

Abstract
CRISPR/Cas systems constitute a widespread class of immunity systems that protect bacteria and archaea against phages and plasmids, and commonly use repeat/spacer-derived short crRNAs to silence foreign nucleic acids in a sequence-specific manner. Although the maturation of crRNAs represents a key event in CRISPR activation, the responsible endoribonucleases (CasE, Cas6, Csy4) are missing in many CRISPR/Cas subtypes. Here, differential RNA sequencing of the human pathogen Streptococcus pyogenes uncovered tracrRNA, a trans-encoded small RNA with 24 nucleotide complementarity to the repeat regions of crRNA precursor transcripts. We show that tracrRNA directs the maturation of crRNAs by the activities of the widely conserved endogenous RNase III and the CRISPR-associated Csn1 protein; all these components are essential to protect S. pyogenes against prophage-derived DNA. Our study reveals a novel pathway of small guide RNA maturation and the first example of a host factor (RNase III) required for bacterial RNA-mediated immunity against invaders.
Organisms of all kingdoms of life have evolved RNA-guided immunity mechanisms to protect themselves against genome invaders1-6. In bacteria and archaea, CRISPR/Cas (clustered, regularly interspaced short palindromic repeats/CRISPR-associated proteins) constitutes an adaptive RNA-mediated defence system which targets invading phages or plasmids in three steps: (1) adaptation via integration of viral or plasmid DNA-derived spacers into the CRISPR locus, (2) expression of short guide crRNAs (CRISPR RNAs) consisting of unique single repeat-spacer units and (3) interference with the invading cognate foreign genomes by mechanisms that are yet to be fully understood7-27. A key event in CRISPR activation is the maturation of the active crRNAs from the CRISPR precursor transcript (pre-crRNA)28,29. Three Cas proteins, Cse3 (CasE), Cas6 and Csy4, have been identified as endoribonucleases that cleave within the repeat sequences of pre-crRNA to generate the mature cRNAs28-31. However, their homologues are missing in many CRISPR/Cas subtypes, suggesting the existence of alternate crRNA maturation pathways involving other Cas proteins and/or fundamentally different RNA processing events. Here, our study of the human pathogen Streptococcus pyogenes uncovered a novel pathway of CRISPR activation wherein a trans-encoded small RNA, the host endoribonuclease III and the CRISPR-associated Csn1 protein are responsible for the production of the active crRNAs.
CRISPR/Cas systems in S. pyogenes
Our analysis of S. pyogenes genome sequences revealed the presence of two CRISPR/Cas loci of two different subtypes, CRISPR01 (system II, Nmeni/CASS4 subtype) and CRISPR02 (system I-C, Dvulg/CASS1 subtype)32,33, each having a distinct set of repeats and cas genes (Fig. 1a, Supplementary Fig. 1 and Supplementary Table 1). Almost all of the associated CRISPR spacers show homology to chromosomal prophage sequences34-38 (Supplementary Tables 2-5), indicating that the CRISPR/Cas systems of S. pyogenes target lysogenic phages.
Figure 1. A newly identified tracrRNA is required for crRNA maturation in S. pyogenes.

a, dRNA-seq reveals expression of tracrRNA and crRNAs. Sequence reads of cDNA libraries derived from untreated and TEX-treated total RNA are shown. Vertical axis, relative amounts of sequenced cDNAs. The absence of ~75 nt tracrRNA form and 39-42 nt crRNA fragments in the TEX-treated cDNA library indicates that they are generated by processing. Genomic organization of tracrRNA and CRISPR01/Cas (csn1-cas1-cas2-csn2) loci. Transcriptional start sites and a terminator are indicated. (Left) tracrRNA (red) is encoded on the minus strand and detected as 171, 89 and ~75 nt tracrRNA species. Black rectangle, 36 nt sequence stretch complementary to CRISPR01 repeat. (Right) pre-crRNA is encoded on the plus strand. Rectangles, CRISPR01 repeats; diamonds, CRISPR01 spacers; 511, 66 and 39-42 nt, pre-crRNA and processed crRNAs. b, Base-pairing of tracrRNA with a CRISPR01 repeat is represented. Cleavages observed by dRNA-seq and leading to the formation of short overhangs at the 3′ ends of the processed RNAs are indicated by black arrows. Open arrow, cleavage in the spacer sequence. c, tracrRNA and pre-crRNA are co-processed in vivo. Northern blot analysis of S. pyogenes total RNA: strains and probes are indicated (Supplementary Figs 2 and 4). Left panel: Processing of tracrRNA into the ~75 nt form is abolished in Δpre-crRNA and re-established upon complementation with pre-crRNA. Right panel: Processing of pre-crRNA into mature crRNA forms (39-42 nt) is abrogated in ΔtracrRNA. Trans complementation of ΔtracrRNA with 171 or 89 nt tracrRNA restores the processing.
To examine the in vivo expression of CRISPR01 and CRISPR02, we analysed S. pyogenes strain SF370 (M1 serotype) by differential RNA sequencing (dRNA-seq)39. The most abundantly recovered small RNA species were CRISPR01 crRNAs originating from a ~511 nt pre-crRNA (Fig. 1a; Supplementary Fig. 2a, b and Supplementary Table 6), confirming that the CRISPR01 locus is active. In contrast, the CRISPR02 locus seems not to be expressed (Supplementary Fig. 3).
We detected six crRNAs from CRISPR01 which were 39 to 42 nt in length and likely processed species, as judged by their depletion in the dRNA-seq library for primary transcripts. The individual crRNAs appeared to result from double cleavage, one within the repeat and the other within the spacer, and carry a 20 nt spacer-derived 5′-guide sequence and a 19-22 nt repeat-derived 3′-sequence (Supplementary Fig. 2a, b). The latter sequence is distinct from the crRNA-tag (8 nt of the upstream repeat sequence) located in 5′ of mature crRNAs produced by the Cse3 (CasE) and Cas6-encoding CRISPR/Cas subtypes of Escherichia coli, Pyrococcus furiosus and Staphylococcus epidermidis28,31, providing evidence for the diversity of crRNA-tags among CRISPR/Cas systems and perhaps also for the underlying crRNA maturation and immunity mechanisms
tracrRNA directs pre-crRNA processing
Strikingly, dRNA-seq also detected abundant RNA species transcribed 210 nt upstream, on the opposite strand of the CRISPR01-associated genes and the leader-repeat-spacer array (Fig. 1a, b; Supplementary Fig. 4a, b and Supplementary Table 6); we refer to these abundant transcripts as tracrRNA (trans-activating CRISPR RNA). Northern blot probing detected four tracrRNA forms with approximate lengths of 171, 89, 75 and 65 nt, all of which were present throughout growth, notwithstanding a slightly decreased abundance of the longer transcripts in late stationary phase (Fig. 1c and Supplementary Fig. 4b-d). According to our dRNA-seq data, the 171 and 89 nt forms corresponded to primary transcripts whereas the shorter ~75 nt species resulted from processing of those longer tracrRNAs (Fig. 1a and Supplementary Fig. 4b).
Remarkably, both the 171 and 89 nt tracrRNAs contain a 25 nt stretch with almost perfect (one mismatch) complementarity to all repeats of CRISPR01 (Fig. 1b and Supplementary Fig. 5), predicting their potential base-pairing with pre-crRNA. Moreover, the tracrRNA and pre-crRNA processing sites detected by dRNA-seq fell in the putative RNA duplex region, indicative of co-processing of the two RNAs upon pairing. In support of this prediction, tracrRNA processing into the ~75 nt form was absent in a Δpre-crRNA mutant. Conversely, we did not detect mature crRNAs in a ΔtracrRNA strain, suggesting that tracrRNA is essential for the processing of pre-crRNA (Fig. 1c, Supplementary Figs. 2c and 4c). Trans complementation with the long tracrRNA species restored pre-crRNA processing in ΔtracrRNA bacteria, and revealed that the 89 nt form of tracrRNA suffices for co-processing (Fig. 1c and Supplementary Fig. 2c). Together, these findings reveal a novel function of a bacterial non-coding RNA such that a trans-encoded small RNA (tracrRNA) directs the maturation of another non-coding RNA (pre-crRNA) to yield the active species (crRNAs).
crRNA maturation requires RNase III and Csn1
According to our dRNA-seq data, the co-processed tracrRNA and pre-crRNA carry short 3′ overhangs reminiscent of cleavage by the endoribonuclease RNase III22,40-44 or the related eukaryotic Dicer and Drosha enzymes1,4-6,45. Since none of the Cas proteins of CRISPR01 contains an RNase III-like motif, we hypothesized that the endogenous RNase III—a general RNA processing factor40,42,46 encoded by the conserved rnc gene of the host—was recruited to cleave tracrRNA and pre-crRNA upon base-pairing. In support of our prediction, tracrRNA and pre-crRNA co-processing was abrogated in an Δrnc mutant of S. pyogenes (Fig. 2), yet restored by trans-complementation of RNase III expression (Supplementary Fig. 6).
Figure 2. Co-processing of tracrRNA and pre-crRNA requires both endogenous RNase III and Csn1 in vivo.

Northern blot analysis of tracrRNA (a) and pre-crRNA (b) expression: strains and probes are indicated (Supplementary Figs 6 and 8). Processing of tracrRNA (a) into a ~75 nt form and pre-crRNA (b) into 39-42 nt mature crRNA forms is abolished in Δrnc, Δcas/csn and Δcsn1 (refer to Supplementary Figs 6 and 8).
To directly demonstrate that the paired RNAs are substrates of this nuclease, tracrRNA and pre-crRNA were synthesized, annealed in vitro and incubated with E. coli RNase III. Whereas neither of the two RNAs alone was cut by the nuclease, their annealing promoted the expected singular RNase III cleavage in either RNA (Fig. 3a-c). Consistent with their shared complementarity to CRISPR01 repeats, both the 171 nt and 89 nt tracrRNAs promoted RNase III cleavage of pre-crRNA within the repeat to produce intermediate crRNA species, and both were converted to the ~75 nt tracrRNA species in the process (Fig. 3b, c). Mutations in the complementarity regions of tracrRNA or pre-crRNA hindered co-processing with the respective wild-type RNA partner, yet RNase III cleavage was fully restored when the compensatory tracrRNA and crRNA mutants were combined (Supplementary Fig. 7), corroborating that RNA duplex formation underlies the observed processing. Others have noticed that the repeats of CRISPR/Cas subtype II (Nmeni/CASS4) lack the potential to form stem-loop structures47; our findings suggest that tracrRNA overcomes this deficiency by providing an inter-molecular RNA structure for pre-crRNA processing. Taken together, RNase III serves as a host factor in tracrRNA-mediated crRNA maturation, and constitutes the first example of a non-Cas protein that is recruited to CRISPR activity.
Figure 3. tracrRNA directs pre-crRNA cleavage by RNase III in vitro.

a, Schematic representation of tracrRNA89 corresponding to 89-nt long tracrRNA, and crRNA213 and crRNA148 corresponding to a 213-nt long leader-repeat-spacer1-repeat-spacer2 fragment and a 148-nt long spacer1-repeat-spacer2-repeat-spacer3 fragment, respectively. b, Identification of tracrRNA89 binding sites on crRNA148*. 5′ end-labeled crRNA148* (~10 nM) was subjected to lead(II), RNase III and RNase T1 cleavage in the absence (lanes 1, 4, 7) and presence of cold tracrRNA89 (final concentration in lanes 2, 5, and 7: ~50 nM; lanes 3, 6, and 9: ~500 nM). Lane C: untreated crRNA148*; Lane T1: RNase T1 digest of crRNA148* under denaturating conditions; Lane OH: alkaline ladder; cleaved G residues are labeled. Vertical bars: crRNA148 region protected by tracrRNA89. Arrows denote specific RNase III cleavages in the two repeat regions of crRNA148 in the presence of tracrRNA89. c, Identification of crRNA148 and crRNA213 binding sites on tracrRNA89. 5′ end-labeled tracrRNA89* (~10 nM) was subjected to RNase T1, lead(II) and RNase III cleavage in the absence (lanes 1, 6, 11) and presence of cold crRNA148 or crRNA213 (final concentration in lanes 2, 4, 7, 9, 12 and 14: ~50 nM; lanes 3, 5, 8, 10, 13 and 15: ~500 nM). Lanes C, T1 and OH, positions of cleaved Gs and vertical bars: as above but referring to tracrRNA89* in the presence of cold crRNA148 or crRNA213.
Next, we entertained the possibility that—in addition to RNase III—Cas proteins facilitate the co-processing of the duplex RNA in vivo. Intriguingly, deletion of the csn1-cas1-cas2-csn2 operon impaired the processing of both tracrRNA and pre-crRNA (Fig. 2 and Supplementary Fig. 8a). In-frame deletions of any of the operon’s four genes then revealed Csn1 as the only Cas protein required for the production of mature crRNAs and concomitant tracrRNA cleavage. This was further supported by restored tracrRNA and pre-crRNA processing upon ectopic expression of Csn1 in Δcas-csn or Δcsn1 mutants (Fig. 2 and Supplementary Fig. 8b-d).
Csn1 (or COG3513) is a large, likely multi-domain protein32,33 of unknown function except that it is essential for CRISPR-mediated immunity in Streptococcus thermophilus8. Here, we propose a model wherein Csn1 acts as a molecular anchor facilitating the base-pairing of tracrRNA with pre-crRNA for subsequent recognition and cleavage of pre-crRNA repeats by the host RNase III (Fig. 4). Because Csn1 has predicted motifs of RuvC-like (RNase H fold) and McrA/HNH nucleases32,33, it might also mediate the second cleavage to occur at a fixed distance within the spacers. Furthermore, Csn1 might help protect tracrRNA and pre-crRNA against other host ribonucleases, as suggested by the strongly reduced accumulation of tracrRNA in the absence of csn1 (Fig. 2 and Supplementary Fig. 8a, b). Collectively, our results show that in the absence of Cse3 (CasE), Cas6 or Csy4 proteins, CRISPR01 crRNA maturation is achieved by the concerted action of three novel factors, a trans-encoded small RNA, a host-encoded ribonuclease and a Cas protein previously not implicated in pre-crRNA cleavage.
Figure 4. Model for tracrRNA-mediated crRNA maturation involving RNase III and Csn1.
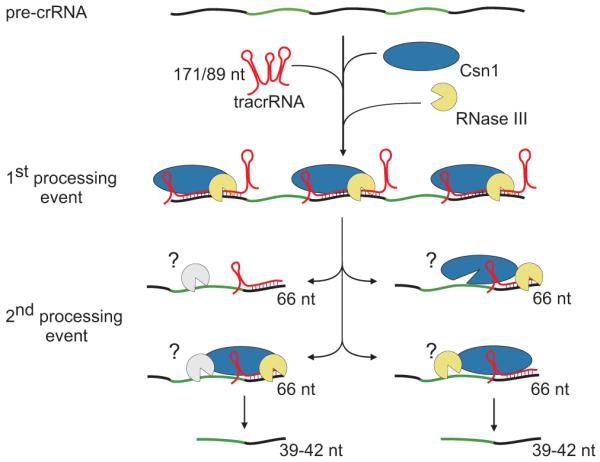
Black, repeat; green, spacer. tracrRNA can bind with almost perfect complementarity to each repeat sequence of the pre-crRNA. The resulting RNA duplex is recognized and site-specifically diced by RNase III in the presence of Csn1, releasing the individual repeat-spacer-repeat units (1st processing event). The generated units undergo further processing within the spacer sequences resulting in mature crRNA species consisting of unique spacer-repeat sequences (2nd processing event) by a yet-to-be elucidated mechanism. Csn1 may also be involved in the silencing of invading sequences.
CRISPR immunity against prophage sequences
To further investigate the role of tracrRNA in CRISPR01-mediated immunity, we developed a plasmid-based read-out system that mimics infection with protospacer-containing lysogenic phages (a protospacer is a DNA target sequence that matches a CRISPR spacer). We assayed transformation rates of a plasmid carrying a protospacer of the speM exotoxin gene, expected to be a target because of 100% identity to the second spacer of CRISPR01 (Spyo1h_002; Supplementary Table 2). Consistent with this protospacer being recognized by CRISPR01, wild-type S. pyogenes was protected from plasmids containing the speM gene, with or without its endogenous promoter region. Protection was specific since the wild-type strain was readily transformed with variants of the parental backbone plasmid as control (Fig. 5 and Supplementary Fig. 9). Importantly, in contrast to the wild-type strain, the Δpre-crRNA, ΔtracrRNA, Δrnc and Δcsn1 mutants invariably tolerated the speM plasmid (Fig. 5 and Supplementary Fig. 9). Together, these results demonstrate that tracrRNA, RNase III and Csn1 are essential in CRISPR01-mediated immunity of S. pyogenes against lysogenic phages, and further suggest that the tracrRNA/CRISPR01/Cas system, in concert with the host RNase III, limits horizontal virulence gene transfer among pathogenic streptococcal species34-38.
Figure 5. Both tracrRNA and pre-crRNA confer immunity against acquisition of a protospacer gene derived from a lysogenic phage.
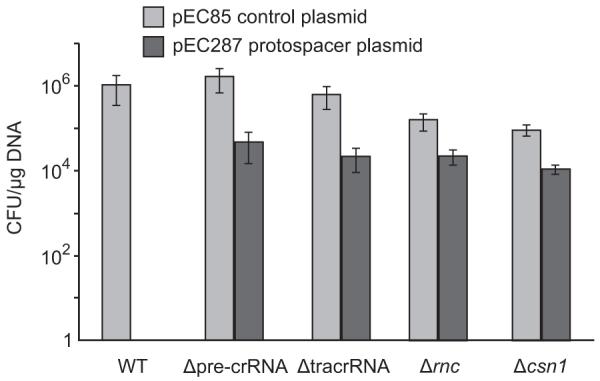
Transformation efficiencies of S. pyogenes with speM protospacer containing plasmid (pEC287) and reference “backbone” plasmid (pEC85) (Supplementary Fig. 9). Strains: S. pyogenes WT (SF370), Δpre-crRNA, ΔtracrRNA, Δrnc and Δcsn1. Graph bars, mean values of colony forming units (CFU) per μg of plasmid DNA; error bars, standard deviation (SD); n≥3. pEC287 is tolerated by the Δpre-crRNA, ΔtracrRNA, Δrnc and Δcsn1 mutants but not by the WT strain. As control, transformants in all strains were obtained with the backbone plasmid (Supplementary Fig. 9).
tracrRNA homologues in CRISPR/Cas systems
How widely spread is the tracrRNA-mediated CRISPR activation? Sequence analysis revealed anti-CRISPR repeat sequences, thus candidate tracrRNA homologues, in the vicinity of system II (Nmeni/CASS4) CRISPR/Cas loci of other bacterial genomes (Supplementary Table 7). We probed selected loci of Listeria innocua, Neisseria meningitidis, Streptococcus mutans and S. thermophilus (Fig. 6 and Supplementary Figs 12-16) and consistently observed both expression and processing of the homologous tracrRNAs and respective pre-crRNAs (Supplementary Figs 12-16). In addition, our analysis indicates potential co-evolution of the tracrRNA anti-repeat and CRISPR repeat sequences (Supplementary Table 7 and Supplementary Fig. 11). Thus, RNA base-pairing might generally determine crRNA maturation in type II CRISPR/Cas systems, and based on RNA probing results, these systems seem to be constitutively activated to target and affect the maintenance of invader genomes.
Figure 6. tracrRNA-mediated crRNA maturation is conserved among different bacterial species.
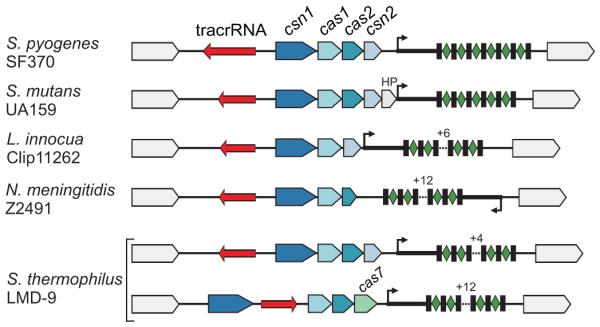
tracrRNA-mediated crRNA maturation is inherent to the type II (Nmeni/CASS4) CRISPR/Cas systems. Type II (Nmeni/CASS4) loci from S. pyogenes SF370, S. mutans UA159, L. innocua Clip11262, N. meningitidis Z2491 and S. thermophilus LMD-9 (Nmeni/CASS4a); red, tracrRNA; rectangles, repeats; diamonds, spacers.
No putative tracrRNA homologue was found in the vicinity of other CRISPR/Cas subtypes, and the two additional degenerated repeats identified near the type III-A (Mtube/CASS6) CRISPR/Cas locus in S. epidermidis RP62a25 lacked a corresponding tracrRNA homologue (Supplementary Fig. 17). Thus, the requirement of a trans-encoded small RNA for pre-crRNA processing into active crRNAs is a general RNA maturation mechanism shared by the type II (Nmeni /CASS4) CRISPR/Cas systems that lack the cse3 (casE), cas6 or csy4 gene but possess csn1. Whether all of the type II CRISPR/Cas loci require RNase III as a host factor remains to be seen.
In summary, trans RNA-mediated activation of crRNA maturation to confer sequence-specific immunity against parasite genomes represents a novel RNA maturation pathway, and highlights the unfolding diversity and complexity of molecular mechanisms of CRISPR/Cas systems9-14,26,28,29. Importantly, CRISPR loci have been generally regarded as autonomous genetic units, encoding all the proteins and RNAs required for their activity. Our identification of RNase III as the first host factor in CRISPR activity raises the exciting possibility that a recruitment of non-Cas proteins from the host chromosome might contribute to the rapid evolutionary diversification of CRISPR/Cas systems.
We suggest that Csn1 together with RNase III forms a microprocessor complex responsible for tracrRNA-mediated pre-crRNA processing (Fig. 4). As such, the requirement of RNase III in the process seems reminiscent of the key roles of related nucleases (Dicer, Drosha) in eukaryotic RNA-protein complexes that mediate the production of small interfering RNAs and maturation of microRNAs. However, the eukaryotic pathways employing RNase III-like enzymes for pre-RNA processing do not rely on trans-encoded RNA factors. More studies are needed to determine whether an RNase III-mediated activation of a small effector RNA by co-processing with a trans-acting non-coding RNA is also used in other biological systems.
METHODS SUMMARY
Details of bacteria (culture conditions, transformation), DNA (biocomputational analysis, plasmid construction, in-frame gene deletion mutants) and RNA manipulation (cDNA library construction [vertis Biotechnologie AG], RNA expression analysis, RNA structure probing and hydrolysis) are provided as Supplementary Information34,39,48. In brief, half of DNase I-treated SF370 total RNA was enriched for primary transcripts by treatment with the Terminator™ 5′-phosphate-dependent exonuclease (TEX) (Epicentre), which degrades RNAs with a 5′P (processed RNAs) but not primary transcripts with a 5′PPP RNA39. cDNA librairies were constructed from both untreated and TEX-treated RNA39. Following 454 pyrosequencing, cDNAs were mapped to the SF370 genome and visualized using the Affymetrix Integrated Genome Browser39. Strains, plasmids and primers are listed in Supplementary Tables 8, 9 and 10, respectively.
Supplementary Material
Acknowledgements
We thank D. Veit for technical help and R. Reinhardt for initial sequencing. This work was funded by the European Community (FP6, BACRNAs-018618; E.C.), the Austrian Science Fund (FWF, P17238-B09 (E.C.) and W1207-B09 (E.C., K.C.)), the Austrian Agency for Research Promotion (FFG, 812138-SCK/KUG; E.C.), the Theodor Körner Fonds (E.C.), Umeå University (E.C.), the Swedish Research Council (K2010-57X-21436-01-3; E.C.), IMPRS-IDI (Y.C.), the German Research Council (DFG Priority Program “Sensory and Regulatory RNAs in Prokaryotes” SPP1258, Vo875/4; J.V.), and the German Ministry of Education and Research (BMBF, 01GS0806/JV-BMBF-01 and 0315836; J.V.).
Appendix
METHODS
Bacterial strains and culture conditions
Bacterial strains used in the study are listed in Supplementary Table 8. Streptococcus pyogenes and Streptococcus mutans were cultured in THY medium (Todd Hewitt Broth (THB, Bacto, Becton Dickinson) supplemented with 0.2% yeast extract (Oxoid)) or on TSA (trypticase soy agar, BBL, Becton Dickinson) supplemented with 3% sheep blood. BHI broth (brain heart infusion) and BHI agar were used for growth of Listeria innocua and Staphylococcus epidermidis. Streptococcus thermophilus was grown in M17 broth supplemented with 0.5% (wt/vol) lactose (LM17)49 or on BHI agar. Escherichia coli was cultured in Luria-Bertani (LB) medium and agar. Neisseria meningitidis cells were grown on GC-agar and in PPM liquid medium including 1% vitamin-mix and 0.5% NaHCO3. Cultures of S. mutans, S. pyogenes and S. thermophilus were incubated at 37°C (S. mutans, S. pyogenes) or 42°C (S. thermophilus) in an atmosphere supplemented with 5% CO2 without shaking. Strains of E. coli, L. innocua, N. meningitidis and S. epidermidis were grown aerobically at 37°C with shaking. Whenever required, suitable antibiotics were added to the medium at the following final concentrations: ampicillin, 100 μg/ml for E. coli; kanamycin, 25 μg/ml for E. coli and 300 μg/ml for S. pyogenes. Bacterial cell growth was monitored periodically by measuring the optical density of culture aliquots at 620 nm using a microplate reader (SLT Spectra Reader).
Bacterial transformation
Transformation of E. coli with plasmid DNA was carried out according to standard protocols50. Transformation of S. pyogenes was performed as previously described51 with some modifications. For the transformation read-out experiment, S. pyogenes electro-competent cells were equalized to the same OD620 nm and subsequently used for electroporation with 500 ng of plasmid. Transformations were done in triplicate with the same batch of electro-competent cells. Each transformation was plated three to four times. Experiments were performed at least three times independently for statistical analysis. Transformation efficiencies were calculated as cfu/μg of DNA. Mock and control transformations were done with sterile dH2O and backbone “empty” plasmid pEC85, respectively.
DNA manipulations
DNA manipulations including DNA preparation, amplification, digestion, ligation, purification, agarose gel electrophoresis and Southern blot analysis were performed according to standard techniques50 with some modifications. Site-directed mutagenesis in plasmid inserts was done using QuickChange® II XL kit (Stratagene). Kits (Peqlab Biotechnology GmbH and Qiagen) were used for plasmid preparation and DNA purification with minor modifications. Synthetic oligonucleotides used in this study were supplied by VBC-Biotech Services GmbH and Sigma-Aldrich and are listed in Supplementary Table 10. When required (e.g., further use in cloning experiments, verification of in-frame gene deletion mutants), PCR-generated DNA fragments and plasmid inserts were sequenced at VBC-Biotech Services GmbH and Agowa Genomics. All plasmids generated in this study are listed in Supplementary Table 9.
In-frame gene deletion in S. pyogenes
The following procedure was used for the generation of ΔtracrRNA, Δpre-crRNA, Δcsn1-cas1-cas2-csn2, Δcsn1, Δcas1, Δcas2, Δcsn2 and Δrnc mutants in S. pyogenes wild-type (WT) SF370 (M1 serotype). Briefly, we constructed the shuttle vector pEC214 that includes a thermo-sensitive origin of replication (pWV01-repAts) specific for Gram-positive bacteria48, the ColE1 replicon specific for E. coli52, a kanamycin resistance cassette (aphIII) for plasmid selection in both E. coli and Gram-positive bacteria52, a thermostable ß-galactosidase cassette (bgaB) under the control of the Staphylococcus aureus clpB promoter for plasmid loss selection53 and a multiple-cloning site (MCS)52 (Supplementary Table 9). For each in-frame gene deletion, DNA fragments located upstream and downstream of the gene of interest were amplified by PCR using genomic DNA of S. pyogenes SF370 and primers containing flanking restriction sites (Supplementary Table 10). The amplified fragments were then cloned in the MCS of pEC214, thus generating thermosensitive plasmids containing a DNA region with in-frame deletion of the gene of interest (Supplementary Table 9). Each recombinant plasmid was introduced in S. pyogenes SF370 by electroporation at the permissive temperature (28°C). A series of incubation at the non-permissive temperature (37°C) and the permissive temperature (28°C)48 led to the seletion of in-frame gene deletion mutants sensitive to kanamycin (Supplementary Table 8). Kanamycin sensitive wild-type (WTts) clones issued from the same temperature shift procedure were also kept for comparative analysis. In this study, the phenotypes of all control WTts strains were identical to that of the wild-type (WT) strain. This indicated that the phenotypes observed in the in-frame gene deletion mutants were not caused by mutations, which would have occurred somewhere else on the chromosome during the recombination procedure (Supplementary Table 8). The correct deletion and non-deletion events in selected clones were verified by PCR, sequencing and Southern blot analyses (Supplementary Table 10). RT-PCR analysis of the in-frame Δrnc, Δcsn1, Δcas1, Δcas2 and Δcsn2 mutants demonstrated that each deletion of the single genes did not affect the expression of the other genes in the operons (operon rnc-smc, Supplementary Fig. 6, and operon csn1-cas1-cas2-csn2, Supplementary Fig. 8) (data not shown). RT-PCR analysis also showed rnc expression in Δcsn1 and csn1 expression in Δrnc (data not shown).
Construction of plasmids for complementation studies in S. pyogenes
Plasmid pEC85 containing an origin of replication specific for Gram-positive bacteria (repDEG-pAMβ1)54, the ColE1 replicon specific for E. coli52, a kanamycin resistance cassette (aphIII) for selection in both E. coli and Gram-positive bacteria52, and an expanded MCS54 was used as vector for complementation studies. For tracrRNA, CRISPR01 pre-crRNA and rnc complementation plasmids, a RNA-(tracrRNA and pre-crRNA) or protein-(rnc) encoding DNA fragment of interest under the control of the native promoter was cloned in pEC85 (Supplementary Tables 9, 10). For csn1, cas1, cas2 and csn2 complementation plasmids, a DNA fragment containing the coding sequence of the gene of interest under the control of the promoter of the csn1-cas1-cas2-csn2 operon was cloned in pEC85 (Supplementary Tables 9, 10). The generated recombinant plasmids were introduced in S. pyogenes mutant strains selecting for kanamycin resistant clones (Supplementary Table 8). The integrity of plasmids was verified by plasmid digestion and sequencing analysis. In addition, RT-PCR analysis was conducted to verify that the genes (encoding RNAs or proteins) in the inserts of the recombinant plasmids were expressed in the recombinant S. pyogenes strains (data not shown).
Construction of plasmids for transformation studies in S. pyogenes
Plasmid pEC85 described above was used as vector for transformation studies. A control DNA fragment and DNA fragments containing WT and mutated protospacers were cloned in pEC85 (Supplementary Tables 9, 10). The integrity of the cloned plasmids was verified by enzymatic digestion and sequencing analysis. The generated recombinant plasmids were introduced in S. pyogenes WT and mutant strains, selecting for kanamycin resistant clones (Supplementary Table 8). The integrity of plasmids was verified in selected clones by plasmid digestion and northern blot analysis of protospacer expression (Supplementary Table 10, Supplementary Fig. 9).
RNA preparation
Total RNA from streptococcal, staphylococcal, listerial and neisserial cells was prepared from culture samples harvested at different time points during growt48,54,55, using the TRIzol reagent (Sigma-Aldrich) with some modifications. The total RNA samples were treated with DNAse I (Fermentas) according to the manufacturer’s instructions. The concentration of RNA in each sample was further normalized following spectrophotometry measurements using Nanodrop.
cDNA library for differential RNA sequencing (dRNA-seq) and 454 pyrosequencing
In brief, total RNA from S. pyogenes SF370 (M1 serotype) cells grown until mid-logarithmic phase was treated with DNase I to remove any residual genomic DNA39. The RNA was submitted to a new treatment protocol to deplete processed transcripts and thereby enrich for primary transcripts by using the Terminator™ 5′-phosphate-dependent exonuclease (TEX) (Epicentre)39. A cDNA library from the TEX-treated and untreated RNA was constructed by vertis Biotechnology AG, Germany (http://www.vertis-biotech.com/). A total of 38,468 cDNAs was sequenced using a Roche 454 GS20 machine at the Max Planck Institute for Molecular Genetics (Berlin, Germany). After 5′end-linker and polyA-tail clipping, cDNAs longer than 17 nt (87%) were mapped to the S. pyogenes M1 GAS genome (NC_02737) using WU-BLAST 2.0 (http://blast.wustl.edu/) as previously described56. Afterwards mapping data were visualized using the “Integrated Genome Browser” (Affimetrix, Inc.). Small RNA candidates were selected on the basis of the following criteria: encoded in intergenic regions, size ranging from 50 to 500 nucleotides, presence of a putative promoter (BPROM software) and/or Rho-independent transcription terminator (TransTermHP (v2.04) program) (see below for references).
Northern blot analysis
Northern blot analysis was carried out essentially as described elsewhere55,57,58. RNAs separated on 10 or 12% polyacrylamide gels containing 8 M urea were blotted onto Hybond-N+ or Hybond-XL membranes (General Electric Healthcare) and then hybridized with specific probes. In general, 32P-labeled primers specific to the RNAs of interest were used as probes (Supplementary Table 10). For primer labeling, 40 pmol of oligonucleotide were denaturated for 5 min at 95°C and then mixed with 20 μCi of 32P-γ-ATP and 10 Units of T4 polynucleotide kinase (Fermentas). Labeled probes were purified prior hybridization using G25 columns (GE Healthcare) in order to remove unincorporated nucleotides. For all northern blots, 5S rRNA served as loading control.
RNA metabolic stability analysis
The metabolic stability of RNAs was determined as previously described59 with minor modifications. Cells were grown until mid-logarithmic phase, at which time point rifampicin (250 μg/ml, Sigma-Aldrich) was added. Aliquots of cells were collected before treatment (control), immediately after (0 min) and at defined time points after addition of rifampicin. A mixture of ice-cold acetone-ethanol (vol:vol, 1:1) was added and cells were frozen immediately at −80°C. Total RNA was prepared and analyzed by northern blot analysis as described above. RNA half-life (t1/2) was calculated by automated pixel counting using the ImageQuant v5.1 software, referring to 5S rRNA as loading control. To ensure absence of rifampicin-resistant bacteria, serial dilutions of the rifampicin-treated cells were plated on TSA blood plates with or without rifampicin (250 μg/ml), incubated overnight at 37°C and then visualized for the possible appearance of colonies.
RT-PCR analysis
RT-PCR analysis of RNA samples was done using the OneStep RT-PCR Kit (Qiagen) according to the manufacturer’s instructions and primers listed in Supplementary Table 10. For the semi-quantitative analysis, RNA samples were serial-diluted prior to reverse transcription and PCR. 8 ng to 1 μg of DNAse I treated RNA samples (TURBO DNA-free, Ambion) were used. Quantification of RT-PCR fragments was done using 5S rRNA or tmRNA as loading control.
RNA mapping
Head-to-tail RNA circularization and/or primer extension were used to map the RNA termini. (i) Head-to-tail RNA circularization: this method and its suitability to simultaneously map the 5′ and 3′ ends of RNAs was described elsewhere60. Briefly, total RNA was treated with Tobacco Acid Pyrophosphatase (TAP, Epicentre® Biotechnologies) according to the supplier’s instructions and the treated RNA was purified by acid:phenol:chloroform extraction and precipitation. RNAs were circularized using T4 RNA Ligase I (New England Biolabs) followed by purification using acid phenol:chloroform (Ambion) extraction and precipitation. The purified circularized RNAs were reverse transcribed and amplified by PCR using inward RNA-specific primers (Supplementary Table 10) and the OneStep RT-PCR Kit (Qiagen). The generated PCR fragments were cloned into pCR®2.1-TOPO vector using TOPO TA cloning kit (Invitrogen) and the inserts of three to five clones were sequenced. (ii) Primer extension: 5 to 10 μg of total RNA were denaturated in presence of 5′ radiolabeled reverse primer. Reverse transcription was done using ThermoScript Reverse Transcriptase (Invitrogen) following the manufacturer’s instructions. The RT reaction was subsequently separated on 8% polyacrylamide / 8 M urea gel and analyzed in reference to the PhiX174/HinfI ladder (Fermentas).
In vitro transcription, purification and 5′ end labeling of RNA
DNA templates carrying a T7 promoter sequence for in vitro transcription were generated by PCR using genomic DNA of S. pyogenes SF370 and primers listed in Supplementary Table 10. RNA was in vitro transcribed, gel-purified and quality-checked as described earlier61. 5′ labeling of RNA was done according to a previously published protocol62.
In vitro RNA structure mapping and footprinting
Secondary structure probing and mapping of RNA complexes was conducted on 5′-end-labeled RNA (~0.1 pmol) in 10 μl reactions. RNA was denatured 1 min at 95°C and chilled on ice for 5 min, upon which 1 μg yeast RNA and 10X structure buffer (0.1 M Tris pH 7, 1 M KCl, 0.1 M MgCl2, Ambion) were added. Concentrations of unlabeled tracrRNA/crRNA fragments added to the reactions are given in the figure legends. Following incubation for 10 min at 37°C, 2 μl of a fresh solution of lead(II) acetate (25 mM; Fluka), or 2 μl of RNase T1 (0.05 u/μl; Ambion) were added and incubated for 1 and 3 min at 37°C, respectively. RNase III cleavage reactions contained 1 mM DTT and 0.0026 units (1 μl of a 1:500 dilution of the 1.3 u/μl enzyme stock) enzyme (RNase III from E. coli, New England Biolabs), and were incubated for 3 min at 37°C. Reactions were stopped by direct addition of 12 μl loading buffer on ice. RNase T1 ladders were obtained by incubating labeled RNA (~0.2 pmol) in 1X sequencing buffer (Ambion) for 1 min at 95°C. Subsequently, 1 μl RNase T1 (0.1 u/μl) was added and incubation was continued at 37°C for 5 min. OH ladders were generated by 5 min incubation of 0.2 pmol labeled RNA in alkaline hydrolysis buffer (Ambion) at 95°C. Reactions were stopped with 12 μl loading buffer. Samples were denatured for 3 min at 95°C prior to separation on 6% or 8% polyacrylamide/7 M urea sequencing gels in 1X TBE. Gels were dried and analyzed using a PhosphorImager (FLA-3000 Series, Fuji) and AIDA software (Raytest).
Computational analysis of DNA and protein sequences
Gene annotations are according to NCBI genome browser and KEGG (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/). DNA sequence analysis for genetic locus organization and plasmid generation was done using the Vector NTI software (Invitrogen). Comparative sequence analysis of DNA and putative proteins was done using BLAST (http://www.ncbi.nlm.nih.gov/BLAST/). ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html) and AlignX (Invitrogen) were used for sequence alignments. Putative rho-independent transcription terminators (RITs) were annotated using the TransTermHP (v2.04) program (http://transterm.cbcb.umd.edu/)63. Putative promoters were predicted using the BPROM software (http://www.softberry.com/) and BDGP Neural Network Promoter Prediction NNPP version 2.2 (http://www.fruitfly.org/seq_tools/promoter.html).
Computational predictions of RNA structure and co-folding
Computational predictions were done using algorithms of the Vienna RNA package64,65. RNA secondary structures were predicted with RNAfold, co-folding was done using RNAcofold and co-folded secondary structures were drawn using VARNA66.
- 49.Deveau H, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N. Y.: 1989. edn. [Google Scholar]
- 51.Caparon MG, Scott JR. Genetic manipulation of pathogenic streptococci. Methods Enzymol. 1991;204:556–586. doi: 10.1016/0076-6879(91)04028-m. [DOI] [PubMed] [Google Scholar]
- 52.Charpentier E, et al. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004;70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arnaud M, Chastanet A, Debarbouille M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 2004;70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siller M, et al. Functional analysis of the group A streptococcal luxS/AI-2 system in metabolism, adaptation to stress and interaction with host cells. BMC Microbiol. 2008;8:188. doi: 10.1186/1471-2180-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urban JH, Vogel J. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007;35:1018–1037. doi: 10.1093/nar/gkl1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sittka A, et al. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herbert S, Barry P, Novick RP. Subinhibitory clindamycin differentially inhibits transcription of exoprotein genes in Staphylococcus aureus. Infect. Immun. 2001;69:2996–3003. doi: 10.1128/IAI.69.5.2996-3003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pall GS, Hamilton AJ. Improved northern blot method for enhanced detection of small RNA. Nat. Protoc. 2008;3:1077–1084. doi: 10.1038/nprot.2008.67. [DOI] [PubMed] [Google Scholar]
- 59.Roberts C, et al. Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J. Bacteriol. 2006;188:2593–2603. doi: 10.1128/JB.188.7.2593-2603.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Britton RA, et al. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol. Microbiol. 2007;63:127–138. doi: 10.1111/j.1365-2958.2006.05499.x. [DOI] [PubMed] [Google Scholar]
- 61.Sittka A, Pfeiffer V, Tedin K, Vogel J. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol. Microbiol. 2007;63:193–217. doi: 10.1111/j.1365-2958.2006.05489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Papenfort K, et al. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol. Microbiol. 2006;62:1674–1688. doi: 10.1111/j.1365-2958.2006.05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007;8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Denman RB. Using RNAFOLD to predict the activity of small catalytic RNAs. Biotechniques. 1993;15:1090–1095. [PubMed] [Google Scholar]
- 65.Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006;22:1172–1176. doi: 10.1093/bioinformatics/btl023. [DOI] [PubMed] [Google Scholar]
- 66.Darty K, Denise A, Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature .
References
- 1.Aliyari R, Ding SW. RNA-based viral immunity initiated by the Dicer family of host immune receptors. Immunol. Rev. 2009;227:176–188. doi: 10.1111/j.1600-065X.2008.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 4.Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457:405–412. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- 5.Malone CD, Hannon GJ. Small RNAs as guardians of the genome. Cell. 2009;136:656–668. doi: 10.1016/j.cell.2009.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 7.Andersson AF, Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–1050. doi: 10.1126/science.1157358. [DOI] [PubMed] [Google Scholar]
- 8.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 9.Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010;64:475–493. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- 10.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 11.Koonin EV, Makarova KS. CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol. Rep. 2009;1:95. doi: 10.3410/B1-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010;11:181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorek R, Kunin V, Hugenholtz P. CRISPR--a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008;6:181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 14.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009;34:401–407. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol .Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 16.Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 17.Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 18.van der Oost J, Brouns SJ. RNAi: prokaryotes get in on the act. Cell. 2009;139:863–865. doi: 10.1016/j.cell.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 19.Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 20.Mojica FJ, Ferrer C, Juez G, Rodriguez-Valera F. Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol. Microbiol. 1995;17:85–93. doi: 10.1111/j.1365-2958.1995.mmi_17010085.x. [DOI] [PubMed] [Google Scholar]
- 21.Nakata A, Amemura M, Makino K. Unusual nucleotide arrangement with repeated sequences in the Escherichia coli K-12 chromosome. J. Bacteriol. 1989;171:3553–3556. doi: 10.1128/jb.171.6.3553-3556.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makarova KS, Aravind L, Grishin NV, Rogozin IB, Koonin EV. A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis. Nucleic Acids Res. 2002;30:482–496. doi: 10.1093/nar/30.2.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garneau JE, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 25.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hale CR, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brouns SJ, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carte J, Pfister NT, Compton MM, Terns RM, Terns MP. Binding and cleavage of CRISPR RNA by Cas6. RNA. 2010;16:2181–2188. doi: 10.1261/rna.2230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vojtek I, et al. Lysogenic transfer of group A Streptococcus superantigen gene among streptococci. J. Infect. Dis. 2008;197:225–234. doi: 10.1086/524687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fischetti VA. In vivo acquisition of prophage in Streptococcus pyogenes. Trends Microbiol. 2007;15:297–300. doi: 10.1016/j.tim.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banks DJ, Beres SB, Musser JM. The fundamental contribution of phages to GAS evolution, genome diversification and strain emergence. Trends Microbiol. 2002;10:515–521. doi: 10.1016/s0966-842x(02)02461-7. [DOI] [PubMed] [Google Scholar]
- 38.Aziz RK, et al. Mosaic prophages with horizontally acquired genes account for the emergence and diversification of the globally disseminated M1T1 clone of Streptococcus pyogenes. J. Bacteriol. 2005;187:3311–3318. doi: 10.1128/JB.187.10.3311-3318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma CM, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 40.Drider D, Condon C. The continuing story of endoribonuclease III. J. Mol. Microbiol. Biotechnol. 2004;8:195–200. doi: 10.1159/000086700. [DOI] [PubMed] [Google Scholar]
- 41.Huntzinger E, et al. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J. 2005;24:824–835. doi: 10.1038/sj.emboj.7600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicholson AW. Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev. 1999;23:371–390. doi: 10.1111/j.1574-6976.1999.tb00405.x. [DOI] [PubMed] [Google Scholar]
- 43.Vogel J, Argaman L, Wagner EG, Altuvia S. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr. Biol. 2004;14:2271–2276. doi: 10.1016/j.cub.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Opdyke JA, Fozo EM, Hemm MR, Storz G. RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J. Mol. Biol. 2010 Dec 13; doi: 10.1016/j.jmb.2010.12.009. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carmell MA, Hannon GJ. RNase III enzymes and the initiation of gene silencing. Nat. Struct. Mol. Biol. 2004;11:214–218. doi: 10.1038/nsmb729. [DOI] [PubMed] [Google Scholar]
- 46.Condon C. Maturation and degradation of RNA in bacteria. Curr. Opin. Microbiol. 2007;10:271–278. doi: 10.1016/j.mib.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 47.Kunin V, Sorek R, Hugenholtz P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007;8:R61. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mangold M, et al. Synthesis of group A streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol. Microbiol. 2004;53:1515–1527. doi: 10.1111/j.1365-2958.2004.04222.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.