

Abstract
RNA containing 5-fluorouridine (F5U) had previously been used to examine the mechanism of the pseudouridine synthase TruA, formerly known as pseudouridine synthase I [Gu et al. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 14270–14275]. From that work, it was reasonably concluded that the pseudouridine synthases proceed via a mechanism involving a Michael addition by an active site aspartic acid residue to the pyrimidine ring of uridine or F5U. Those conclusions rested on the assumption that the hydrate of F5U was obtained after digestion of the product RNA and that hydration resulted from hydrolysis of the ester intermediate between the aspartic acid residue and F5U. As reported here, 18O labeling definitively demonstrates that ester hydrolysis does not give rise to the observed hydrated product and that digestion generates not the expected mononucleoside product but rather a dinucleotide between a hydrated isomer of F5U and the following nucleoside in RNA. The discovery that digestion products are dinucleotides accounts for the previously puzzling differences in the isolated products obtained following the action of the pseudouridine synthases TruB and RluA on F5U in RNA.
The pseudouridine synthases (Ψ synthases)1 catalyze the most common post-transcriptional modification in RNA, the isomerization of uridine to pseudouridine (Ψ). Based on sequence data, the Ψ synthases were grouped into six families that share no global statistically significant sequence similarity.(1) Several conserved motifs were identified, however, and one included the only absolutely conserved amino acid residue, an Asp that is essential for activity.(2–6) The structural elucidation of members of each family revealed that the Ψ synthases share the same overall fold and are thus related by divergent rather than convergent evolution.(1)
Given their same overall fold and largely conserved constellation of active site residues (including the essential Asp), the families all likely follow the same catalytic mechanism. Two alternatives seem likely from chemical precedent and mechanistic work. In the first (Figure 1, panel A), the essential Asp nucleophilically attacks the pyrimidine ring in a Michael addition to make an ester intermediate.(7) After breakage of the glycosidic bond, the pyrimidine ring rotates about the new ester bond to reposition C5 near C1′ to form the characteristic C-glycosidic linkage of Ψ. Deprotonation of C5 and protonation of N1 complete the reaction. In the second mechanism (Figure 1, panel B), the essential Asp nucleophilically attacks the ribose rather than the pyrimidine ring to form an acylal intermediate.(2) The detached pyrimidine ring rotates to allow formation of a bond between C5 and C1′ to generate the C-glycoside, the deprotonation of which generates Ψ.
Figure 1.

The two mechanisms proposed for Ψ synthases. A, The catalytic Asp is a nucleophile in a Michael addition to the pyrimidine ring. B, The catalytic Asp is a nucleophile in the formation of an acylal intermediate. The stereochemistry at C5 and C6 in intermediates and products reflects that observed in the cocrystal of TruB and [F5U]TSL.(8) The depiction of the catalytic Asp as the general base for the final deprotonation rests on the interpretation of the pH dependence of the TruB reaction (20) and differs from conclusions drawn by Phannacet et al. (15), as has previously been discussed in some detail (12).
To examine the mechanism of the Ψ synthases, Santi and co-workers employed the mechanistic probe 5-fluorouridine (F5U) in the in vitro transcript of Escherichia coli tRNAPhe (abbreviated [F5U]tRNA) and TruA (formerly called Ψ synthase I).(2, 7) These two species form a covalent adduct as judged by the irreversible inhibition of the enzyme and a shifted band containing both TruA and RNA in denaturing gels. The adduct was heat-disrupted, and the RNA was isolated. The mass spectrum of the oligonucleotides resulting from digestion of the RNA by RNase A (which cuts after U and C) clearly showed that F5U had become hydrated. The RNA was also digested with nuclease P1 and alkaline phosphatase to generate the component nucleosides, which were examined by chromatography (for these experiments, tritiated F5UTP was used to prepare the [F5U]tRNA so that the small amount of material could be sensitively tracked). The component nucleosides were examined by cellulose thin layer chromatography, and the nucleoside product of F5U behaved very similarly to the two isomers of authentic hydrated F5U, but the TLC conditions did not resolve the hydrated F5U isomers from each other. The nuclease P1/alkaline phosphatase digestion products were also examined by reverse phase HPLC, which cleanly resolves the two isomers of photohydrated F5U. As judged by comparison of the UV trace with the tritium content of collected fractions, the nucleoside product of F5U co-eluted from the reverse phase HPLC column with one isomer of authentic hydrated F5U. From these results, Santi and co-workers (7) reasonably concluded that TruA operates via the mechanism involving Michael addition of the active site Asp to the pyrimidine ring, with the enzyme “getting stuck” at the first ester intermediate, which neatly explains the irreversible inhibition and covalent adduct. When heated, the intermediate underwent ester hydrolysis to generate hydrated F5U (Figure 2).
Figure 2.
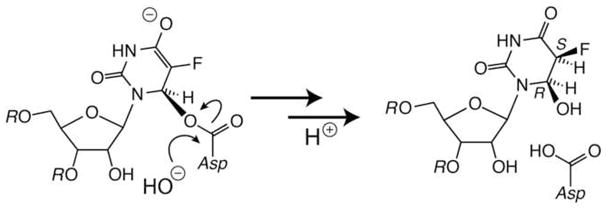
The hydrolysis of the initial Michael adduct between F5U in RNA and the catalytic Asp proposed by Gu et al. (7) followed by implicit protonation of C5.
This general scenario seemed to be supported by the co-crystal structure of the Ψ synthase TruB with an RNA stem-loop corresponding to its natural substrate but containing F5U in place of the isomerized U.(8) A covalent adduct was not observed, but the product of F5U was hydrated; surprisingly, that product was also rearranged to the C-glycoside as U is rearranged to generate Ψ. The crystallographers reasonably concluded that the enzyme proceeded beyond the initial ester intermediate to the C-glycoside, the ester group of which underwent slow hydrolysis during crystal growth and data acquisition.(8) Subsequent examination of TruB and its [F5U]stem-loop revealed evidence for neither potent inhibition nor the formation of a covalent adduct but instead indicated that TruB handles the F5U as a substrate and converted it into two products that are partially resolvable by reverse phase HPLC.(9) MALDI-MS analysis of the oligonucleotides resulting from the digestion of product stem-loops with RNase T1 (which cuts after G) demonstrated that the product of F5U was hydrated. Labeling studies with [18O]water definitively showed, however, that the hydrated products did not arise from ester hydrolysis.(10) Although disallowing the reasonable conclusion that hydrated products indicated that the Michael mechanism was followed, the labeling results were consistent with both proposed mechanisms.
A similar suite of experiments with the Ψ synthase RluA revealed that it behaved similarly to TruA in its stoichiometric irreversible inhibition by [F5U]RNA with apparent covalent adduct formation.(9) A cocrystal of RluA and [F5U]RNA, however, did not reveal the expected covalent linkage, but the adduct was shown to be sensitive to X-irradiation and so likely decomposed in the crystal.(11) After heat disruption of the adduct and S1 nuclease/phosphatase digestion, HPLC analysis revealed a single product peak of F5U from the action of RluA that did not co-elute with either product of F5U from the action of TruB, and MALDI-MS analysis of the oligonucleotides resulting from the RNase T1 digestion of product stem-loop confirmed that RluA causes the hydration of F5U.(12) 18O labeling studies confirmed that hydration did not result from ester hydrolysis.(12)
TruA, TruB, and RluA each belong to different Ψ synthase families, and even though they share the same overall fold and active site architecture, it remained possible that interfamilial variation could lead to different outcomes from the same suite of experiments. The labeling studies were extended to TruA to examine whether or not ester hydrolysis occurs during the hydration of F5U. Further characterization of the products of F5U from the action of each enzyme was undertaken to ascertain whether or not Ψ synthases of different families handle F5U differently. In particular, the behavior of the nuclease P1/alkaline phosphatase digestion products on reverse phase HPLC was examined to check the assignment of the product of F5U from TruA action as the hydrate of the parent nucleoside (with the C–N glycosidic bond intact as opposed to the C–C glycosidic bond observed in the TruB cocrystal) and the apparent formation of two products of F5U by TruB action as opposed to a single different product by RluA action.
Materials and Methods
General
Chemicals were purchased from Fisher Scientific (Pittsburgh, PA) or its Acros Organics division (Pittsburgh, PA) unless otherwise noted. Alkaline phosphatase and S1 nuclease2 were purchased from Promega (Madison, WI). Radiant Red RNA stain was purchased from Biorad (Hercules, CA).3 SYBR® Gold nucleic stain was obtained from Invitrogen (Carlsbad, CA). T7 RNA polymerase was purchased from Epicentre Biotechnologies (Madison, WI), and MEGAshortscript™ T7 transcription kits were purchased from Applied Biosystems (Foster City, CA). Electro-elution of RNA from polyacrylamide gels was accomplished using an Elutrap device (Whatman Inc., Piscataway, NJ) following the manufacturer’s protocol. “UV shadowing” was used to locate RNA-containing gel bands; the gel was laid on plastic wrap over a silica TLC plate impregnated with a fluorescent indicator, and the gel was illuminated with a handheld UV lamp, resulting in a “shadow” on the plate beneath the RNA-containing band (because the RNA absorbs UV light). All experiments were performed with water (≥18.2 MΩ) from a MilliQ Synthesis system (Millipore, Billerica, MA). Reaction buffer is 50 mM HEPES, pH 7.5, containing ammonium chloride (100 mM), dithiothreitol (5 mM), and EDTA (1 mM).
HPLC
HPLC was performed either on an Agilent 1100 system composed of a binary pump, micro vacuum degasser, variable-wavelength detector, and thermostated autosampler with an extended volume upgrade kit run by ChemStation software (Agilent Technologies, Santa Clara, CA) or on a Beckman System Gold system composed of a 125 gradient pump module, 508 autosampler, and 168 photodiode array detector run by 32Karat XP software (Beckman Coulter, Fullerton, CA). Analytical runs of S1 nuclease/alkaline phosphatase digestion products were performed as described previously.(12) Preparative runs of the products of F5U are described in detail below.
NMR
NMR spectra were acquired using INOVA 500 NMR spectrometers (Varian Inc., Palo Alto, CA) equipped with either a 5 mm inverse HFX z-PFG probe (Wang NMR, Inc., Palo Alto, CA) or a 5 mm direct broadband probe (Varian). Spectra were processed with VNMRJ v2.1c (Varian) or MestReC v4.9.9.6 (Mestrelab Research, Santiago de Compostela, Spain).
RNA oligonucleotides and their digestion
Synthetic RNA oligonucleotides with 2′-ACE protecting groups were purchased from Dharmacon (Boulder, CO), deprotected according to the manufacturer’s protocol, and desalted as previously described.(12) The RNA oligonucleotide corresponding to the anticodon stem-loop of Escherichia coli tRNAPhe with F5U in place of the isomerized U, [F5U]ASL, is GGGGAF5UUGAAAAUCCCC. The RNA oligonucleotide corresponding to the T-arm stem-loop of Saccharomyces cerevisiae tRNAPhe with F5U in place of the isomerized U, [F5U]TSL, is CUGUGUF5UCGAUCCACAG.
Digestion of RNA with S1 nuclease and alkaline phosphatase was achieved as described previously with the latter enzyme added directly to the sample in nuclease buffer.(12) For large scale sample preparation, glycerol was removed from S1 nuclease and alkaline phosphatase before their use by exchanging them into the manufacturer’s S1 nuclease buffer by dialysis, ultrafiltration, or spin size-exclusion chromatography.
Synthesis of F5U derivatives
5-Fluorouridine (Acros), F5U, was phosphorylated by the method of Darlix et al. (13) to afford F5UMP, which was converted to F5UTP enzymatically using the method of Tolbert and Williamson.(14) F5U was photohydrated to the 5S,6R and 5R,6S isomers of 5,6-dihydro-6-hydroxy-5-fluorouridine according to the method of Gu et al.(7) Detailed procedures and characterization data can be found in the supplementary material.
In vitro transcription of [F5U]tRNA
The in vitro transcription of E. coli tRNAPhe was performed either as described previously (3) with the substitution of F5UTP (4 mM) for UTP or using MEGAshortscript T7 transcription kits according to the manufacturer’s protocol with F5UTP (2.25 mM) substituted for UTP. The resulting crude [F5U]tRNA was purified by urea-PAGE (5% gel, 30 min, 160 V or 6% gel, 80 min, 150 V; both at room temperature); the band was located by UV shadowing for excision and subsequent electro-elution. All batches of [F5U]tRNA were characterized by MALDI-MS after digestion with RNase A and/or RNase T1. Concentration was determined by A260 using an extinction coefficient of 5 × 105 M−1cm−1, and aliquots were quick-frozen and stored at −80 °C. Before use, aliquots were quick-thawed, incubated at 100 °C for 5 min then snap-cooled.
Overexpression and purification of Ψ synthases
TruB was overexpressed, purified, and stored as previously described (3) with an additional polishing step over a column (4.6 × 100 mm) of POROS HQ anion exchange resin (Applied Biosystems), eluting with a gradient (0–1.5 M) of sodium chloride in 50 mM Tris·HCl buffer, pH 8.5. RluA was overexpressed, purified, and stored as previously described.(11)
TruA was overexpressed in E. coli BLR(DE3) pLysS cells (Novagen, Madison, WI) from a plasmid (pBH500) with truA inserted in-frame into pET15b (Novagen) at the NdeI site to cause expression of TruA with an N-terminal His6·tag immediately preceding Met-1 of the native enzyme. The cells were lysed and cell extract treated and subjected to chromatography over a column of Ni-NTA resin (Novagen) as described previously for TruB and RluA. (3) The TruA-bearing fractions were combined and dialyzed against 50 mM potassium phosphate buffer, pH 6.5, then subjected to chromatography over a column (4.6 × 100 mm) of POROS HS cation exchange resin (Applied Biosystems), eluting with a gradient (0–1.5 M) of potassium chloride in the same buffer. TruA-bearing fractions were tested for RNase activity, and clean fractions were combined and dialyzed into 20 mM Tris·HCl buffer, pH 7.5 containing EDTA (0.1 mM), sodium chloride (300 mM), β-mercaptoethanol (5 mM), and glycerol (10% v/v). TruA was typically stored at 4 °C and used within one month; for longer storage at −20 °C, glycerol was added to 50% (v/v). An extinction coefficient at 280 nm of 41 840 M−1cm−1 was calculated using A280 and the protein concentration determined by biuret assay using bovine serum albumin as the standard, as previously described (3).
Formation and denaturing gel analysis of the TruA-[F5U]RNA adduct
TruA (8–40 μM) and [F5U]tRNA (1–16 μM) were incubated for 5 min to 48 h at either 20 °C or 37 °C in 20 mM Tris·HCl buffer, pH 8.0, containing ammonium chloride (100 mM) and dithiothreitol (2 mM).(2) The various concentrations of protein and RNA in the numerous experiments ranged from a stoichiometry of 10:1 to 2:5 TruA:[F5U]tRNA. To measure the extent of adduct formation, aliquots of the incubations were subjected to SDS-PAGE (10% gel) or urea-PAGE (5% or 6% gel; 8 M urea) both with and without prior incubation of the samples at 100 °C for 5 min. Gels were stained first with Radiant Red or SYBR Gold to detect RNA3 and then with Coomassie Blue to detect protein. Complete adduction was very rarely observed. Suspecting that in-gel decomposition might be reducing the apparent yield of adduct, minigels (7 cm × 8 cm or 10 cm × 10 cm) were run at 4 °C and lower voltage (60 V versus 160 V), or larger format (16 cm × 16 cm) gels were run in a chamber (Hoefer SE 600; GE Healthcare) with a heat exchanger connected to a circulating bath to maintain constant temperature over the gel throughout the run. These measures only modestly improved the yield. In the end, the best results were obtained with an excess of TruA over [F5U]RNA (5:2), which typically led to 65–70% of (limiting) RNA found in the adduct based on urea-PAGE analysis.
18O labeling studies
To determine whether oxygen from solvent was incorporated into the active site Asp-60 of TruA or into the hydrated product of F5U, the TruA-[F5U]tRNA adduct was formed in buffer containing 50% [18O]water, purified, and heat-disrupted, followed by the separate MALDI-MS analysis of protein and RNA after digestion with trypsin and RNase T1, respectively, to determine the location of the isotopic label. The adduct between TruA and [F5U]tRNA was formed by incubating [F5U]tRNA (10 μM) for 5 h at room temperature with TruA (40 μM) in 20 mM Tris·HCl buffer, pH 8.0, containing ammonium chloride (100 mM) and dithiothreitol (2 mM) (2). Adduction was confirmed by SDS-PAGE analysis of a small aliquot. The bulk of the reaction mixture was diluted with an equal volume of neat formamide and subjected to urea-PAGE. The adduct band was located by UV shadowing and excised, and the adduct was electro-eluted and concentrated in a Microcon-3 ultrafiltration device (Millipore). The sample was then incubated for 5 min at 100 °C, and precipitated protein was pelleted by centrifugation. The supernatant was washed with phenol/chloroform and the RNA precipitated with ethanol. The digestion and MALDI-MS analysis of the TruA and RNA from the disrupted adduct followed the procedure described in detail for the similar study of the adduct between RluA and [F5U]RNA (12) except that 6-aza-2-thiothymine was used as the matrix for MALDI-MS of oligonucleotides; the procedure includes the addition of alkaline phosphatase after RNase to remove 3′-phospho groups, which themselves incorporate an oxygen atom from solvent during the digestion. For comparison, the adduct was formed separately in both labeled and unlabeled buffer, and after gel purification, heat disruption of the adduct was conducted in both labeled and unlabeled buffer. All four combinations were covered: both formation and disruption in unlabeled buffer; formation in unlabeled and disruption in labeled buffer; formation in labeled and disruption in unlabeled buffer; both formation and disruption in labeled buffer.
Large scale preparation of the products of F5U from the action of TruB
TruB (10 μM) and [F5U]TSL (100 μM) were incubated in reaction buffer (400 μL). After 3 h at 37 °C, an aliquot (3.5 μL) was removed and subjected to digestion and HPLC analysis in order to verify that the reaction was complete. The protein was removed from the aqueous phase by vortex mixing with an equal volume of phenol saturated with TE buffer, pH 4.3, and then with an equal volume of chloroform:isoamyl alcohol (24:1). The RNA in the aqueous layer was precipitated by adding first sodium acetate buffer, pH 4.6 (to 300 mM), and then an equal volume of cold absolute ethanol (−20 °C). After incubation for at least 3 h at −20 °C, the precipitate was collected by cold centrifugation (30 min, 13 100g). The pellet was gently washed twice with cold aqueous ethanol (70% v/v; −20 °C) and air dried. The RNA pellet was dissolved in S1 nuclease buffer and digested with S1 nuclease (450 U) and alkaline phosphatase (9 U).
The resulting nucleoside products were purified by reverse phase HPLC over a preparatory Ultrasphere C18 column (5 μm, 10 × 250 mm; Beckman-Coulter) with an acetonitrile gradient by use of the following program (the first number is the percentage of aqueous acetonitrile (40% v/v); the second number is the elapsed time in minutes): 0, 0; 0, 3; 5, 8; 15, 18; 15, 23; 30, 26; 50, 27.5; 50, 29; 100, 30; 100, 31; 0, 32; 0, 39; 0, 40. All gradient steps were linear. Fractions (1.5 mL) were subjected to analytical HPLC to locate the products of F5U. Product-bearing fractions were combined, taken to dryness in vacuo, redissolved in 50 mM sodium phosphate buffer in D2O (500 μL), pD 7.9, containing sodium 4,4-dimethyl-4-silapentane-1-sulfonate-d6 (0.12 mM; kindly provided by T. Fan, U. of Louisville), and subjected to NMR and nESI-MS analysis.
Large scale preparation of the products of F5U from the action of RluA
RluA (200 μM) and [F5U]ASL (150 μM) were incubated for 3 h at 37 °C in standard assay buffer (1.6 mL), with the completion of reaction verified by SDS-PAGE gel shift analysis. The [F5U]ASL–RluA adduct was concentrated by using an Amicon Ultra-10 centrifugal ultrafiltration device (Millipore), and to remove unreacted RluA and [F5U]ASL, the crude adduct sample was purified as previously described (11) with slight variation. The adduct was subjected to anion exchange chromatography over a column of POROS HQ resin (Applied Biosystems). Elution was achieved with a linear gradient of sodium chloride (0–1 M) in 30 mM Tris·HCl buffer, pH 8.5, containing EDTA (1 mM) and DTT (1 mM). Adduct-bearing fractions were identified by SDS-PAGE analysis, combined, and concentrated in an Amicon Ultra-10 ultrafiltration device. To achieve heat disruption of the adduct, the sample was incubated for 5 min at 100 °C in a dry heating block. Heat-precipitated RluA was pelleted by centrifugation (10 min, 18 000g), and the supernatant was spin-filtered (0.22 μm, cellulose acetate; Corning, Upton, NY).
The modified [F5U]ASL was simultaneously digested and dephosphorylated by the addition of glycerol-free S1 nuclease (450 units) and calf intestine alkaline phosphatase (9 units). After overnight at 37 °C, the reaction was judged complete from the reverse phase HPLC analysis of an aliquot (2.5 μL). The product of F5U was then purified by reverse phase HPLC over a preparatory C18 column as described above for the products of F5U from TruB action. Product-bearing fractions were combined, taken to dryness in vacuo, and redissolved in 50 mM sodium phosphate buffer in D2O (500 μL), pD 7.9, containing sodium 4,4-dimethyl-4-silapentane-1-sulfonate-d6 (0.12 mM) for analysis by NMR and nESI-MS.
Mass spectrometry
Matrix-assisted laser desorption ionization spectrometry, MALDI-MS, was performed on either a Ominflex (Bruker Daltonics, Inc., Billerica, MA) or a Voyager DE-PRO (Applied Biosystems) MALDI-TOF mass spectrometer as described previously.(12) Nano-electrospray ionization mass spectrometry, nESI-MS, was performed using an LTQ-FT mass spectrometer (Thermo Scientific, Waltham, MA) equipped with a TriVersa NanoMate direct infusion nano-ESI ion source (Advion BioSystems, Ithaca, NY). Aliquots (0.25–0.5 μL) of the NMR samples of the products of F5U from the action of TruB and RluA were diluted into water (25 μL), to which methanol (25 μL; Optima* LC/MS grade, Fisher) was then added. The mass spectra were acquired after at least 10 min at room temperature. The products of F5U from the action of TruA were analyzed similarly by dilution of the collected HPLC peaks with water and methanol; the quality of the mass spectrum was lower than for the other two cases, presumably because of ion suppression by buffer in the HPLC eluate. To increase sensitivity, a narrow range (± 3 m/z) around the ion of interest was isolated in the linear ion trap and then injected into the FT-ITCR to acquire high-resolution and high-accuracy spectra from 100–1000 m/z. The selected ranges (259–265, 277–83, 338–344, 356–362, 563–569, and 581–587) encompassed the candidate products (rearranged F5U, hydrated F5U, the monophosphate of F5U, the monophosphate of hydrated F5U, the dinucleotide of rearranged F5U and the following C, and the dinucleotide of hydrated F5U and the following C).
Results
Synthesis of F5UTP and [F5U]tRNA
F5UTP was obtained by the chemical phosphorylation of F5U, isolation of the resulting F5UMP, its subsequent enzymatic conversion to F5UTP, and purification by anion exchange chromatography. The F5UTP was used in place of UTP during the in vitro transcription of E. coli tRNAPhe.
Adduct formation
TruA and [F5U]tRNA were incubated together under many permutations of relative concentration, absolute concentration, temperature, and time, but the limiting partner was very rarely driven completely into the adduct as judged by denaturing gel analysis (Figure 3, which shows a rare near quantitative adduction). The routine yield of adduct was 65–70%. Running the gels at lower temperature did not significantly improve the apparent yield of adduct, strongly suggesting that decomposition in the gel does not account for the apparently incomplete reaction of limiting RNA or limiting enzyme. Huang et al. (2) reported complete adduction of [F5U]tRNA after overnight incubation with 25-fold excess TruA; their yield of 75% after 5 h is consistent with our typical results. Incomplete adduction does not compromise the results reported here since the [F5U]tRNA–TruA adduct was gel-purified before subjecting it to the characterization methods described below.
Figure 3.

Gel shift results implying a covalent adduct between TruA and [F5U]tRNA. Adduct was formed by incubating TruA (20 μM) and [F5U]RNA (10 μM) at 20 °C for 20 h. A, SDS-PAGE, stained for RNA with SYBR Gold (left) then for protein with Coomassie Blue (right). a, TruA; b, [F5U]tRNA; c, adduct; d, heat-disrupted adduct. RNA in the band consistently appears to decrease the efficiency of Coomassie Blue staining, leading to an adduct band (54.6 kD MWmeasured; 57.0 kD MWpredicted) that is lighter than expected based on the staining of an equal amount of loaded TruA (31.7 kD MWmeasured; 32.5 kD MWpredicted) and [F5U]tRNA (lowest band). The faint staining of protein by SYBR Gold is apparent. The band that runs at slightly lower MWapparent than the main adduct band is presumed to be a conformational isomer or a product of limited hydrolysis of TruA or [F5U]tRNA; the low abundance of the band (<10% of the total adduct) precluded further characterization. B, Urea-PAGE, stained for RNA with SYBR Gold. mw, RNA markers; a, TruA; b, [F5U]tRNA; c, adduct; d, heat-disrupted adduct. The adduct band (167 nt measured) runs slower than [F5U]tRNA (70 nt measured; 76 nt predicted). Coomassie Blue faintly stains the adduct and reveals that TruA mostly collects at the well bottom and enters the gel as a diffuse streak (data not shown).
Characterization of the adduct components by MALDI-MS
The gel-purified adduct was disrupted by heating; the denatured TruA was pelleted by centrifugation, and the RNA was ethanol-precipitated from the removed supernatant. The pelleted TruA was digested with trypsin, and the precipitated [F5U]tRNA was digested with RNase T1 (cuts after G). The resulting oligopeptides and oligonucleotides were analyzed by MALDI-MS as described previously.(9, 10, 12) The tryptic peptides covered 41% of the TruA sequence, including 59TDAGVHGTGQVVHFETTALR78 (2096.05 m/zpredicted; 2095.98 m/zobserved), which contains the active site Asp-60. Digestion with RNase T1 and alkaline phosphatase produced the expected oligonucleotides, which were compared to the oligonucleotides from digestion of [F5U]tRNA not incubated with TruA. The oligonucleotide containing F5U39 (35AAAA(F5U)CCCCG44; 3142.47 m/zpredicted; 3142.50 m/zobserved) shifted +18.05 m/z after adduct formation/disruption, clearly indicating that F5U39 suffered hydration (35AAAA(F5U-H2O)CCCCG44; 3160.46 m/zpredicted; 3160.55 m/zobserved).
18O labeling
To determine the source of oxygen added to F5U during adduct formation/disruption, the adduct was formed by incubating [F5U]RNA with TruA in buffer that contained 50% [18O]water. The gel purification was run entirely in unlabeled buffer, and the purified adduct was put back into buffer containing 50% [18O]water before heat disruption. The precipitated TruA and product [F5U]tRNA were separated, digested, and analyzed by MALDI-MS as was the unlabeled sample.
The mass spectra (Figure 4) clearly indicate that 18O ends up not in Asp-60 but in F5U39. A control in which the trypsinolysis was performed in buffer containing 50% [18O] water (so that 50% of the newly generated peptide C-terminal carboxylates will contain 18O rather than 16O) confirms that 18O in Asp-60 would be immediately detectable (data not shown). Other controls described previously (10, 12) to test for exchange of oxygen between solvent and carboxylate groups demonstrated that no such exchange occurs in TruA under any of the experimental conditions. The complementary analysis of the product oligonucleotides from [F5U]tRNA confirm that 18O is incorporated directly into F5U (Figure 4), and controls (10, 12) demonstrate that the incorporated oxygen does not exchange with solvent after the hydration event.
Figure 4.

Partial MALDI mass spectra showing incorporation of 18O from solvent into RNA rather than protein. A, The oligopeptide (59TDAGVHGTGQVVHFETTALR78; 2096.05 m/zpredicted) containing the catalytic Asp-60 after TruA from heat-disrupted adduct was subjected to trypsinolysis; upper trace, adduct was formed and disrupted in unlabeled buffer; lower trace, adduct was formed and disrupted in buffer containing 50% [18O]water. B, The oligonucleotide (35AAAA(F5U-H2O)CCCG43; 3160.46 m/zpredicted) containing the reactive F5U after [F5U]tRNA from heat-disrupted adduct was subjected to digestion with RNase T1; upper trace, adduct was formed and disrupted in unlabeled buffer; lower trace, adduct was formed and disrupted in buffer containing 50% [18O]water.
To probe whether hydration of F5U occurs during adduct formation or disruption, the analysis was repeated with adduct prepared in labeled buffer and disrupted in unlabeled buffer and vice versa. As expected, 18O was not incorporated into Asp-60 in either case (data not shown). Informatively, the isotopic content of the product oligonucleotides containing F5U39 matched the isotopic content of the buffer in which heat disruption was performed. In other words, adduct prepared in unlabeled buffer and disrupted in buffer containing [18O]water produced an identical mass spectrum to adduct both prepared and disrupted in labeled buffer (data not shown). Conversely, adduct prepared in labeled but disrupted in unlabeled buffer yielded a mass spectrum identical to the case in which [18O]water was never present (data not shown).
NMR analysis of F5U products
The appearance of two partially resolved peaks in the C18 HPLC analysis of the RNA digestion products after incubation of [F5U]TSL with TruB indicates that the enzyme converts F5U two products (10), but a similar analysis of [F5U]ASL after incubation with RluA gives a single product peak.(12) To start the definitive chemical identification of these distinct products, they were prepared and purified on a scale sufficient to allow the acquisition of their NMR spectra (Figure 5). First, [F5U]TSL was incubated with TruB (0.1 eq); after HPLC analysis revealed that reaction was complete, the protein was extracted with phenol/chloroform. The RNA was ethanol-precipitated and digested with S1 nuclease (non-sequence specific; generates nucleoside 5′-monophosphates) and alkaline phosphatase. The 1H NMR spectrum (Figure 5, panel A) of the products of F5U from the action of TruB (“TruB products” for ease of reference) unexpectedly reveals the presence of twice the expected ribose protons (3.5–4.6 ppm) and of a pair of cytidine protons (identified by chemical shift and JH5-H6 values) in the same ratio as the integration of the major and minor product peaks in the HPLC trace. This result immediately suggested that the major and minor products were both dinucleotides of the hydrated F5U products and the C that follows it in [F5U]TSL rather than the expected mononucleosides. The 31P NMR spectrum (data not shown) consisted of two resonances in the same ratio as the cytidine peaks in the 1H NMR spectrum and the integration of the HPLC trace, indicating that both products contained a bridging phospho group.
Figure 5.

1H NMR spectra of the products of F5U from Ψ synthase action. A, The TruB products. B, The RluA products. The appearance of cytidine and uridine resonances in the TruB and RluA products, respectively, indicates that digestion by S1 nuclease produced dinucleotides between the product of F5U and the nucleoside that follows it in the RNA. Both samples consist of a major and a minor product, accounting for the appearance of two sets of cytidine and uridine resonances (pyrimidine protons, left insets).
To obtain a sufficient quantity of the product of F5U from the action of RluA on [F5U]ASL (“RluA product”), the adduct formed by incubation of the two was prepared on a large scale and purified by anion exchange chromatography. After heat disruption, the precipitated protein was pelleted by centrifugation, and the RNA was digested with S1 nuclease and alkaline phosphatase. The nucleoside products were separated by preparative HPLC. Although the “RluA product” was contained in a single peak, NMR analysis (1H: Figure 5, panel B; 31P: not shown) clearly revealed that two dinucleotide products were present in roughly the same ratio as seen between the major and minor TruB products but with the hydrated F5U products followed by U (rather than C) as expected from the sequence of [F5U]ASL.
Mass spectrometric analysis of F5U products
An earlier report of the mass spectra of the TruB products stated that both were hydrated mononucleosides, but the mass spectrum itself was not included.(15) Given that the NMR results indicated dinucleotide products, resolution of this apparent discrepancy was imperative. A small portion of the NMR samples of the TruB products and the RluA products were therefore subjected to nESI-MS analysis (Figure 6). The observed molecular ions matched dinucleotides of hydrated F5U and C in the case of the TruB products and of hydrated F5U and U in the case of the RluA products.4 The mass spectra lacked peaks corresponding to either the nucleoside or nucleoside monophosphate of hydrated F5U. Furthermore, the mass spectra contained no peaks corresponding to unhydrated F5U, either alone or as a dinucleotide with the following residue, which indicates that all products of F5U are hydrated in agreement with the MALDI-MS data from digestion to oligonucleotides. The mass difference between the TruB and RluA products corresponds to the 1 Da difference between C and U, indicating that the major and minor products from the action of both enzymes are composed of isomers of hydrated F5U.
Figure 6.
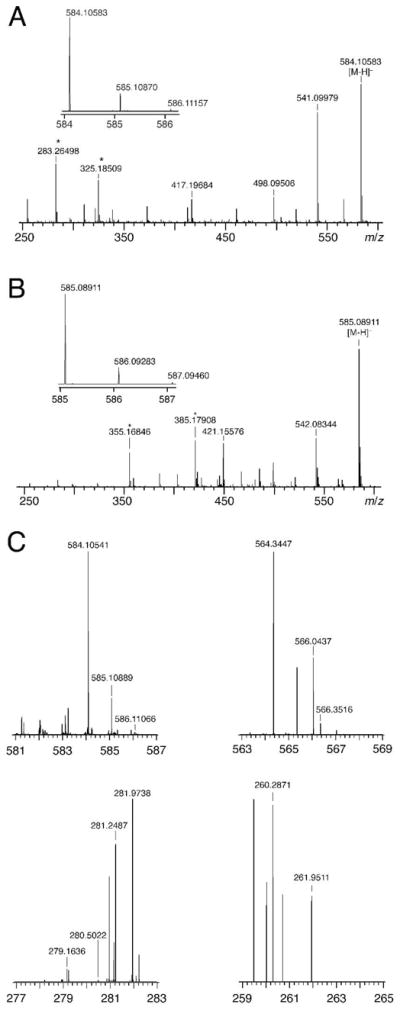
nESI-MS analysis of the products of F5U. A, The TruB products (584.10414 m/zpredicted for the dinucleotide of hydrated, rearranged F5U and C; 584.10583 m/zobserved). B, The RluA products (585.08816 m/zpred for the dinucleotide of hydrated, rearranged F5U and U; 585.08911 m/zobserved). C, The TruA products; mass spectra are from a 6 m/z range of ions spanning candidate products that were isolated in a linear ion trap: 581–587 (the dinucleotide of hydrated, rearranged F5U and C, 584.10414 m/zpredicted, 584.10541 m/zobserved), 563–569 (the dinucleotide of rearranged F5U and C, 566.0936 m/zpredicted, not observed), 277–283 (hydrated F5U, 280.0707 m/zpredicted, not observed), 259–265 (rearranged F5U, 262.0601 m/zpredicted, not observed); ranges acquired but not shown: 356–362 (hydrated F5UMP, 359.0292 m/zpredicted, not observed) and 338–344 (F5UMP, 341.0186 m/zpredicted, not observed). Measured m/z values have an uncertainty of ±2.5 ppm, or ±0.0014 for 566 m/z and ±0.0007 for 262 m/z; no peaks match candidate products within this tolerance other than the dinucleotide of hydrated, rearranged F5U and C. Asterisks denote background peaks present in dummy runs.
Although the technical difficulties of purifying suitable quantities of the adduct between TruA and (full length) [F5U]tRNA precluded the acquisition of NMR spectra of the “TruA products”, the lower material demands of mass spectrometry and HPLC analysis (see below) made feasible the analysis of the TruA products by these two methods. The adduct between TruA and [F5U]tRNA was therefore prepared, gel-purified, and heat-disrupted. The RNA was ethanol-precipitated and digested with S1 nuclease and alkaline phosphatase. The nESI-MS analysis of the TruA products reveals them to be the dinucleotides between hydrated F5U isomers and the C that follows the reactive F5U in the sequence of [F5U]tRNA.
HPLC analysis of the F5U products
As previously reported, the TruB products are partially resolved under the HPLC conditions (9), but the RluA products elute as a single peak (12) with a different retention time than either TruB product (Figure 7, panel A). The TruA products co-elute with the TruB products (Figure 7, panel B) as expected from the sequence of [F5U]tRNA, in which C follows F5U39. Furthermore, the TruB (and therefore, by extension, the TruA products) products co-elute with one isomer of photohydrated F5U. In concordance with the formation of dinucleotide products, the integration of the HPLC traces of component nucleosides in the S1 nuclease/alkaline phosphatase digestion of [F5U]RNA after incubation with all three Ψ synthases indicate that the nucleoside following the reactive F5U falls one equivalent short of the amount predicted by the sequence (Table 1). Dinucleotide products also explain the previously unsettling ability to detect them by monitoring A260; as expected, the spectra of the product peaks (observed with a diode array detector) matches the spectra of the following cytidine or uridine residue (data not shown).
Figure 7.

HPLC analysis of the products of F5U. A, Comparison of the reverse phase HPLC traces of the TruB (—; 2.42, 2.49 min) and RluA (– – –; 3.43 min) products. B, Comparison of the TruB (– – –) and TruA (- - -) products (retention time = 2.8 min); because of the slight difference in the retention times of U and F5U (and G and A, not pictured) and the somewhat decreased resolution of the column compared to earlier runs (panels A and C), a co-injection (—) was performed to verify that the TruB and TruA products behaved identically. C, Comparison of the TruB products (– – –; 1.82, 1.92 min) and the RluA products (—; 1.82, 1.95 min) from [F5U]ASL in which C replaces the U that follows F5U; peaks from the different storage buffers of the two enzymes are marked with asterisks.
Table 1.
Quantitation of peaks from the HPLC traces of digested [F5U]RNA.a
| C | U | F5Ub | G | A | products | |
|---|---|---|---|---|---|---|
| [F5U]TSL | 5.2 | 3.9 | 0.67 | 4.1 | 5.0 | 0.0 |
| [F5U]TSL + TruB | 4.3 | 3.8 | 0.10 | 4.0 | 5.0 | 0.7 |
| [F5U]ASL | 4.0 | 2.0 | 0.72 | 5.0 | 5.0 | 0.0 |
| [F5U]ASL + RluA | 4.3 | 1.3 | 0.10 | 5.2 | 5.0 | 0.6 |
| [F5U]tRNA | 20.8 | – | 11.1 | 22.3 | 15.0 | 0.0 |
| [F5U]tRNA + TruA | 20.4 | – | 10.8 | 23.1 | 15.0 | 0.67 |
| [F5U]ASL(U⇑C)c | 4.7 | 1.0 | 0.7 | 5.0 | 4.9d | 0 |
| [F5U]ASL(U⇑C)c + RluA | 4.0 | 0.9 | 0.2 | 5.0 | 5.0d | 0.5 |
Relative integration (A260) was calculated by adjusting the arbitrary integration units using ε260 for each species; for products, ε260 of the nucleotide following F5U (either C or U) in the RNA sequence was used. Adenosine was used to normalize all values (except as notedd), which have an error of ±0.2. Bold type indicates the “missing” nucleoside integration (beyond the expected decrease of F5U) after incubation with the Ψ synthase that is accounted for by dinucleotide products.
Quantitation of F5U is difficult even with great care because it is highly sensitive to pH variations in the injected samples, presumably due to its lower pKa value relative to the other nucleosides.
[F5U]ASL in which the U following the F5U is replaced with C.
The lot of S1 nuclease used for this set of experiments was contaminated by adenosine deaminase, which led to the substantial hydrolysis of adenosine to inosine. The sum of adenosine and inosine is reported in the “A” column, and guanosine was used to normalize the values.
If the differential HPLC retention behavior of the TruB and TruA products relative to the RluA products truly arises from the difference in the 3′ nucleoside of the dinucleotide (C for TruB and TruA; U for RluA), then the mutation of the nucleoside following F5U to an identical nucleoside should lead to the formation of identical products by the three Ψ synthases. To recognize RNA as a substrate, TruB requires a C to follow the isomerized U (or, in this case, F5U), which precludes the experiment in that direction.(16) RluA, however, will accept an RNA with the isomerized U followed by either U or C, so [F5U]ASL with C (in place of U) following the F5U was incubated with RluA to form an adduct, which was heat disrupted and digested with S1 nuclease and alkaline phosphatase. As expected, the HPLC analysis revealed products that coelute with TruB products with only a modest difference in the ratio of the major to the minor product (Figure 7, panel C).
Discussion
The initial detailed mechanistic investigations of the Ψ synthases was the examination by Santi and co-workers of the interaction between [F5U]tRNA and TruA (formerly Ψ synthase I).(2, 7) The two appeared to form a covalent adduct based on comigration in denaturing gels and irreversible inhibition of enzymatic activity by the fluorinated tRNA. The adduct was heat labile, and the investigators did the maximum characterization of the product of F5U with the limited amount of material available. They demonstrated conclusively that F5U was hydrated during the formation or heat disruption of the adduct. They further used radiolabeled F5U to locate the elution position of the reaction product, which was in a collected fraction that also contained one isomer of photohydrated F5U. Based on this evidence, Santi and coworkers very reasonably concluded that TruA follows a mechanism involving a Michael adduct (Figure 1, panel A), with the initial adduct being sufficiently stabilized by the electron-withdrawing properties of the fluoro group so that the enzyme accumulated in this state. Upon heating, hydrolysis of the ester linkage between the active site Asp-60 and F5U was hypothesized to give rise to the hydrated product as depicted in Figure 2.(7)
Subsequent studies of the interaction between TruB and RluA and [F5U]RNA revealed that the former does not appear to form a covalent adduct but the latter does.(9) F5U becomes hydrated by the action of both enzymes, but 18O labeling studies definitively rule out hydrolysis of an ester linkage between the active site Asp and F5U as the mechanism of hydration.(10, 12) Furthermore, TruB action generated two products from F5U based on two partially resolved peaks in the reverse phase HPLC analysis (9), but a single peak in the analysis of the RluA products (12) suggested that this enzyme generates only one product that differs from either of the TruB products (Figure 7, panel A). Cocrystals of both TruB (8) and RluA (11) with [F5U]RNA did not show a covalent linkage between RNA and protein, though both could reflect post-reaction events rather than productive states.5 The TruB cocrystal unambiguously showed that the F5U was both rearranged to the C-glycoside and hydrated.(8) Given the high degree of similarity of the active sites of all of the Ψ synthases, it seems unlikely that F5U would be handled so differently to give rise to at least three different products. The current work extends the 18O labeling studies to TruA to determine whether hydration of F5U occurs by hydrolysis of a Michael intermediate, and the additional characterization of the products of F5U generated by the action of TruA, TruB, and RluA untangles the knot of seemingly conflicting threads of evidence concerning their generation.
18O labeling
If hydration of F5U occurred because of the hydrolysis of an ester intermediate involving the essential Asp-60 (whether the initial or the rearranged Michael adduct), then oxygen from solvent will be incorporated into Asp-60, and an oxygen atom from the side chain carboxylate group will end in the pyrimidine ring. The results (Figure 4) unambiguously indicate that the pyrimidine ring bears an oxygen atom from solvent and so is directly hydrated. Ester hydrolysis does not occur, a result in agreement with similar studies of TruB and RluA.(10, 12)
Unlike in the cases of TruB (which handles [F5U]TSL as a substrate rather than making a tight or covalent adduct) and RluA (which makes a stoichiometric complex that was suitable for direct analysis when one equivalent of protein is incubated with [F5U]ASL), it was necessary to purify the [F5U]tRNA-TruA adduct from free protein and RNA before analysis. This inconvenience, however, made it trivial to perform an additional experiment: testing whether incorporation of oxygen from solvent occurred during adduct formation or during its disruption. The incorporation of 18O from solvent into F5U or its rearranged isomer during adduct formation would strongly imply that the adduct does not sport a covalent linkage between RNA and protein but instead is a very tight noncovalent complex. The results show conclusively that the oxygen content of the products of F5U reflects the isotopic composition of the solvent during heat disruption rather than adduct formation.
These results are consistent with the scheme proposed following the TruB and RluA studies in which F5U is rearranged by either mechanism, both of which then falter because the fluoro group cannot be abstracted by an enzymic base in the way that the proton of the natural substrate is (Figure 8).(10, 12) The enzyme binds the rearranged F5U tightly because the sp3 center at C5 marks it as an intermediate rather than the product (Ψ with a planar pyrimidine ring). The idiosyncratic geometry of some Ψ synthase active sites—such as TruA and RluA—allow the essential Asp to approach C6 closely enough to make a covalent bond, which is detected by comigration of RNA and protein in denaturing gels; in other Ψ synthases—such as TruB—C6 lies just beyond the reach of the Asp, and no covalent adduct is made. The rearranged intermediate eventually dissociates from TruB and undergoes spontaneous hydration in solution; the isomer with a cis relationship between the fluoro group and the new hydroxyl group predominates, as suggested by the appearance of that product in the cocrystal of TruB and [F5U]TSL (8). Upon heating, the covalent adducts with TruA and RluA suffer elimination (rather than hydrolysis) to regenerate rearranged F5U, which undergoes spontaneous hydration once it finds itself in solution as the enzyme denatures. The data currently cannot exclude alternatives, including other idiosyncratic features leading to very tight binding of the rearranged F5U by TruA and RluA but not TruB, access of water to the TruB but not TruA or RluA active sites to effect hydration of the bound intermediate, or the greater susceptibility to denaturation by SDS and urea of the complex between TruB and product [F5U]RNA. Hydration of the pyrimidine ring of rearranged F5U from either face (as shown in Figure 8) is an attractive proposition for a molecule free in aqueous solution and neatly explains the appearance of two isomeric products, but other processes that generate skeletal isomers or other stereoisomers cannot be excluded.
Figure 8.

A scheme for the handling of F5U in RNA by Ψ synthases. The lower sets of arrows indicate the equilibrium position for adduction and the result of heat disruption for TruB and for RluA and TruA.
Identity of the products of F5U
The foregoing argument implicitly assumes that the three enzymes convert F5U into the same products, or at least that RluA and TruA convert F5U into one of the two TruB products. The HPLC analyses of the products after enzymatic digestion that nominally would produce nucleoside products suggested that RluA (12) formed a single product distinct from the TruB products (9), and the TruA product (7) was earlier reported as a particular isomer of hydrated F5U (with the C–N glycosidic bond intact). These apparent discrepancies were examined in an attempt to resolve them.
Upon large scale isolation of the TruB products, NMR analysis revealed that the two products of F5U were both dinucleotides with the cytidine residue that follows F5U in [F5U]TSL (Figure 5). This finding was confirmed by mass spectrometry (Figure 6), and a literature search revealed that S1 nuclease has very low activity towards phosphodiester linkages to the 3′ side of nonplanar nucleobases such as a hydrated pyrimidine ring.(17, 18) Dinucleotide products of F5U immediately explain the disparate HPLC behavior of the TruB and RluA products, for uridine rather than cytidine follows F5U in [F5U]ASL. Indeed, NMR (Figure 5) and MS (Figure 6) analysis subsequently verified that RluA generates two products that are both dinucleotides with uridine, indicating that the two RluA products comigrate under the HPLC conditions. The 1H NMR spectra of the TruB and RluA products are very similar (except for the pyrimidine protons of cytidine and uridine), which suggests that both enzymes generate the same two isomeric products from F5U itself (only the 3′ nucleoside differs). In agreement with this conclusion, when RluA acts on [F5U]ASL in which the uridine following F5U was replaced with cytidine, products are obtained that behave identically to the TruB products under the HPLC conditions.
With the resolution of the discrepancy in behavior between the TruB and RluA products, we turned to the TruA products. If all Ψ synthases generate the same products of F5U as we hypothesize, then the TruA products should match the TruB products (or, perhaps, only one of them) because the reactive F5U in [F5U]tRNA is followed by cytidine. The TruA products were prepared on a scale that allowed their detection by absorbance to provide better resolution in the HPLC analysis than was possible with the collection of fractions for scintillation counting used in earlier work. The HPLC (Figure 7) and MS (Figure 6) analysis revealed that TruA does, indeed, generate two products from F5U and that they behave identically to the TruB products. One isomer of photohydrated F5U migrates very closely to the TruA products (data not shown), which accounts for the earlier report leading to the assignment of the product as a particular isomer of hydrated F5U.6
Although the definitive characterization of the TruB and RluA products remains a work in progress, several features of the NMR spectra support the conclusion that F5U is rearranged in both products. The coupling constants for the 3′ cytidine and uridine components of the dinucleotides match those of the authentic nucleosides free in solution, strongly suggesting that the difference in the two products lies in the F5U moiety of each dinucleotide. If one product were hydrated F5U rather than its rearranged C-glycoside isomer, then a proton would be expected on C5 with geminal coupling to the fluoro group, which is on the order of 45–50 Hz.(19) No proton with such coupling is observed. If one product were rearranged but not hydrated F5U, then the proton on C6 would have a chemical shift that reflects its attachment to an sp2 carbon, but no such proton is evident. Instead, the resonance of H6 in all products has been assigned between 5.20 and 5.34 ppm by a suite of 2D NMR experiments (E. Miracco, unpublished observations). Furthermore, the mass spectra of the TruA, TruB, and RluA products (Figure 6) do not show peaks consistent with rearranged but unhydrated F5U, and they all manifest identical and intriguingly unusual fragmentation patterns (B. Bogdanov, manuscript in preparation), further suggesting that all three enzymes handle F5U to generate the same products.
Conclusions
All current data point to the identical handling of F5U by TruB, RluA, and TruA to give two isomeric products. Differences in the behavior of those products under identical HPLC conditions is now surely ascribed to the generation of dinucleotide rather than the expected mononucleoside products during enzymatic digestion. The striking similarity of the 1H NMR spectra of the TruB and RluA products strongly suggest that both of these Ψ synthases generate identical products from F5U itself, and the identical behavior of the TruA and TruB products in HPLC and MS analysis suggest that TruA action generates the same products from F5U. All of the data are consistent with a mechanism involving a Michael adduct (Figure 1, panel A) or an acylal intermediate (Figure 1, panel B) since both generate rearranged F5U. This species can be released into solution (as TruB appears to do) where it undergoes spontaneous hydration, or rearranged F5U can (re)combine with the active site Asp to make a covalent adduct (as RluA and TruA appear to do). Upon heating, this adduct suffers elimination to release rearranged F5U, which is hydrated spontaneously in solution. Although this scheme (Figure 8) is consistent with the current data, variations involving hydration at the active site and other stereoisomers (or even skeletal ones) cannot yet be excluded and are the subject of active investigation.
Supplementary Material
Acknowledgments
We thank B. Bogdanov, N. Stolowich, and S. Aramgum (University of Louisville) and S. Bai (University of Delaware) for technical assistance and P. DiMaria (Delaware State University) for allowing us to acquire MALDI-MS data when our home instrument was down. We acknowledge the helpful discussions of R. Wittebort and A. Lane.
Footnotes
This work was supported by NIH grant GM059636 (to E.G.M.), the Commonwealth of Kentucky Research Challenge Trust Fund (“Bucks for Brains”), and the Charles L. Bloch, M. D. Professorship. nESI-MS was performed at the CREAM facility at U. of Louisville, which is supported by NSF/EPSCoR grant EPS-0447479.
Abbreviations: Ψ, pseudouridine; F5U, 5-fluorouridine; [F5U]tRNA, the in vitro transcript of E. coli tRNAPhe containing F5U in place of U; TSL, T-arm stem-loop; [F5U]TSL, TSL containing F5U in place of the U isomerized by TruB; ASL, anticodon stem-loop; [F5U]ASL, ASL containing F5U in place of the U isomerized by RluA. nESI, nano-electrospray ionization; MALDI, matrix-assisted laser desorption ionization; TOF, time of flight; TruB products, the products of F5U resulting from the action of TruB on [F5U]TSL; RluA products, the products of F5U resulting from the action of RluA on [F5U]ASL; TruA products, the products of F5U resulting from the action of TruA on [F5U]tRNA.
S1 nuclease has the same enzymatic activity and specificity as nuclease P1, which we used previously (21) to get digestion products that displayed identical HPLC behavior as the ones reported here with S1 nuclease. The change in nuclease, therefore, does not account for the difference in products reported by Santi and co-workers (7) and Huang and co-workers (15).
Radiant Red ceased to be commercially available during the course of this work.
The mass spectra cannot, of course, distinguish between hydrated F5U and its skeletal or stereochemical isomers. The isotopic equilibration of the deuterium on the products in the NMR samples with the vast excess of unlabeled water (and methanol) is expected to be very fast and should be easily achieved in the time between dilution and acquisition of the mass spectra. In support of full equilibration, no change in the monoisotopic masses was observed during replicate runs of the samples (data not shown), and the products of TruB action have the same monoisotopic mass as those from the action of TruA, which were never exposed to D2O.
The RluA-[F5U]ASL adduct detected in denaturing gels was shown to be sensitive to X-irradiation of its crystal, so the final observed noncovalent complex may be a result of radiolysis.
As explained more fully in the supplementary material, the NMR spectrum of the isomer of hydrated F5U that comigrates with the TruA and TruB products matches that of 5R,6S-5,6-dihydro-6-hydroxy-5-fluorouridine (19) rather than the 5S,6R isomer reported by Gu et al.(7) Regardless of the assignment of isomers, the comigration of one hydrated F5U isomer and the TruA products resolves the apparent discrepancy between the results.
Supporting Information Available. Detailed synthetic protocols and characterization for F5UTP and the photohydration products of F5U. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mueller EG, Ferré-D’Amaré AR. Pseudouridine formation, the most common transglycosylation in RNA. In: Grosjean H, editor. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Landes Bioscience; Austin, TX: 2009. pp. 363–376. [Google Scholar]
- 2.Huang LX, Pookanjanatavip M, Gu XG, Santi DV. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry. 1998;37:344–351. doi: 10.1021/bi971874+. [DOI] [PubMed] [Google Scholar]
- 3.Ramamurthy V, Swann SL, Paulson JL, Spedaliere CJ, Mueller EG. Critical aspartic acid residues in pseudouridine syntheses. J Biol Chem. 1999;274:22225–22230. doi: 10.1074/jbc.274.32.22225. [DOI] [PubMed] [Google Scholar]
- 4.Raychaudhuri S, Conrad J, Hall BG, Ofengand J. A pseudouridine synthase required for the formation of two universally conserved pseudouridines in ribosomal RNA is essential for normal growth of Escherichia coli. RNA. 1998;4:1407–1417. doi: 10.1017/s1355838298981146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conrad J, Niu LH, Rudd K, Lane BG, Ofengand J. 16S ribosomal RNA pseudouridine synthase RsuA of Escherichia coli: Deletion, mutation of the conserved Asp102 residue, and sequence comparison among all other pseudouridine synthases. RNA. 1999;5:751–763. doi: 10.1017/s1355838299990167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaya Y, Ofengand J. A novel unanticipated type of pseudouridine synthase with homologs in bacteria, archaea, and eukarya. RNA. 2003;9:711–721. doi: 10.1261/rna.5230603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu XR, Liu YQ, Santi DV. The mechanism of pseudouridine synthase I as deduced from its interaction with 5-fluorouracil-tRNA. Proc Natl Acad Sci U S A. 1999;96:14270–14275. doi: 10.1073/pnas.96.25.14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoang C, Ferré-D’Amaré AR. Cocrystal structure of a tRNA Ψ55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell. 2001;107:929–939. doi: 10.1016/s0092-8674(01)00618-3. [DOI] [PubMed] [Google Scholar]
- 9.Spedaliere CJ, Mueller EG. Not all pseudouridine synthases are potently inhibited by RNA containing 5-fluorouridine. RNA. 2004;10:192–199. doi: 10.1261/rna.5100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG. The pseudouridine synthases: Revisiting a mechanism that seemed settled. J Am Chem Soc. 2004;126:12758–12759. doi: 10.1021/ja046375s. [DOI] [PubMed] [Google Scholar]
- 11.Hoang C, Chen JJ, Vizthum CA, Kandel JM, Hamilton CS, Mueller EG, Ferre-D’Amare AR. Crystal structure of pseudouridine synthase RluA: Indirect sequence readout through protein-induced RNA structure. Mol Cell. 2006;24:535–545. doi: 10.1016/j.molcel.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 12.Hamilton CS, Greco TM, Vizthum CA, Ginter JM, Johnston MV, Mueller EG. Mechanistic investigations of the pseudouridine synthase RluA using RNA containing 5-fluorouridine. Biochemistry. 2006;45:12029–12038. doi: 10.1021/bi061293x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darlix JL, Fromageot HPM, Fromageot P. An improved method for the preparation of 52-monophosphoderivatives of the common ribonucleosides. Biochim Biophys Acta. 1967;145:517–519. doi: 10.1016/0005-2787(67)90073-1. [DOI] [PubMed] [Google Scholar]
- 14.Tolbert TJ, Williamson JR. Preparation of specifically deuterated RNA for NMR studies using a combination of chemical and enzymatic synthesis. J Am Chem Soc. 1996;118:7929–7940. [Google Scholar]
- 15.Phannachet K, Elias Y, Huang RH. Dissecting the roles of a strictly conserved tyrosine in substrate recognition and catalysis by pseudouridine 55 synthase. Biochemistry. 2005;44:15488–15494. doi: 10.1021/bi050961w. [DOI] [PubMed] [Google Scholar]
- 16.Gu XR, Yu M, Ivanetich KM, Santi DV. Molecular recognition of tRNA by tRNA pseudouridine 55 synthase. Biochemistry. 1998;37:339–343. doi: 10.1021/bi971590p. [DOI] [PubMed] [Google Scholar]
- 17.Weinfeld M, Liuzzi M, Paterson MC. Selective hydrolysis by exonucleases and endonucleases of phosphodiester bonds adjacent to an apurinic site. Nucleic Acids Res. 1989;17:3735–3745. doi: 10.1093/nar/17.10.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinfeld M, Soderlind KJM, Buchko GW. Influence of nucleic acid base aromaticity on substrate reactivity with enzymes acting on single-stranded DNA. Nucleic Acids Res. 1993;21:621–626. doi: 10.1093/nar/21.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visser GWM, Herder RE, Noordhuis P, Zwaagstra O, Herscheid JDM, Dekanter FJJ. Reaction of acetyl hypofluorite with pyrimidines. 3 Synthesis, stereochemistry, and properties of 5-fluoro-5,6-dihydropyrimidine nucleosides. J Chem Soc Perkin Trans. 1988;1:2547–2554. [Google Scholar]
- 20.Hamilton CS, Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG. The roles of the essential Asp-48 and highly conserved His-43 elucidated by the pH dependence of the pseudouridine synthase TruB. Arch Biochem Biophys. 2005;433:322–334. doi: 10.1016/j.abb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Mueller EG, Buck CJ, Palenchar PM, Barnhart LE, Paulson JL. Identification of a gene involved in the generation of 4-thiouridine in tRNA. Nucleic Acids Res. 1998;26:2606–2610. doi: 10.1093/nar/26.11.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.