calculated from at least 8 measurements.
Results and Discussion
Polyester Synthesis and Characterization
The preparation of crosslinked polyesters via polycondensation reactions allows for the synthesis of a wide range of materials. Initially, four polymer compositions were synthesized from their corresponding prepolymers containing a diacid and a polyol. The first two prepolymers were synthesized by the reactions of glycerol with a calculated excess of GA (PE1) or AA (PE2). To alter the crosslink density (and the resulting degradation and thermal transitions) of the polyester materials made, glycerol was substituted with the tetrafunctional alcohol, pentaerythritol was used in place of glycerol resulting in two additional compositions, PE3 and PE4, again containing excess GA or AA, respectively. Further compositional diversity was afforded by incorporating a combination of glycerol and pentaerythritol with each diacid yielding PE5 (with GA) and PE6 (with AA). While 13C NMR confirmed the formation of ester linkages (Figure 1), GPC analysis indicated prepolymer molecular weights < 1000 g/mol. Similarly, accurate estimation of molecular weight could not be determined via end group analysis due to the highly branched structure of the prepolymers. However, the inclusion of polyfunctional alcohols allows for sufficient crosslinking even at low prepolymer molecular weights.
Figure 1.

13C NMR spectra of A) glutaric acid, B) glycerol, and C) PE1 prepolymer in methyl sulfoxide – d6.
As with other crosslinked aliphatic polyesters,28 thermal curing over a wide range of temperatures alters the degree of crosslinking and crosslink density, resulting in diverse transition temperatures and degradation properties. The effect of curing temperature on the properties of compositionally identical polyester formulations is thus not discussed further. To increase throughput, the minimum temperatures required for complete curing after 24 h were considered optimal. As expected, differences in prepolymer melting temperatures and the total functionality of the polyol crosslinker resulted in a range of curing temperatures (Table 2). Higher curing temperatures were necessary for materials containing adipic acid over glutaric acid due to the inherently greater melting temperature of adipic acid (151 vs 96 °C for AA and GA, respectively). Additionally, the increase in molecular functionality upon switching the polyol precursor from glycerol to pentaerythritol facilitated crosslinking, thereby decreasing the necessary curing temperatures compared to materials containing the trifunctional crosslinker.
Table 2.
Melt and curing temperatures for polyesters.
| Polyester | Melt Temperature (°C) | Curing Temperature (°C) |
|---|---|---|
| PE1 | 100 | 120 |
| PE2 | 160 | 160 |
| PE3 | 100 | 75 |
| PE4 | 160 | 120 |
| PE5 | 100 | 120 |
| PE6 | 160 | 120 |
As reported by others,28 the curing of crosslinked aliphatic polyesters significantly influence the thermal transitions inherent to a material. Flexible polymers at physiological temperatures are often advantageous over more rigid materials due to facilitated surgical introductions and improved tissue incorporation.29 As a result, the optimal glass transition temperatures (Tg) for the materials synthesized in this study would be below physiological temperatures (37 °C). Likewise, thermal degradation of the polyesters would be minimal under normal storage conditions to prolong the lifetime of these materials. The observed glass transition temperatures ranged from −25.5 °C to 3.2 °C for PE3 and PE4, respectively (Table 3). Additionally, the polyesters synthesized did not exhibit appreciable thermal degradation at temperatures below 200 °C. As expected, AA-based compositions (PE2, PE4) resulted in faster degradation and greater glass transition temperatures than the corresponding GA-based compositions (PE1, PE3). We attribute this observation to the higher degrees of crosslinking previously linked to elevated curing temperatures.28 Polymers that contained both polyols in addition to their respective diacid (PE5, PE6) did not follow this trend. Rather, the GA-based composition (PE5) had both higher glass transition and degradation temperatures than the AA-based composition (PE6), attributable to a higher crosslink density caused by the smaller diacid constituent and similar curing temperatures.
Table 3.
Thermal analysis of polyesters PE1– PE6.
| Polyester | 5% wt loss (°C) | 10% wt loss (°C) | Tg (°C) |
|---|---|---|---|
| PE1 | 242 | 289 | −25.3 |
| PE2 | 328 | 359 | −14.0 |
| PE3 | 252 | 412 | −25.5 |
| PE4 | 419 | 437 | 3.2 |
| PE5 | 295 | 368 | 0.6 |
| PE6 | 221 | 269 | −17.0 |
In vitro degradation
As cytotoxicity of degradation products is an important consideration for all degradable biomaterials, these materials were designed so that the products of hydrolysis would be metabolic intermediates or FDA approved compounds. Although the compatibility of degradation products is essential for such a material, the degradation rate is also an important consideration for the application of the material. Rate of degradation is influenced by material composition, water uptake, and degree of crosslinking.30 Due to differences in the composition and curing temperatures used in the preparation of the polyesters in this study, a wide variation of degradation rates was observed (Figure 2). As expected, the polyesters containing glycerol (PE1 and PE2) degraded more rapidly than those containing pentaerythritol (PE3 and PE4) due to the decreased crosslink density associated with the trifunctional alcohol. Additionally, the degradation rate data indicated that the GA-based polyesters were more susceptible to hydrolysis than their counterparts composed of AA, matching trends identified in previous studies relating increased aliphatic chain length to slowed hydrolysis rates.31 The rate of degradation for PE4 was much slower than anticipated due to the high crosslink density of the material. Through 10 weeks, no appreciable degradation was observed for PE4. Even at extended periods (~20 weeks) when all other polyesters had degraded appreciably, only 15% mass loss was noted for PE4 materials. Polymers containing both glycerol and pentaerythritol (PE5 and PE6) had faster degradation rates than PE4, but slower than the other polyesters due to crosslinking effects facilitated by the presence of both polyols and curing. As expected the higher crosslink density of PE5 resulted in inhibited hydrolysis and slower degradation compared to PE6. Overall, degradation of all samples except PE4 over the first 10 weeks of analysis was zero-order and linear as would be expected for cured, amorphous elastomers (Supporting Information).7 Complete degradation of PE1, PE2, PE3 was observed after 9, 10, and 14 weeks, respectively. Although complete degradation of PE5 and PE6 was not observed over the maximum period investigated (15 weeks), extrapolation of the zero-order degradation model based on degradation rates over the initial 10 weeks predicts complete degradation after approximately 103 and 45 weeks, respectively. Due to extremely slow degradation rates for PE4, extrapolation of a zero-order degradation model was not attempted.
Figure 2.

Degradation rates of PE1 – PE6 as mass lost (%) vs. time (■ – PE1, ● – PE2, ▲ – PE3, ▼ - PE4, ◆- PE5, ◀ – PE6).
Polyester Functionalization and Nitrosation
The availability of free terminal carboxylic acids resulting from the excess GA or AA in the prepolymer melt phase permitted the coupling of amine-functionalized molecules via facile NHS/EDC techniques. In this case, coupling of cysteamine and penicillamine to the polymer was investigated as a means to store NO. Due to the hydrolytic instability of ester-based materials in acidic or basic solutions, and the pH-dependent reaction kinetics of amide bond formation using NHS and EDC, reaction conditions were optimized for control of both polyester degradation and aminothiol conjugation. Upon formation of polymer-bound activated esters via exposure of the polyesters to aqueous NHS and EDC, the materials were rinsed with chilled water and soaked in solutions containing cysteamine in water (pHs 4–10). The unadjusted alkilinity of cysteamine dissolved in water (pH 10) proved to be excessively harsh for the functionalization step as the polyester was observed to degrade extensively. Less basic solutions (pH 8 and 9) were also not suitable due to decomposition of the polyester matrix after 24 h, albeit less pronounced than at pH 10. While polyesters in slightly acidified solutions (pH 4–6) did not degrade appreciably relative to the basic solutions, the subsequent aminothiol coupling resulted in decreased NO release (data not shown). A neutralized solution of cysteamine afforded the optimal pH for functionalization, as determined by minimal mass lost due to degradation and maximum NO release capabilities (Figure 3).
Figure 3.

Functionalization efficiency of PE1 as % degradation (●) and NO Release (□).
S-nitrosothiols are most commonly prepared via reactions of free thiols with solutions of acidifed or organic nitrites.21 To prevent uptake of potentially harmful organic nitrites into the polyester materials, nitrosothiols were formed via exposure to aqueous solutions of acidified nitrite. Due to the susceptibility of aliphatic polyesters to hydrolysis at acidic pH, the effects of solution acidity and reaction duration on polyester stability during nitrosation were investigated. As expected, the rate of polymer degradation increased substantially in strongly acidic solutions (>1 M HCl) for even short periods of time (i.e, 5–10 min). With decreasing acid concentration, the time necessary to induce noticeable material degradation increased markedly. The nitrosothiol formation observed for functionalized polyesters submerged in solutions of 0.25 M HCl containing 10 mg/mL NaNO2 after 30 min combined with minimal mass loss due to hydrolysis indicated that the S-nitrosothiol reaction conditions were appropriate for further experimentation. The procedures used for the functionalization and nitrosation of PE1–PE6 did not appreciably change the degradation and thermal characteristics of these materials from those of the preliminary polyesters (Supporting Information).
NO Release Analysis
The combination of compositional diversity, degradation rates, and thermal transitions provide scaffolds with the capacity to store and release variable amounts of NO. Due to the numerous NO release mechanisms inherent to S-nitrosothiols,21 the total NO storage was characterized via multiple analyses. As shown in Figure 4, the kinetics of NO release were accelerated upon exposure to light, supporting the use of high wattage broad spectrum irradiation to rapidly liberate all of the NO from the scaffold and determine NO release. As provided in Table 4, the total NO storage for the six polyester materials varied by almost two orders of magnitude. Predictably, the total NO release for polyesters containing cysteamine residues was larger than their penicillamine counterparts. The decreased NO storage for penicillamine has been attributed to the increased steric crowding of the tertiary thiol functionality compared to that of primary thiols (cysteamine).21 Although these steric effects give rise to nitrosothiols with increased stabilization compared to primary nitrosothiols, these effects decrease the thiol to nitrosothiol conversion efficiency resulting in less NO storage at analogous nitrosation times. Extended nitrosation times were not evaluated due to the necessity to limit polymer exposure to hydrolytic conditions. The two-component polyesters containing GA also stored more NO than those with AA. This characteristic may be attributed to the temperatures required for thermal curing. As mentioned above, curing temperatures for the polyester prepolymers were selected based on the melting temperatures of the diacid precursors. As a result, AA-containing materials were cured at higher temperatures than their GA counterparts giving rise to higher degrees of crosslinking. Increased crosslinking decreases the number of available acid functional groups limiting further coupling of cysteamine or penicillamine, and ultimately NO storage capacity. This trend remained consistent for the three-component polyesters as PE5 was characterized by a higher degree of crosslinking than PE6.
Figure 4.

Nitric oxide release comparison of NPE1A with light trigger (—, 200 W broad spectrum) and thermal trigger (---, 37 °C, shielded from light).
Table 4.
Nitric oxide release characteristics for nitrosated polyesters.
| Polyester | t[NO]a (μmol) | [NO]mb (pmol cm−2 sec−1) | t1/2c (h) | Duration Above 21 pmol cm−2 sec−1 (h) |
|---|---|---|---|---|
| NPE1A | 2.28 ± 0.21 | 429 ± 93 | 5.37 ± 0.09 | 16.31 ± 0.10 |
| NPE1B | 0.80 ± 0.10 | 210 ± 73 | 0.97 ± 0.44 | 0.61 ± 0.10 |
| NPE2A | 1.13 ± 0.02 | 481 ± 47 | 1.35 ± 0.23 | 2.93 ± 0.51 |
| NPE2B | 0.14 ± 0.06 | 136 ± 62 | 0.18 ± 0.03 | 0.43 ± 0.09 |
| NPE3A | 1.86 ± 0.14 | 384 ± 96 | 2.55 ± 0.20 | 4.60 ± 0.74 |
| NPE3B | 0.72 ± 0.29 | 215 ± 102 | 3.99 ± 0.98 | 0.60 ± 0.23 |
| NPE4A | 0.53 ± 0.26 | 347 ± 87 | 0.63 ± 0.18 | 1.09 ± 0.11 |
| NPE4B | 0.04 ± 0.01 | 53 ± 15 | 0.11 ± 0.05 | 0.08 ± 0.01 |
| NPE5A | 0.92 ± 0.16 | 324 ± 108 | 0.76 ± 0.14 | 1.79 ± 0.39 |
| NPE6A | 0.99 ± 0.04 | 697 ± 46 | 0.90 ± 0.25 | 1.16 ± 0.10 |
t[NO] = total NO released
[NO]m = maximum NO flux;
t1/2 = half life of NO release.
Although the total NO storage of a material is important to assess its utility as a potential therapeutic, many of the bioregulatory roles of NO, including vasodilation, bacterial killing, and wound healing, are flux dependent.32–36 As such, understanding NO release as a function of time is important. Hetrick et al. previously determined that a 65% reduction of P. aeruginosa adhesion to a surface was achievable using NO fluxes of ~21 pmol cm−2 sec −1.35 The maximum NO fluxes released from the polyesters prepared herein all exceeded the 21 pmol cm−2 sec −1 threshold when immersed in PBS solutions at 37 °C (Table 4). The magnitude of NO surface fluxes displayed similar trends to the total NO released, with all cysteamine-functionalized materials releasing greater amounts of NO than their penicillamine-functionalized counterparts. Additionally, both the NO flux and total NO release proved tunable by varying the extent of aminothiol coupling to the polyester backbone (Figure 5).
Figure 5.

Total NO release triggered by direct light irradiation for NPE1A as a function of cysteamine concentration in functionalization solutions (— 2 mM cysteamine, --- 1 mM cysteamine).
Bacterial Adhesion
Although NO-releasing materials have been shown to inhibit bacterial adhesion at their surfaces in a flux-dependent manner,35 differences in surface chemistry between materials undoubtedly influence the extent of adhesion and propagation of bacteria. As such, the ability of the NO-releasing polyesters to inhibit bacterial adhesion was investigated and compared to control polyesters. Indeed, the NO-releasing NPE3A, NPE4A, and NPE6A polyesters significantly reduced P. aeruginosa adhesion to the material surface in a dose-dependent manner compared to both unmodified and cysteamine-modified controls (Figure 6, Table 5). The reduction in bacterial adhesion mirrors the total NO release for the three polyesters investigated with NPE3A > NPE6A > NPE4A. Interestingly, NPE3A caused only a 25% greater reduction in P. aeruginosa adhesion compared to NPE4A, despite releasing more NO. This behavior may be attributed to similarities in the maximum NO flux values for these two substrates. Previous studies have indicated that greater concentrations of NO over a short time period prevent bacterial adhesion to model substrates more effectively than lower fluxes over extended periods.35, 37 Nevertheless, the improved performance of NPE3A over NPE4A is likely due to the increased duration of NO release (t1/2 NPE3A: 2.55 ± 0.20 h, t1/2 NPE4A: 0.63 ± 0.18 h). The utilization of slowly degrading polyesters ensures that reduced bacterial adhesion due to interfacial degradation of the polyesters over the duration of the experiment is an unlikely source of error.
Figure 6.
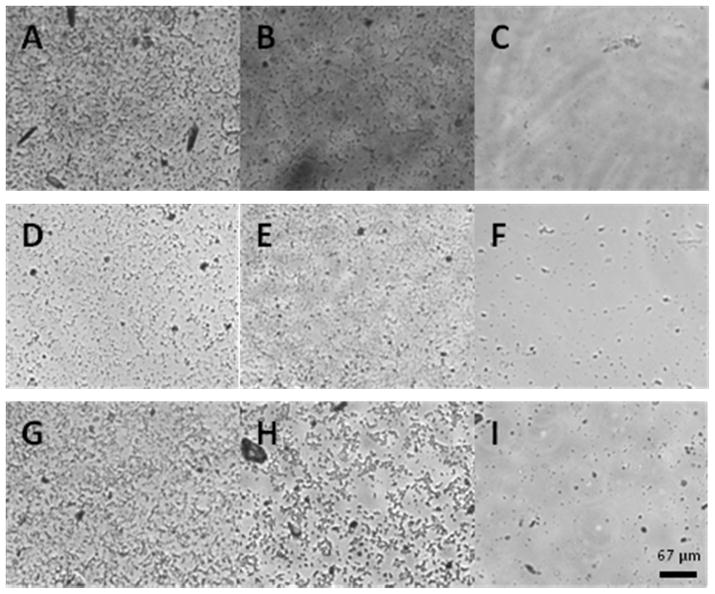
Representative bright field optical micrographs of P. aeruginosa surface coverage on polyester substrates. A) PE3, B) FPE3A, C) NPE3A, D) PE4, E) FPE4A, F) NPE4A, G) PE6, H) FPE6A, and I) NPE6A. Dark spots are bacteria.
Table 5.
Reduction of P. aeruginosa adhesion to representative NO-releasing polyester substrates.
| % Bacterial coverage | % Bacterial coverage of unmodified controls | % Reduction | ANOVA P- value | % Bacterial coverage of cysteamine modified controls | % Reduction | ANOVA P- value | |
|---|---|---|---|---|---|---|---|
| NPE3A | 3.98 ± 2.40 | 20.04 ± 7.77 | 80 | 3.51 × 10−20 | 20.94 ± 5.88 | 81 | 3.51 × 10−30 |
| NPE4A | 11.19 ± 6.94 | 24.89 ± 8.15 | 55 | 1.45 × 10−9 | 25.82 ± 10.61 | 57 | 1.03 × 10−9 |
| NPE6A | 7.03 ± 3.07 | 20.28 ± 8.41 | 65 | 5.92 × 10−15 | 27.63 ± 6.03 | 75 | 1.17 × 10−33 |
In vitro toxicity of polyesters
To assess the utility of these polymers for biomedical applications, the cytotoxicity of the starting materials and their degradable components was evaluated against healthy mammalian cells. L929 mouse fibroblasts were employed for these studies due to the prevalence of fibroblasts in both the extracellular and wound healing environments.38 As shown in Figure 7, the PE1 and PE2 materials exhibited the greatest cytotoxic response (34 ± 2 and 53 ± 20% viable cells, respectively) through 24 h. Despite this, the cell viabilities observed for all other material compositions were = 65% indicating minimal cytotoxicity. The rapid degradation of PE1 and PE2 likely resulted in elevated toxicity due to larger amounts of low-molecular weight degradation products in the soak media compared to the slower degrading materials (e.g., PE3 and PE4). Of note, the functionalized (e.g., FPE1A and FPE2A) and nitrosated (e.g., NPE1A and NPE2A) counterparts of PE1 and PE2 elicited much less cytotoxicity (Figure 7, Table 6), suggesting that appreciable unpolymerized starting material may also be leaching from PE1 and PE2. The soaking of these polyesters in aqueous solutions (required for subsequent functionalization) reduced such cytotoxicity.
Figure 7.

Viability of L929 mouse fibroblasts exposed to polyester leachables and degradation products for 24 h.
Table 6.
Statistical analysis of cell viability of L929 mouse fibroblasts exposed to polyester degradation products
| % Cell Viability | % Cell viability of unmodified controls | ANOVA P- value | % Cell viability of cysteamine modified controls | ANOVA P- value | |
|---|---|---|---|---|---|
| NPE1A | 68.17 ± 11.00 | 33.79 ± 2.11 | 3.25 × 10−9 | 74.17 ± 20.71 | 0.481 |
| NPE2A | 87.59 ± 17.10 | 52.56 ± 20.21 | 0.198 | 65.95 ± 11.10 | 0.954 |
| NPE3A | 93.53 ± 19.96 | 94.00 ± 27.45 | 0.637 | 92.01 ± 1.80 | 0.664 |
| NPE4A | 87.27 ± 16.60 | 102.09 ± 13.96 | 0.033 | 85.11 ± 8.26 | 0.852 |
Conclusion
The thermal polycondensation of diacids with polyols was demonstrated as a versatile route for the formation of degradable NO-releasing polyesters. The degradation rates and thermal properties of the synthesized polyesters varied widely based on polymer composition and curing conditions. These polymers were easily modified by controlling the stoichiometry of the prepolymer melt-phase reaction and by using aqueous phase coupling with the terminal acid functionalities on the crosslinked polyester. Total incorporation of NO-donating moieties proved dependent on the thermal properties of the polymers as materials with lower glass transition temperatures have been shown to store and release more NO than materials with higher transition temperatures. The cysteamine- and penicillamine-modified polyesters prepared released between 0.01 – 0.81 μmol NO cm−2 for up to 6 days (pH 7.4, 37 °C). Nitric oxide release from these polyesters reduced bacterial adhesion to model substrates by up to 80% compared to controls. The synthesis of absorbable nitric oxide-releasing polyesters represents a significant advancement in the development of implantable materials with enhanced antifouling properties. Future studies are planned to evaluate the performance of these polymers in vivo.
Supplementary Material
Scheme 1.

Representative synthesis of NO-releasing polyesters derived from glutaric acid and glycerol.
Table 1.
Composition of synthesized polyesters.
| Polyester | Diacid | Polyol(s) | Thiol | Nitrosated (y/n) |
|---|---|---|---|---|
| PE1 | Glutaric Acid | Glycerol | - | n |
| FPE1A | Glutaric Acid | Glycerol | Cysteamine | n |
| NPE1A | Glutaric Acid | Glycerol | Cysteamine | y |
| FPE1B | Glutaric Acid | Glycerol | Penicillamine | n |
| NPE1B | Glutaric Acid | Glycerol | Penicillamine | y |
| PE2 | Adipic Acid | Glycerol | - | n |
| FPE2A | Adipic Acid | Glycerol | Cysteamine | n |
| NPE2A | Adipic Acid | Glycerol | Cysteamine | y |
| FPE2B | Adipic Acid | Glycerol | Penicillamine | n |
| NPE2B | Adipic Acid | Glycerol | Penicillamine | y |
| PE3 | Glutaric Acid | Pentaerythritol | - | n |
| FPE3A | Glutaric Acid | Pentaerythritol | Cysteamine | n |
| NPE3A | Glutaric Acid | Pentaerythritol | Cysteamine | y |
| FPE3B | Glutaric Acid | Pentaerythritol | Penicillamine | n |
| NPE3B | Glutaric Acid | Pentaerythritol | Penicillamine | y |
| PE4 | Adipic Acid | Pentaerythritol | - | n |
| FPE4A | Adipic Acid | Pentaerythritol | Cysteamine | n |
| NPE4A | Adipic Acid | Pentaerythritol | Cysteamine | y |
| FPE4B | Adipic Acid | Pentaerythritol | Penicillamine | n |
| NPE4B | Adipic Acid | Pentaerythritol | Penicillamine | y |
| PE5 | Glutaric Acid | Glycerol, Pentaerythritol | - | n |
| FPE5A | Glutaric Acid | Glycerol, Pentaerythritol | Cysteamine | n |
| NPE5A | Glutaric Acid | Glycerol, Pentaerythritol | Cysteamine | y |
| PE6 | Adipic Acid | Glycerol, Pentaerythritol | - | n |
| FPE6A | Adipic Acid | Glycerol, Pentaerythritol | Cysteamine | n |
| NPE6A | Adipic Acid | Glycerol, Pentaerythritol | Cysteamine | y |
Acknowledgments
This work was supported by the National Institutes of Health (NIH EB000708). We thank Birgit Andersen, College of Textiles, North Carolina State University, for assistance with product analysis.
Footnotes
Supporting Information Available: Kinetic plots for degradation experiments, NO-release temperature dependence, thermal analysis data for functionalized and nitrosated polyesters, and 1H and 13C NMR spectra for prepolymers. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nair LS, Laurencin CT. Prog Polym Sci. 2007;32:762–798. [Google Scholar]
- 2.Darouiche RO. N Engl J Med. 2004;350:1422–1429. doi: 10.1056/NEJMra035415. [DOI] [PubMed] [Google Scholar]
- 3.Stamm WE. Ann Intern Med. 1978;89:764–769. doi: 10.7326/0003-4819-89-5-764. [DOI] [PubMed] [Google Scholar]
- 4.Jarvis WR. Infect Control Hosp Epidemiol. 1996;17:552–557. doi: 10.1086/647371. [DOI] [PubMed] [Google Scholar]
- 5.Ramcharitar S, Serruys PW. Am J Cardiovasc Drugs. 2008;8:305–314. doi: 10.2165/00129784-200808050-00003. [DOI] [PubMed] [Google Scholar]
- 6.Lim TY, Poh CK, Wang W. J Mater Sci: Mater Med. 2009;20:1669–1675. doi: 10.1007/s10856-009-3727-z. [DOI] [PubMed] [Google Scholar]
- 7.Amsden B. Soft Matter. 2007;3:1335–1348. doi: 10.1039/b707472g. [DOI] [PubMed] [Google Scholar]
- 8.Wei G, Ma PX. Adv Funct Mater. 2008 doi: 10.1002/adfm.200800662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waris E, Ashammakhi N, Lehtimaki M, Tulamo RM, Kellomaki M, Tormala P, Konttinen YT. Biomaterials. 2008;29:683–691. doi: 10.1016/j.biomaterials.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 10.Kuijer R, Jansen EJP, Emans PJ, Bulstra SK, Riesle J, Pieper J, Grainger DW, Busscher HJ. Biomaterials. 2007;28:5148–5154. doi: 10.1016/j.biomaterials.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Smith AW. Adv Drug Delivery Rev. 2005;57:1539–1550. doi: 10.1016/j.addr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 12.An YH, Friedman RJ. J Biomed Mater Res: Appl Biomater. 1997;43:338–348. [Google Scholar]
- 13.Liu VWT, Huang PL. Cardiovasc Res. 2008;77:19–29. doi: 10.1016/j.cardiores.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murohara T, Asahara T. Antioxid Redox Signal. 2002;4:825–831. doi: 10.1089/152308602760598981. [DOI] [PubMed] [Google Scholar]
- 15.Witte MB, Barbul A. Am J Surg. 2002;183:406–412. doi: 10.1016/s0002-9610(02)00815-2. [DOI] [PubMed] [Google Scholar]
- 16.Fang FC. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hetrick EM, Shin JH, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:2782–2789. doi: 10.1016/j.biomaterials.2009.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, Janczuk AJ. Chem Rev. 2002;102:1091–1134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 19.Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135–1154. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 20.Miller MR, Megson IL. Br J Pharmacol. 2007;151:305–321. doi: 10.1038/sj.bjp.0707224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams DLH. Acc Chem Res. 1999;32:869–876. [Google Scholar]
- 22.Seabra AB, Rocha LLD, Eberlin MN, Oliveira MGD. J Pharm Sci. 2005;95:994–1003. doi: 10.1002/jps.20314. [DOI] [PubMed] [Google Scholar]
- 23.Saavedra JE, Southan GJ, Davies KM, Lundell A, Markou C, Hanson SR, Adrie C, Hurford WE, Zapol WM, Keefer LK. J Med Chem. 1996;39:4361–4365. doi: 10.1021/jm960616s. [DOI] [PubMed] [Google Scholar]
- 24.Marxer SM, Rothrock AR, Nablo BJ, Robbins ME, Schoenfisch MH. Chem Mater. 2003;15:4193–4199. [Google Scholar]
- 25.Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:4494–4502. doi: 10.1016/j.biomaterials.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jun HW, Taite L, West JL. Biomacromolecules. 2005;6:838–844. doi: 10.1021/bm049419y. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds MM, Hrabie JA, Oh BK, Politis JK, Citro ML, Keefer LK, Meyerhoff ME. Biomacromolecules. 2006;7:987–994. doi: 10.1021/bm060028o. [DOI] [PubMed] [Google Scholar]
- 28.Barrett DG, Yousaf MN. Macromolecules. 2008;41:6347–6352. [Google Scholar]
- 29.Lendlein A, Langer R. Science. 2002;296:1673–1676. doi: 10.1126/science.1066102. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Webb AR, Ameer GA. Adv Mater. 2004;16:511–516. [Google Scholar]
- 31.Nagata M, Kiyotsukuri T, Ibuki H, Tsutsumi N, Sakai W. React Funct Polym. 1996;30:165–171. [Google Scholar]
- 32.Lowenstein CJ, Dinerman JL, Snyder SH. Ann Intern Med. 1994;120:227–237. doi: 10.7326/0003-4819-120-3-199402010-00009. [DOI] [PubMed] [Google Scholar]
- 33.Wink DA, Mitchell JB. Free Radical Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 34.Lee RH, Efron D, Tantry U, Barbul A. J Surg Res. 2001;101:104–108. doi: 10.1006/jsre.2001.6261. [DOI] [PubMed] [Google Scholar]
- 35.Hetrick EM, Schoenfisch MH. Biomaterials. 2007;28:1948–1956. doi: 10.1016/j.biomaterials.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Robbins ME, Hopper ED, Schoenfisch MH. Langmuir. 2004;20:10296–10302. doi: 10.1021/la048368n. [DOI] [PubMed] [Google Scholar]
- 37.Hetrick EM, Shin JH, Stasko NA, Johnson CB, Wespe DA, Holmuhamedov E, Schoenfisch MH. ACS Nano. 2008;2:235–246. doi: 10.1021/nn700191f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong T, McGrath JA. Br J Dermatol. 2007;156:1149–1155. doi: 10.1111/j.1365-2133.2007.07914.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.