

Abstract
The recent discovery of 5-hydroxymethyl-cytosine (5hmC) in embryonic stem cells and post-mitotic neurons has triggered the need for quantitative measurements of both 5-methyl-cytosine (5mC) and 5hmC in the same sample. We have developed a method using liquid chromatography electrospray ionization tandem mass spectrometry with multiple reaction monitoring (LC-ESI-MS/MS-MRM) to simultaneously measure levels of 5mC and 5hmC in digested genomic DNA. This method is fast, robust and accurate, and is more sensitive than the current 5hmC quantitation methods such as end-labeling with thin-layer chromatography and radio-labeling by glycosylation [1; 2]. Only 50 ng of digested genomic DNA is required to measure the presence of 0.1% 5hmC in DNA from mouse embryonic stem cells. Using this procedure we show that human induced pluripotent stem cells exhibit a dramatic increase in 5mC and 5hmC levels compared to parental fibroblast cells, suggesting a dynamic regulation of DNA methylation and hydroxymethylation during cellular reprogramming.
INTRODUCTION
The pattern of methylated cytosine residues in DNA provides an inheritable epigenetic code that regulates gene expression during development. The covalent addition of a methyl group at the 5-position of cytosine primarily occurs in the CpG dinucleotide, and is catalyzed by a family of DNA methyltransferases (Dnmts) including maintenance Dnmt1 and de novo Dnmt3a and Dnmt3b. DNA methylation is involved in various biological processes such genomic imprinting, silencing of retroviral transposons, X chromosome inactivation, and cellular differentiation. Mechanistically, promoter methylation can lead to transcriptional repression directly by inhibiting transcriptional binding, or indirectly by recruiting various proteins including methyl CpG binding proteins (MBDs), co-repressors and histone modification enzymes involved in chromatin remodeling [3; 4; 5; 6; 7; 8]. Importantly, many studies have shown that DNA methylation is a dynamic process in cellular proliferation and differentiation, and is tightly regulated in normal development. Aberrant DNA methylation patterns and mechanisms are deleterious to the developing central nervous system (CNS) [8; 9; 10; 11].
Recently there has been renewed interest in another, related, mammalian DNA modification, 5-hydroxymethyl-cytosine (5hmC). Significant levels of 5hmC are found in the developed murine central nervous system and in embryonic stem cells [1; 12; 13;14]. In vivo addition of a hydroxyl group onto 5-methyl-cytosine (5mC) is catalyzed by 2-oxoglutarate oxygenase Tet1, Tet2, and Tet3 [1; 14]. There are also reports that 5hmC can be formed by other mechanisms beside Tet pathway, including UV irradiation of 5mC in aerated aqueous solution [15] and DNA methyltransferase reaction of cytosine with formaldehyde [16]. To date, only the Tet pathway has been demonstrated to produce 5hmC in mammalian genomic DNA.
Speculation that 5hmC is involved in the DNA demethylation pathway comes from the two reported mechanisms of converting 5hmC into C. Bacterial DNA methyltranferases catalyze the removal of formaldehyde from 5hmC, thus converting 5hmC to C [16]. Another deformylation mechanism involves the photochemical hydration of 5hmC in basic solution [15]. However, these two possible DNA demethylation mechanisms have yet to be confirmed in mammalian models.
Some of the most commonly used methods for profiling and quantification of DNA methylation, such as bisulfite sequencing and methylation-sensitive enzyme-based assays, are unable to distinguish between 5hmC and 5mC [1; 17]. Several methods have been used to measure the 5hmC levels in the genome: these include end-labeling followed by thin layer chromatography [1], high performance liquid chromatography (HPLC) with UV detection [16], enzymatic radioactive glycosylation labeling [2], and single molecule, real-time sequencing [18]. The thin layer chromatography method has the advantage of being low cost and simple, but requires the availability of radioactive substrates and the accuracy is not comparable to other available methods. The specificity of UV detection relies heavily on the chromatographic separation to avoid co-elution of other components, including other DNA and RNA nucleotides that may be present in biological samples. The glycosylation method is based on enzymatic incorporation of radio-labeled glucose into genomic 5hmC, with quantification by radioactive counting. However a complete enzymatic reaction cannot be readily assured and 5mC levels cannot be measured simultaneously. Independent measurement of 5hmC is possible with next generation sequencing, but the technology has yet to be perfected for accurate quantitation of many low abundant nucleotides including 5hmC.
Previous work demonstrated the precision, selectivity and sensitivity of liquid chromatography tandem mass spectrometry for measuring 5mC in biological samples, and as a diagnostic tool for cancer [19; 20; 21; 22; 23; 24; 25]. Using this technique all known DNA (excluding 5hmC) and RNA components, have been separated, distinguished and independently quantitated [24]. This approach allows DNA methylation to be measured both at the global [20; 21; 23; 24] and gene promoter regions [22]. However, none of the previous reports include 5hmC. We were prompted to develop a fast, sensitive and accurate method to measure both 5mC and 5hmC levels to support ongoing work on epigenetic control of stem cells and neural development. Here we report the use of liquid chromatography electrospray ionization tandem mass spectrometry with multiple reaction monitoring (LC-ESI-MS/MS-MRM) for the determination of genomic DNA methylation and hydroxymethylation. Separation of the deoxyribonucleosides is achieved within 6 minute using sub-two micron particle size reverse phase chromatography columns. In addition, mass-based detection discriminates between the three nucleoside bases of interest- 5hmC, 5mC and cytosine (C). The combination of LC and MS minimizes any possible cross-talk between the measurements of low abundant molecules (5hmC and 5mC) in the face of a chemically similar abundant species (C). Together, our data indicate that the MRM method provides unambiguous and independent quantification of 5hmC, 5mC, and C with high reproducibility and low limits of detection of around 0.5 fmol per sample. This limit of detection can be equated to 50 ng of digested genomic DNA to measure 5hmC levels at the 0.1% level. Furthermore, the method is relatively fast, requiring less than 48 hours from extracting genomic DNA (few hours to a day), to digesting genomic DNA into nucleoside components (1–2 hours), and measuring the 5hmC and 5mC levels using the MRM method (6 minutes per sample).
MATERIALS AND METHODS
Cell culture
mESCs of wild-type J1, Dnmt1−/− (cc), double knockout Dnmt3a−/−;Dnmt3b−/− (DKO), and triple knockout Dnmt1−/−; Dnmt3a−/−;Dnmt3b−/− (TKO), were maintained on gelatin-coated plates in mESC medium containing DMEM (Invitrogen), 10% fetal bovine serum (FBS, Gibco), 100 µM 2-mercaptoethanol (Sigma), 0.1 mM nonessential amino acids (Gibco), 1 mM sodium pyruvate, 1X L-glutamine (Gibco), 1X penicillin/streptomycin (MP Biomedicals), and 100 units/mL of leukemia inhibitory factor (LIF).
Human BJ fibroblasts from neonatal foreskin were purchased from American Type Culture Collection (ATCC). These were maintained in medium containing DMEM, 1X penicillin/streptomycin, 1X glutamine, and 10% fetal bovine serum. Fibroblasts were passaged every 3 ~ 5 days using 0.05 % trypsin (Invitrogen).
Undifferentiated hESCs (HSF1) were maintained on a feeder layer of mitomycin C (Sigma)-treated mouse embryonic fibroblasts (MEF) in hESC medium containing DMEM/F12 (Invitrogen) supplemented with 20% Knockout Serum Replacement (KSR, Gibco), 1X glutamax, 1X non-essential amino acids, 0.11 mM β-mercaptoethanol (Sigma), 1X penicillin/streptomycin and 10 ng/ml of bFGF (PeproTech). hESCs were passaged every 5 ~ 7 days using Collagenase IV (Gibco) and Dispase (Gibco) at a final concentration of 1 mg/ml in hESC medium. All cells were cultured under a protocol approved by the Chancellor’s Animal Research Committee (ARC) and Embryonic Stem Cell Research Oversight (ESCRO) Committee at UCLA.
Derivation of human induced pluripotent stem cells (iPSCs)
The production of human iPSCs followed a published protocol [26] with slight modifications. Specifically, retroviruses containing OCT4, SOX2, KLF4 and c-MYC were produced in the Platinum-E (PLAT-E) retrovirus packaging cell line (Cell Biolabs). Viral supernatants were collected 48 hours and 72 hours after transfection and filtered through a 0.45 µM PVDF filter (Millipore). The Slc7a1-expressing human BJ fibroblasts were plated at 1.5 X 105 cells per well on a 6-well plate on day 1. On day 2, each retroviral supernatant was added into the fibroblasts in the presence of 4 µg/ml of polybrene (Sigma). A second round of transduction was performed on day 3. Infection efficiency was monitored by fluorescence microscopy of cells transduced by retrovirus carrying GFP. On day 5, cells were trypsinized and re-plated in a density of 1 X 105 per 10 cm plate on mitomycin (Sigma)-treated MEFs. On day 6, the medium was changed to hESC medium. iPS cell colonies were picked 3 weeks after infection. The picked colonies were cultured and passaged according to standard culturing protocols. All plasmids including pMXs-OCT4, pMXs-SOX2, pMXs-KLF4, pMXs-c-MYC and pLenti6/UbC/mSlc7a1 used for the derivation of iPSCs were purchased from Addgene.
DNA extraction and hydrolysis
hESC and iPSC colonies were harvested and passed through a cell strainer (BD falcon) to remove the feeder cells. Colonies were washed with PBS, treated with 500 µL of DNA lysis buffer (100 mM Tris-HCl pH 8.0, 5 mM EDTA, 200 mM NaCl, 0.2% SDS), 5 µL of proteinase K (100 mg/mL, Roche), and 5 µL of RNase A (10 mg/mL, Roche), and incubated overnight at 37 °C in a shaking incubator. Genomic DNA was purified by a standard phenol/chloroform extraction followed by precipitation with two volumes of cold 100% ethanol. Subsequently the extracted genomic DNA was redissolved in TE buffer (10 mM Tris-HCl and 1 mM EDTA, pH 8.0). Genomic DNA was quantified spectrophotometrically at 260 nm (Thermo Scientific NanoDrop).
DNA hydrolysis was performed by using DNA Degradase Plus (Zymo Research). Briefly, 1 µg of genomic DNA was mixed with 2.5 µL 10X DNA Degradase Reaction buffer, 1 µL DNA Degradase Plus and water to make a total reaction volume of 25 µL. The reaction mixture was incubated in 37 °C for more than an hour. Finally, the reaction was inactivated by adding 175 µL of 0.1% formic acid to yield a final concentration of 5 ng of digested DNA/µL.
DNA standards
Three 897bp DNA standards, each homogenous for either unmodified C, 5mC, or 5hmC, were purchased (Zymo, Irvine, CA), and used to generate a calibration curve. The standards were prepared by PCR using the appropriate nucleotides and were spin-column purified by the manufacturer to obtain 50 ng/µL solutions. By MRM criteria these standards were all more than 99.6% pure (Supplementary Figure 1).
Multiple Reaction Monitoring (MRM) Quantitation
DNA hydrolysis samples (10 µl typically containing 50 ng of digested DNA) were injected onto a reverse phase UPLC column (Eclipse C18 2.1 × 50 mm, 1.8 µ particle size, Agilent) equilibrated and eluted (100 µL/min) with water/methanol/formic acid (95/5/0.1, all by volume). The effluent from the column was directed to an electrospray ion source (Agilent Jet Stream) connected to a triple quadrupole mass spectrometer (Agilent 6460 QQQ) operating in the positive ion multiple reaction monitoring mode using previously optimized conditions, and the intensity of specific MH+→fragment ion transitions were recorded (5mC m/z 242.1→126.1, 5hC 258.1→142.1 and dC m/z 228.1→112.1). The measured percentage of 5mC and 5hmC in each experimental sample was calculated from the MRM peak area divided by the combined peak areas for 5mC plus 5hmC plus C (total cytosine pool).
With each batch of experimental samples a series of standard samples was simultaneously prepared using the 897bp DNA standards, and run. The standard samples contained increasing amounts of 5mC and 5hmC in the presence of the same amount of C (0–10% for 5mC and 0–2% for 5hmC). Calibration curves were constructed for 5mC and 5hmC from the data obtained from the standard samples (measured 5mC or 5hmC peak area/total cytosine pool plotted against actual percentage of either 5mC or 5hmC in the samples). The measured percentage of 5mC and 5hmC in each experimental sample was then converted to actual percentage 5mC and 5hmC by interpolation from the calibration curves. This provided a correction for any differences that might exist in the molar MRM responses of the various nucleosides.
TET1 cloning, virus production and infection
The Flag-tagged TET1 catalytic domain (NM_030625) was amplified from hESCs cDNA using PCR with Hotstar Taq polymerase (Qiagen). Primers used for the amplification of TET1 are described in Supplementary Table 1. PCR products were purified and cloned into pCR4-TOPO plasmid using TOPO TA cloning kit (Invitrogen) following the manufacturer’s protocol. TET1 sequences were verified by DNA sequencing. The TET1 catalytic domain was digested and ligated into BamHI and EcoRI sites of lentiviral plasmid, FUIGW (Addgene).
For lentivirus production, 293T cells were plated at 8 X 106 cells per 10 cm plate and incubated at 37 °C overnight. Cells were transfected with 9 µg of FUIGW-Flag-TET1-GFP or FUIGW) along with 4.5 µg of pMLDg/pRRE (Addgene), 1.8 µg of pRSV-Rev (Addgene), and 2.7 µg of pCMV-VSVG (Addgene) by Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol; 48 and 72 hours after transfection, viral supernatants were collected and filtered through a 0.45 µM PVDF filter (Millipore). Fresh viral supernatants were infected into 293T cells in the presence of 4 µg/ml of polybrene (Sigma) overnight. Infected cells were analyzed by immunostaining and extracted for LC-ESI-MS/MS-MRM.
Immunocytochemistry
Antibodies used for immunostaining were: OCT4 (1:20, Santa Cruz), NANOG (1:100, Abcam), TRA1-60 (1:200, Chemicon), SSEA4 (1:200, Chemicon), SOX2 (1:200, Cell Signaling) DNMT3B (1:500, a gift from Dr. En Li). Human ES and iPS cells were plated on sterile coverglasses in 6-well plates and cultured for 24 ~ 48 hr. The medium was aspirated and cells were washed once with PBS and fixed with 4% paraformaldehyde/PBS for 30 min at room temperature. Cells were washed three times with 0.2 % Tween 20/PBS, then permeabilized with 0.2% Triton X-100/PBS for 30 min at room temperature and washed once with 0.2% Tween20/ PBS. Blocking was performed for 1 h at room temperature with 2% BSA/0.1% Tween 20/PBS. Primary antibodies diluted in blocking solution were incubated for 1 h at room temperature. Cells were washed three times with 0.2% Tween 20/PBS. Cy2- and Cy3- conjugated secondary antibodies diluted in blocking solution were incubated at room temperature for 30 min. Cells were washed three times with 0.2% Tween 20/PBS, stained with DAPI and mounted on glass slides (Fisher Scientific). Images were analyzed on a Nikon Eclipse 80i inverted microscope equipped with a CCD camera by using Spot Advance imaging software (Diagnostics Instruments).
Southern blot analysis
Genomic DNA (5 µg) was digested with BstBI (NEB) overnight at 37 °C, separated on a 1% agarose gel, and transferred to a Hybond-N+ membrane (Amersham) in 10X SSC. The membrane was hybridized with P32-end-labeled oligo probes for Sat 2 and Sat 3 in QuikHyb solution (Stratagene) at 42 °C for 2 hours. The hybridized membrane was washed twice in 2X SSC/0.1 % SDS at room temperature and washed once in 0.1XSSC/0.1 % SDS at 60 °C. The membrane was exposed to a Kodak BioMax MS film. Oligo probes are described in Supplementary Table 1.
Bisulfite sequencing
Genomic DNA (2 µg) was subjected to bisulfite conversion using EZ DNA Methylation Kit (Zymo research) following the manufacturer’s protocol. Subsequently, PCR was carried out with HotStar Taq polymerase (Qiagen). Primers (OCT4 and NANOG) and PCR conditions are described in Supplementary Table 1. PCR products were purified by Wizard SV gel and PCR clean-up kit (Promega) and cloned into pCR4-TOPO plasmid using TOPO TA cloning kit (Invitrogen) following the manufacturer’s protocol. Following transformation, 10 ~ 12 colonies were subjected to direct sequencing with the M13 reverse primer, followed by inoculations and minipreps.
RT-PCR
Total RNA was isolated from cells using the RNeasy Mini kit (Qiagen) with QIAshredder (Qiagen) following the manufacturer’s protocol. The small residual amounts of DNA were removed by the RNase-Free DNase Set kit (Qiagen). The concentration of RNA was quantified spectrophotometrically at 260 nm (Thermo Scientific NanoDrop). Total RNA (1 µg) was reverse transcribed into complimentary DNA (cDNA). The reverse transcription (RT) was performed using the iScript cDNA synthesis Kit (Bio-Rad) according to the manufacturer’s protocol. PCR was performed on a MyIQ Thermocycler (Biorad).
RESULTS
Mass spectrometric characterization of 5hmC, 5mC, and C
An equal molar mixture of three commercial 897bp standard DNA fragments (Zymo, Irvine, CA), each homogenous for either unmodified C, 5mC, or 5hmC, was prepared and digested into the nucleoside components. This mixture produced ions during electrospray ionization corresponding to the protonated nucleosides deoxycytidine (dC), 5-methyl-2’-deoxycytidine (5mdC) and 5-hydroxymethyl-2’-deoxycytidine (5hmdC) at m/z 228.1, 242.1and 258.1, respectively. Collisionally induced dissociation (CID) of these protonated nucleosides produced a number of fragments, the most abundant of which correspond to the protonated bases liberated by cleavage of the glycosidic bond at m/z 112.1 (C), 126.1 (5mC) and 142.1 (5hmC). Mass-based distinction between these nucleosides is therefore possible because the parent masses are unique as are the corresponding bases that result from glycosidic cleavage. The gas phase glycosidic cleavage of nucleosides is efficient, and the intensity of transitions of the protonated nucleosides to their corresponding bases can be used in the MRM mode for independent quantification: m/z 228.1→112.1, 242.1→126.1 and 258.1→142.1 for dC to C, 5mdC to 5mC and 5hmdC to 5hmC, respectively.
When the equivalent of 50 ng of DNA was analyzed by LC-ESI-MS/MS-MRM, the sequentially eluting symmetrical peaks corresponding to dC to C, 5mdC to 5mC, and 5hmdC to 5hmC transitions revealed no detectable cross-talk (Figure 1A). Using the same commercial DNA fragments, the linearity of the response was tested by preparing and analyzing samples with varying amount of 5mC and 5hmC in the presence of a constant amount of C containing DNA. Calibration curves constructed from this data set for both 5mC and 5hmC were linear (Figure 1B), and were used to calculate the percent DNA methylation and hydroxymethylation in experimental samples.
Figure 1.
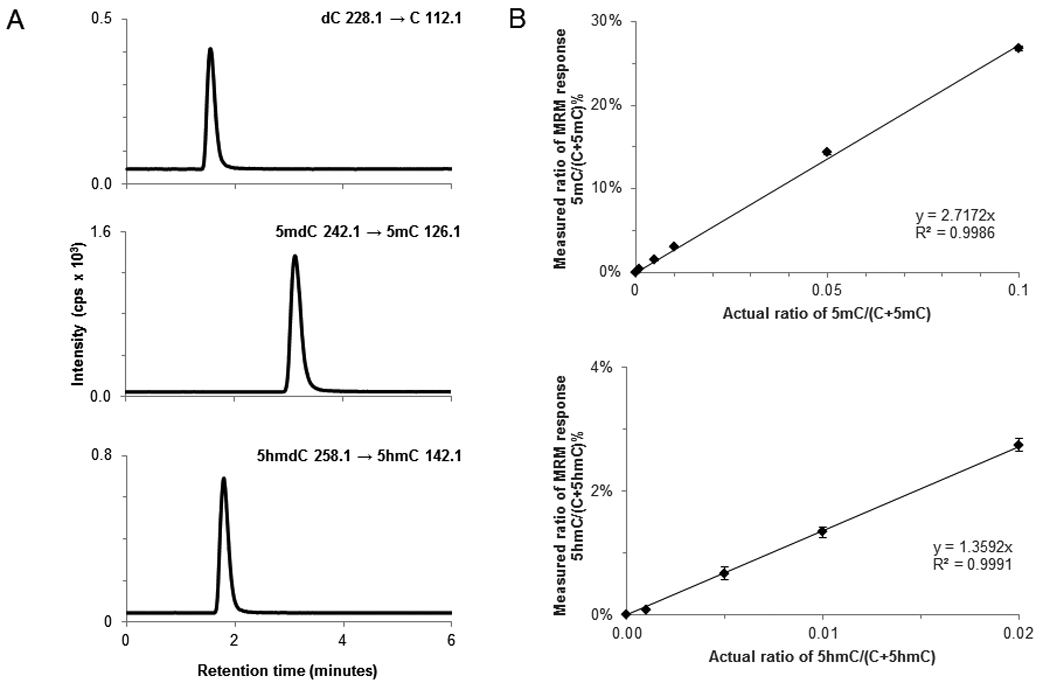
(A) LC-MS/MS-MRM chromatograms of nucleosides derived from an equal molar mixture of three commercial 948bp standard DNA fragments showing peaks corresponding to the response obtained from gas phase transitions of dC to C, 5hmdC to 5hmC, and 5mdC to 5mC. (B) Standard curves for 5mC and 5hmC. Percent DNA methylation and hydroxymethylation is plotted against the known ratios of methylated or hydroxymethylated DNA to the total pool of cytosine in the standard sample. cps, counts per second.
Validation of the MRM method
The method was then used to measure the percentage of 5mC and 5hmC in some mouse embryonic stem cell (mESC) lines. The 5mC level of Dnmt1−/− mESC is about 25% of the wild-type 5mC level. Also, the double knockout, Dnmt3a−/− and Dnmt3b−/−, mESC at passage 35 (P35) has a 5mC level of about 16% of the wild-type (Figure 2). These results are consistent with previous studies that used nearest neighbor analysis and bisulfite next generation sequencing (BS-Seq) [27; 28].
Figure 2.
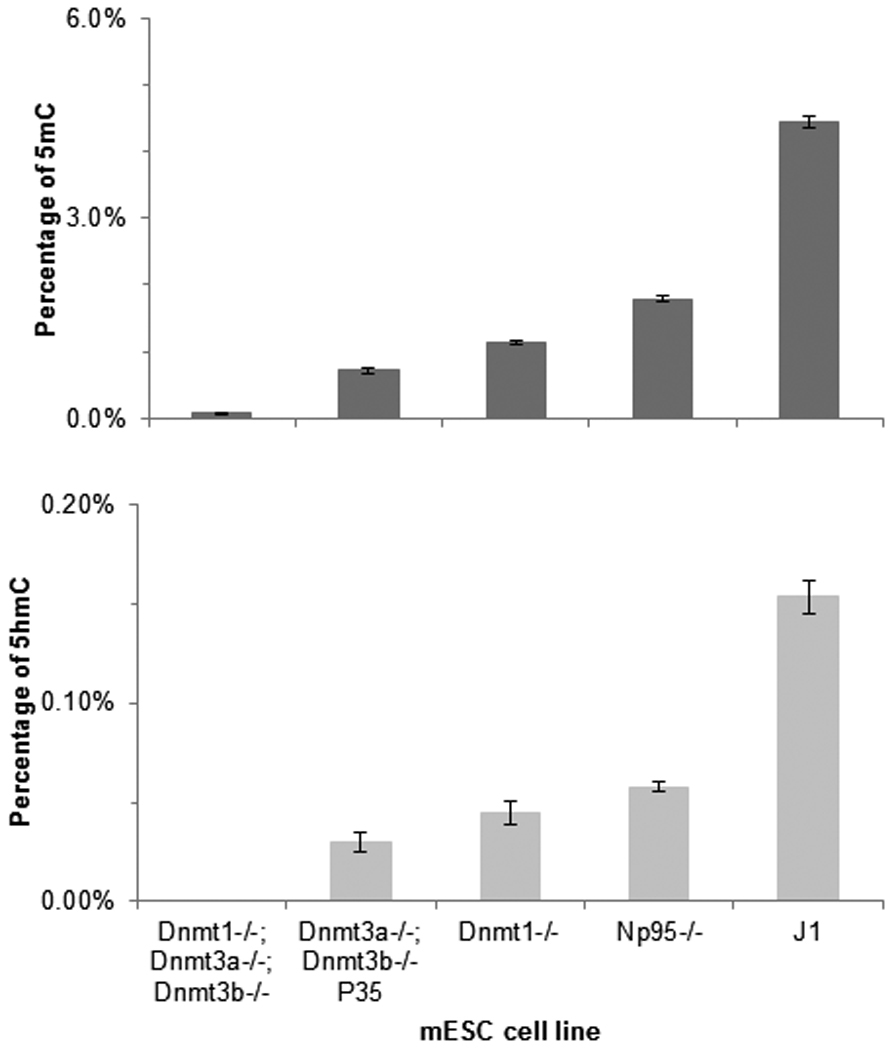
Percentage of 5hmC and 5mC in mESC DNA. 5mC and 5hmC contents are expressed as the percentage of 5mC or 5hmC in the total pool of cytosine. Data are the mean ± s.d. from triplicate analyses.
A comparison of the 5mC and 5hmC levels in various mESC lines shows a strong correlation (Figure 2). This correlation is consistent with the biological conversion of 5mC to 5hmC by oxygenase TET enzyme [1; 14]. A higher 5mC level would favor more 5hmC conversion, and thus raise the global level of 5hmC.
To confirm another previous study, the FLAG-tagged TET1 catalytic domain was over-expressed in 293T cells (Figure 3A). Using the MRM method a drastic increase in 5hmC level was recorded accompanied by about 50% loss of 5mC level compare to control cells (Figure 3B). This observation was consistent with the previous study using 5mC antibody fluorescence immunocytochemistry that showed transfected 293T cells have 55% of the DNA methylation level found in control cells [1].
Figure 3.

Over-expression of human TET1 catalytic domain in 293T cells. (A) 293T cells over-expressing FLAG-catalytic domain of TET1 were co-stained for FLAG antibodies and DAPI. Scale bar, 100 µm. (B) Percentage of 5hmC and 5mC in DNA from TET1 transfected cells, mock-transfected and un-transfected cells. Data are the mean ± s.d. from triplicate analyses.
Measuring 5hmC and 5mC in somatic and induced pluripotent stem cells
BJ fibroblasts were used to generate induced pluripotent stem cells (iPSCs) by retrovirally introducing Oct4, Sox2, Klf4, and c-Myc (Supplementary Figure 2). Two iPSC colonies (BJ iPS #7 and BJ iPS #8) were picked and expanded for further analysis. Both iPS lines showed a significant increase in 5mC after reprogramming from BJ fibroblasts (Figure 4A). This 5mC increase was accompanied by a significant increase in the 5hmC level.
Figure 4.

Reprogramming BJ fibroblast into iPSCs. (A) Percentage of 5mC and 5hmC in DNA from BJ fibroblast and two BJ iPS cell lines, #7 and #8. (B) Southern blot analysis of DNA methylation in BJ and two BJ iPS cell lines. DNA was digested with methyl-sensitive BstBI, separated on agarose gel, transferred to the membrane and hybridized to probes of the repetitive regions of Sat 2 and Sat 3. Small DNA fragments of BJ fibroblast are indicative of DNA hypomethylation in Sat 2 and Sat 3 repetitive region. (C) Bisulfite sequencing of Oct4 and Nanog promoter of BJ fibroblasts and two BJ iPS cell lines. Each row represents one clonal analysis and each box represents a CpG site where the site number is indicated above. The methylation analysis is displayed according to the key. The overall percentage methylation of the gene promoter is indicated for each sample.
Southern blot was performed on Sat 2 and Sat 3 repetitive sequences of BJ fibroblasts, BJ iPS #7 and BJ iPS #8, and showed an increase in DNA methylation at BstBI sites (TTCGAA) in the repetitive regions (Figure 4B). These results are consistent with the MRM result. However the promoter regions of both Nanog and Oct4 underwent DNA demethylation (Figure 4C), suggesting that the 5mC level increase occurs on selective gene regions.
DISCUSSION
We report the details of a fast and reliable method for measuring the relative levels of 5mC and 5hmC in small samples of digested DNA. Through the use of ultra performance liquid chromatography (UPLC) with sub-micron particle size packing, the analysis time is reduced to 6 minutes per sample. Using this method the limit of detection for these two nucleosides is around 0.5 fmol injected on-column. The linearity of the response is demonstrated across one order of magnitude which is more than sufficient for biological samples, and is probably much greater, and the levels of 5mC and 5hmC have been measured in ten different cell lines. Experience has shown that batches exceeding one hundred samples can be analyzed without any noticeable change or deterioration in chromatographic performance and MRM response. The durability of the UPLC columns used in this work is such that hundreds of samples have been analyzed on the same column, although as a precaution high organic washes every 20–30 samples are used to avoid any complications that could arise from the accumulation of materials not eluted during the isocratic analyses.
Both internal [23] and external standards [20; 21; 22; 24] have been used for quantitative measurements of DNA methylation. External standards that mimic the processing of biological samples have been used here. This has been done by preparing pre-mixed standard DNA samples, and then processing them through the entire work-up and digestion. The resulting standard curves reflect the unavoidable errors that arise during sample work-up such as ion suppression that might arise from components used in the reaction solutions. Consistent with the report of Song et al [25], our results reveal no evidence that small variations in the completeness of DNA hydrolysis adversely affects the linearity of the observed responses.
In this report, the DNA methylation levels in iPSCs are not similar to ESCs and fibroblasts. Various findings have already indicated that there are epigenetic differences between normal ESCs and iPSCs, particularly in DNA methylation patterns [29; 30; 31; 32; 33]. Our preliminary results on the comparison between iPSCs and parental somatic cells show a significant number of genes undergo increased DNA methylation during re-programming (Shen et al., unpublished data). It has been reported by others that the epigenetic mechanism of DNA methylation is a limiting factor in the reprogramming process, and that the DNA methylation pattern may not truly emulate the pattern found in ESC [31; 34]. For example, treatment of DNA methyltransferase inhibitor, 5-aza-cytidine, facilitated the transition of partially reprogrammed cells to iPSC [34]. Interestingly, the level of 5hmC in iPSC from reprogrammed fibroblast reported here appears to be restored to the levels found in ESC.
In conclusion, we have established an accurate and robust assay for the simultaneous quantification of 5hmC and 5mC levels in biological samples. LC-ESI-MS/MS-MRM is acknowledged as a gold standard in quantitation methodology, and the method described here will have widespread applicability and is sufficiently flexible for expansion to include other rare nucleosides.
Supplementary Material
Supplementary Table 1 -Primers and its sequence from various experiments.
Supplementary Figure 1 - Individual ion chromatograms of the three nucleosides to base transitions for all three DNA standards used to generate the calibration curve. Value beside the standards for each ion chromatograms is the percent purity of the indicated nucleoside of interest in the total pool of cytosine determined solely on the area of the peaks. Three replicates were done to calculate the purity of the nucleoside.
Supplementary Figure 2 - Immunostaining characterization of the two BJ iPS cell lines using various ESC markers co-stained with DAPI. Scale bars are indicated in the section.
ACKNOWLEDGEMENT
The authors wish to thank Andy Gieschen from Agilent for his technical support with the Agilent 6460 mass spectrometer, Zymo Research for providing reagents and samples, and Dr. Julian Whitelegge for providing suggestions and encouragement for this study.
FUNDING
This project is supported by CIRM RC1-0111 and NIH RO1 NS 051411 to GF.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-ydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cross SH, Meehan RR, Nan X, Bird A. A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nat Genet. 1997;16:256–259. doi: 10.1038/ng0797-256. [DOI] [PubMed] [Google Scholar]
- 4.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 5.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 6.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 7.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 8.Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, Hattori D, Ge W, Shen Y, Wu H, ten Hoeve J, Shuai K, Sun YE. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. 2005;132:3345–3356. doi: 10.1242/dev.01912. [DOI] [PubMed] [Google Scholar]
- 9.Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hutnick LK, Golshani P, Namihira M, Xue Z, Matynia A, Yang XW, Silva AJ, Schweizer FE, Fan G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. 2009;18:2875–2888. doi: 10.1093/hmg/ddp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penn NW, Suwalski R, O'Riley C, Bojanowski K, Yura R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem J. 1972;126:781–790. doi: 10.1042/bj1260781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010 doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Privat E, Sowers LC. Photochemical deamination and demethylation of 5-methylcytosine. Chem Res Toxicol. 1996;9:745–750. doi: 10.1021/tx950182o. [DOI] [PubMed] [Google Scholar]
- 16.Liutkeviciute Z, Lukinavicius G, Masevicius V, Daujotyte D, Klimasauskas S. Cytosine-5-methyltransferases add aldehydes to DNA. Nat Chem Biol. 2009;5:400–402. doi: 10.1038/nchembio.172. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burke DG, Griffiths K, Kassir Z, Emslie K. Accurate measurement of DNA methylation that is traceable to the international system of units. Anal Chem. 2009;81:7294–7301. doi: 10.1021/ac901116f. [DOI] [PubMed] [Google Scholar]
- 20.Kok RM, Smith DE, Barto R, Spijkerman AM, Teerlink T, Gellekink HJ, Jakobs C, Smulders YM. Global DNA methylation measured by liquid chromatography-tandem mass spectrometry: analytical technique, reference values and determinants in healthy subjects. Clin Chem Lab Med. 2007;45:903–911. doi: 10.1515/CCLM.2007.137. [DOI] [PubMed] [Google Scholar]
- 21.Liu Z, Liu S, Xie Z, Blum W, Perrotti D, Paschka P, Klisovic R, Byrd J, Chan KK, Marcucci G. Characterization of in vitro and in vivo hypomethylating effects of decitabine in acute myeloid leukemia by a rapid, specific and sensitive LC-MS/MS method. Nucleic Acids Res. 2007;35:e31. doi: 10.1093/nar/gkl1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z, Wu J, Xie Z, Liu S, Fan-Havard P, Huang TH, Plass C, Marcucci G, Chan KK. Quantification of regional DNA methylation by liquid chromatography/tandem mass spectrometry. Anal Biochem. 2009;391:106–113. doi: 10.1016/j.ab.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinlivan EP, Gregory JF., 3rd DNA methylation determination by liquid chromatography-tandem mass spectrometry using novel biosynthetic [U-15N]deoxycytidine and [U-15N]methyldeoxycytidine internal standards. Nucleic Acids Res. 2008;36:e119. doi: 10.1093/nar/gkn534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song L, James SR, Kazim L, Karpf AR. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:504–510. doi: 10.1021/ac0489420. [DOI] [PubMed] [Google Scholar]
- 25.Yang I, Kim SK, Burke DG, Griffiths K, Kassir Z, Emslie KR, Gao Y, Wang J, Foy CA, Pardos-Pardos AC, Ellison S, Domann PJ, Fujii S, Park SR. An international comparability study on quantification of total methyl cytosine content. Anal Biochem. 2009;384:288–295. doi: 10.1016/j.ab.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 27.Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, Ramsahoye B. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park IH, Yu J, Daley GQ, Eggan K, Hochedlinger K, Thomson J, Wang W, Gao Y, Zhang K. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, McKinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pick M, Stelzer Y, Bar-Nur O, Mayshar Y, Eden A, Benvenisty N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells. 2009;27:2686–2690. doi: 10.1002/stem.205. [DOI] [PubMed] [Google Scholar]
- 33.Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 -Primers and its sequence from various experiments.
Supplementary Figure 1 - Individual ion chromatograms of the three nucleosides to base transitions for all three DNA standards used to generate the calibration curve. Value beside the standards for each ion chromatograms is the percent purity of the indicated nucleoside of interest in the total pool of cytosine determined solely on the area of the peaks. Three replicates were done to calculate the purity of the nucleoside.
Supplementary Figure 2 - Immunostaining characterization of the two BJ iPS cell lines using various ESC markers co-stained with DAPI. Scale bars are indicated in the section.