

Abstract
Seizures induce excitatory shifts in the reversal potential for GABAA-receptor-mediated responses, which may contribute to the intractability of electro-encephalographic seizures and preclude the efficacy of widely used GABAergic anticonvulsants such as phenobarbital. We now report that, in intact hippocampi prepared from neonatal rats and transgenic mice expressing Clomeleon, recurrent seizures progressively increase the intracellular chloride concentration ([Cl−]i) assayed by Clomeleon imaging and invert the net effect of GABAA receptor activation from inhibition to excitation assayed by the frequency of action potentials and intracellular Ca2+ transients. These changes correlate with increasing frequency of seizure-like events and reduction in phenobarbital efficacy. The Na+–K+–2Cl− (NKCC1) cotransporter blocker bumetanide inhibited seizure-induced neuronal Cl− accumulation and the consequent facilitation of recurrent seizures. Our results demonstrate a novel mechanism by which seizure activity leads to [Cl−]i accumulation, thereby increasing the probability of subsequent seizures. This provides a potential mechanism for the early crescendo phase of neonatal seizures.
Introduction
Neurons respond to a variety of stimuli with long-term shifts in the reversal potential for GABAA receptor (GABAA-R)-mediated postsynaptic currents (E GABA). For example, epileptogenic injuries alter neuronal chloride transport, which is the principal determinant of E GABA (Jin et al., 2005; Pathak et al., 2007). E GABA is persistently shifted to more positive (depolarizing) potentials by prolonged seizure activity (Khalilov et al., 2003) or trains of action potentials without (Fiumelli and Woodin, 2007; Brumback and Staley, 2008) or with concomitant GABAA receptor activation (Woodin et al., 2003). In addition, other events, including oxygen-glucose deprivation (Pond et al., 2006) and trauma (van den Pol et al., 1996), shift E GABA to more depolarized potentials. Depolarized values of E GABA reduce the efficacy of inhibition and contribute to seizure activity (Dzhala and Staley, 2003; Khazipov et al., 2004; Dzhala et al., 2005).
Immature neurons express high levels of NKCC1 (Delpire, 2000; Wang et al., 2002; Dzhala et al., 2005), an electroneutral cotransporter that imports Na+, K+, and Cl−. This raises the intracellular chloride concentration, resulting in a depolarized E GABA. This shift in E GABA can initiate a positive feedback cycle in neonatal seizures (Staley and Smith, 2001), and depolarized values of E GABA contribute to the reduced efficacy of GABA-enhancing anticonvulsants (Dzhala et al., 2005, 2008) that are first-line agents in the treatment of neonatal seizures (Rennie and Boylan, 2003; Carmo and Barr, 2005). NKCC1 is selectively blocked by low micromolar concentrations of the loop diuretic bumetanide (Isenring et al., 1998; Hannaert et al., 2002). By reducing intracellular chloride accumulation, this diuretic shifts E GABA to negative potentials, resulting in more effective inhibition (Yamada et al., 2004; Dzhala et al., 2005, 2008; Sipila et al., 2006; Rheims et al., 2008). The increase in inhibition has a significant anticonvulsant effect that is synergistic with anticonvulsants such as barbiturates that prolong the open probability of the GABAA-receptor-mediated chloride channels (Twyman et al., 1989; Dzhala et al., 2005, 2008).
Several recent reports regarding the experimental treatment of neonatal seizures have reported conflicting results (Table 1). For example, if administered soon after the onset of seizures, phenobarbital is relatively more effective (Quilichini et al., 2003; Raol et al., 2009) and bumetanide is less effective (Kilb et al., 2007) as an anticonvulsant. In contrast, experiments in which drugs are administered after many seizures demonstrate reduced phenobarbital efficacy and increased efficacy of drugs such as bumetanide that block NKCC1-mediated shifts in E GABA (Dzhala et al., 2005, 2008; Mazarati et al., 2009). Here we consider the possibility that activity-dependent, NKCC1-dependent changes in E GABA help determine the time course of neonatal seizures. This hypothesis could explain the substantial variations in the efficacy of GABAergic anticonvulsants in the experimental settings described in Table 1. This hypothesis is also of considerable clinical importance because it provides an additional impetus for immediate and aggressive treatment of neonatal seizures: the more seizures that occur, the more E GABA will shift, and the lower the probability that the seizures will respond to available anticonvulsants such as phenobarbital, particularly during the crescendo phase of neonatal seizures (Painter et al., 1999). A corollary of this hypothesis is that, by blocking NKCC1, bumetanide should prevent or reduce the seizure-induced shift in E GABA, potentially ameliorating the crescendo pattern of neonatal seizures. Finally, enhancement of the efficacy of GABAergic anticonvulsants by bumetanide should increase with the duration of the previous seizure activity. These hypotheses are tested in whole hippocampal preparations from neonatal rats and CLM-1 mice.
Table 1.
Efficacy of GABAergic anticonvulsants in animal models of neonatal seizures
| # | Preparation/model | Age | Anticonvulsant (AED) | Concentration | Application | Efficacy | Authors |
|---|---|---|---|---|---|---|---|
| 1 | In vivo/flurothyl | P9, P15 | Phenobarbital | 20 mg/kg | Pretreatment (i.p) | High | (Velísek et al., 1995) |
| 2 | In vivo/pentylenetetrazol | P7, P12 | Clobazam | 0.5–7.5 mg/kg | Pretreatment (i.p) | High | (Haugvicová et al., 1999) |
| 3 | In vitro/0 mm Mg2+ | P7–P8 | Phenobarbital | 30 μm | After 3 ILDs | Low | (Quilichini et al., 2003) |
| Phenobarbital | 100 μm | High | |||||
| 4 | In vitro; in vivo/K+ + gabazine | P8–P12, P17–P21 | Diazepam | 5 μm | Coinjection (before ILD) | High | (Isaev et al., 2007) |
| 5 | In vitro/0 mm Mg2+ | P4–P6 | Phenobarbital | 100 μm | After 7 ILDs | Low | (Dzhala et al., 2008) |
| 6 | In vitro/8.5 mm K+; in vivo/kainic acid | P5–P7 | Phenobarbital | 100 μm | After 7 ILDs pretreatment (i.p) | Low | (Dzhala et al., 2005) |
| P9–P12 | Phenobarbital | 25 mg/kg | Modest | ||||
| 7 | In vivo/kainic acid; flurothyl | P10 | Phenobarbital | 50 mg/kg | Stage 5 seizures (i.p) | Significant | (Raol et al., 2009) |
| P10 | Diazepam | 4–16 mg/kg | Transient |
ILD, Ictal-like discharges; AED, antiepileptic drug.
Materials and Methods
Animals.
All animal-use protocols conformed to the guidelines of the National Institutes of Health and the Massachusetts General Hospital Center for Comparative Medicine on the use of laboratory animals.
In vitro electrophysiology.
Intact hippocampal formations were prepared from neonatal postnatal day 5 (P5) to P7 male and female CLM-1 mice pups and P3–P6 Sprague Dawley rat male pups as described previously (Dzhala et al., 2008). Animals were anesthetized and decapitated. The brain was rapidly removed to oxygenated (95% O2–5% CO2) ice-cold (2–5°C) artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 3.5 KCl, 2 CaCl2, 1.3 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 glucose, pH 7.4. The hemispheres were separated, and, after removing the cerebellum, the frontal part of the neocortex, and surrounding structures, the intact hippocampi were dissected from the septohippocampal complex. The hippocampi were incubated in oxygenated ACSF at room temperature (20–22°C) for 1–2 h before use. For recordings, the hippocampi were placed into a conventional submerged chamber and continuously superfused with oxygenated ACSF at 32°C and at a flow rate 4 ml/min.
Extracellular field potentials were recorded in the intact hippocampal preparations in vitro using tungsten microelectrodes and low-noise multichannel amplifier (bandpass, 0.1 Hz to 10 kHz; 1000×) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Microelectrodes made from coated tungsten wire of 50 μm diameter (California Fine Wire Company) were used for simultaneous recordings of population field activity in EEG band (0.1–100 Hz), ripple (100–200 Hz), and fast ripple (200–500 Hz) oscillations and multiple-unit activity (MUA) (500 Hz high-pass filter). Root mean square (RMS) noise level with an electrode placed in the perfusion solution was typically 3–4 μV, whereas the amplitude of action potentials recorded from the pyramidal cell layer ranged from this noise level up to 60–80 μV. The signals were digitized using an analog-to-digital converter (DigiData 1322A; Molecular Devices). Sampling interval per signal was 100 μs (10 kHz). pClamp 9.2 (Molecular Devices), Mini Analysis 5.6 (Synaptosoft), and Origin 7.5 SR6 (Microcal Software) programs were used for the acquisition and data analysis. Power spectrum analysis was performed after applying a Hamming window function. Power was calculated by integrating the RMS value of the signal in frequency bands from 1 to 1000 Hz.
Group measures are expressed as mean ± SEM; error bars also indicate SEM. Statistical tests of significance were assessed with Student's t, ANOVA, χ2, and Mann–Whitney tests as indicated in Results. The level of significance was set at p < 0.05.
Clomeleon imaging and [Cl−]i determination.
Clomeleon is a fusion protein comprising the Cl−-sensitive yellow fluorescent protein (YFP) and the Cl−-insensitive cyan fluorescent protein (CFP). Transgenic CLM-1 mice expressing Clomeleon were received from Duke University Medical Center (Durham, NC) and housed at Massachusetts General Hospital Center for Comparative Medicine (Charlestown, MA). Intact hippocampi were prepared from neonatal (P5–P7) mice as described previously (Dzhala et al., 2008). The hippocampi were incubated in oxygenated ACSF at room temperature (20–22°C) for at least 1 h before use. For optical imaging and simultaneous extracellular field potential recordings, the hippocampus was placed into a conventional submerged chamber on a precision x–y stage mounted on the microscope and continuously superfused with oxygenated ACSF at 32°C and at a flow rate 4 ml/min. Two-photon Clomeleon imaging was performed on an Olympus Fluoview 1000MPE with pre-chirp excitation optics and a fast acousto-optical modulator mounted on an Olympus BX61WI upright microscope using an Olympus 20× water-immersion objective (XLUMPLFL 20xW; numerical aperture, 0.95). A mode-locked titanium/sapphire laser (MaiTai; Spectra Physics) generated two-photon fluorescence with 860 nm excitation. Emitted light passed through a dichroic mirror (460 nm cutoff) and was bandpass filtered through one of two emission filters 480 ± 15 nm (D480/30) for CFP and 535 ± 20 nm (D535/40) for YFP (FV10MP-MC/Y). Detectors containing two photomultiplier tubes were used. Image size (x–y dimension) was 512 × 512 pixel or unit converted 634.662 × 634.662 μm. Clomeleon expressing neurons were typically sampled 20–200 μm below the surface of the intact hippocampus (z dimension, 0–200 μm; step size, 2.00 μm). In ictal experiments, Clomelon imaging was performed 1 min after seizure termination to minimize pH-related changes in fluorescence (Xiong et al., 2000). Peak ictal changes in pH in vitro are ∼0.02 pH units (Xiong et al., 2000), which would cause ≤2 mm change in our estimates of intracellular chloride concentration ([Cl−]i) (Kuner and Augustine, 2000).
Quantitative measurements on three-dimensional (3D) stacks were performed using NIH ImageJ 1.40 g software. The CFP and YFP images were opened, and their respective background value was subtracted for the 3D volume. Median filtering was applied to all of the 3D planes. Cells were visually identified, and a region of interest was drawn around the cell bodies. The ratio of the YFP/CFP fluorescence intensity was measured for each identified cell. The [Cl−]i was calculated from the following equation:
 |
where R is the measured YFP/CFP ratio, K′D is the apparent dissociation constant of Clomeleon, R max is the ratio when Clomeleon is not bound by Cl−, and R min when it is completely quenched by F − (Kuner and Augustine, 2000; Berglund et al., 2008). The constants K′D, R max, and R min were determined from the calibration of [Cl−]i using solutions of known concentrations of Cl− [20, 80, and 123 mm extracellular chloride concentration ([Cl−]o)]. The K+/H+ ionophore nigericin (50 μm) and the Cl−/OH− antiporter tributyltin chloride (100 μm) were used to remove transmembrane H+/OH− and Cl− gradients. Neurons that experienced a change in YFP/CFP intensity to each [Cl−]o were used for the calibrations. The data points obtained with the different [Cl−]o are described by rewriting the equation above as the ratiometric function:
 |
where R max and K′D were free parameters, whereas R min was determined by quenching Clomeleon with 123 mm F − (Kuner and Augustine, 2000; Duebel et al., 2006). With single-cell resolution, R max and K′D were calculated for 47 individual neurons that were followed across all calibration conditions. The K′D was 91 ± 5.43 mm, R max was 1.026, and R min was 0.268.
The mean of the logarithmic value of the [Cl−]i was used in pseudocolor images, because the ion concentration follows a log-normal distribution. E Cl was calculated using the Nernst equation setting the temperature variable at 33°C.
Two-photon fluorescence Ca2+ imaging.
Both excitatory and inhibitory GABA responses are evident during status epilepticus in the intact hippocampus in vitro. The dense packing of cells in the pyramidal layer makes it difficult to directly measure these differential activity-dependent responses of individual neurons using extracellular recordings of multiple-unit activity. Two-photon fluorescence imaging of bulk-loaded calcium dyes provides an optical proxy to single-cell electrophysiology with the inherent ability to multiplex. Simultaneous investigation of hundreds of neurons with single-cell resolution (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) makes it possible to determine whether the responses to GABA indeed vary from cell to cell and, more importantly, how this response topology shifts after recurrent seizures. The high spatial resolution of this technique allowed us, in many instances, to follow the same cells across multiple conditions, e.g., in control versus during and after spontaneous seizures, allowing us to use an individual cell as its own control.
We loaded hippocampal neurons with a calcium-sensitive dye under visual guidance through the two-photon microscope. A total of 0.8 mm Oregon Green 488 BAPTA-1 AM (OGB) was dissolved in DMSO with 20% pluronic acid and mixed in ACSF containing sulforhodamine 101 (SR-101), a red-fluorescent astrocyte-specific marker (all from Invitrogen). A 2–3 μm patch pipette was filled with this solution and inserted into the intact hippocampus to a depth of 150–300 mm from the surface. The AM dye, combined with SR-101, was pressure ejected from the pipette (5–20 psi for 60–150 s) under continuous visual guidance with multiphoton microscopy. OGB is not fluorescent until fully hydrolyzed inside a cell; as a result, the size of the microinjection was estimated using the red fluorescent signal from SR-101. Full loading of the OGB dye took 0.5–1 h. After confirming loading, the pipette was withdrawn. Several hundred to thousands of cells were loaded from a single injection in a 300-μm-diameter spherical region.
Time-lapsed two-photon calcium imaging was performed on the same microscope used for Clomeleon imaging. For continuous visual guidance during dye loading, an Olympus 40× (numerical aperture, 0.80) was also used. OGB-loaded neurons were typically sampled 50–200 μm below the surface of the intact neonatal hippocampus.
Image analysis.
Time-lapsed images were analyzed in NIH ImageJ (Wayne Rasband, National Institutes of Health, Bethesda, MD), Matlab (MathWorks), and Microsoft Office Excel using custom-written software. Cells were automatically identified, and time course data were extracted for each individual cell. On average, >200 neurons could be imaged simultaneously in a given experiment. Astrocytes could be identified based on their uptake of SR-101. Cells were defined as responsive if there was a significant difference their baseline fluorescence and the fluorescence immediately after electrical stimulation. The peak response of the cell was defined as the maximum ΔF/F immediately after stimulation. Cells were considered to be inhibited by GABA if their response to stimulus during application of kynurenic acid (KYNA) + CGP 55845 [(2S)-3-[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl) (phenylmethyl)phosphinic acid] + SR 95531 [2-(3-carboxypropyl)- 3-amino-6-(4-methoxyphenyl)pyridazinium bromide] (a potent GABAA-R antagonist) significantly exceeded that under KYNA + CGP 55845 alone. In a subset of experiments, we were able to follow the same cells over the course of several hours to permit comparison across seizure and pharmacological conditions for the same cells. Before and after seizures, cells responses in three conditions (control, KYNA + CGP 55845, and KYNA + CGP 55845 + SR 95531) were compared. If cells were inhibited by GABA, there would be a change in binary response probability (from inactive to active) or peak intensity (>15% increase) when comparing the KYNA + CGP 55845 + SR 95531 condition to KYNA only. In total, we were able to analyze 468 cells in this manner. Of those that were inhibited by GABA (n = 423 neurons), we then determined the percentage that changed their polarity (“switched”), i.e., how many cells that were initially inhibited by GABA shifted their response after spontaneous seizures. We did this by comparing the cellular response properties from before and after seizure (again using both binary response probability and peak ΔF/F). Finally, because we had single-cell resolution across conditions, we correlated the probability of a single cell “switching” to the initial response characteristic (peak ΔF/F) during control conditions before spontaneous seizures. We grouped cells based on their initial evoked response to electrical stimulus in control conditions and created a probability distribution based on this simple formula: number of cells that switched/total number of cells. We then determined whether there was a correlation between initial evoked response and the probability of switching.
Phospho-NKCC1 Western blotting.
Lysates were fractionated using 4–15% SDS-PAGE gradient gel electrophoresis (Bio-Rad). Proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad) at 100 V and 4°C for 2 h. The membrane was then blocked in 10% nonfat dried milk in PBS with 0.1% Tween 20 (PBS-T). Primary antibodies, including rabbit R5 anti-phospho-NKCC1 (Flemmer et al., 2002) (gift from Dr. Biff Forbush, Yale University, New Haven, CT) and rabbit anti-β-tubulin (Cell Signaling Technology) were diluted, both at 1:500, in 5% milk in PBS-T and incubated with the membrane for 1 h at room temperature. Blots were washed in PBS with 0.1% Tween 20 and probed with an HRP-conjugated anti-rabbit secondary antibody (Zymed) in 1% milk in PBS-T for 30 min at room temperature. The filter was then washed and chemiluminescence was performed using ECL-Plus (GE Healthcare), following standard protocols.
Drugs.
Reagents were purchased from Sigma-Aldrich and Tocris Cookson, prepared as stock solutions, and stored before use as aliquots in tightly sealed vials at the temperatures and conditions recommended by the manufacturers. Fluorescent probes were from Invitrogen.
Supplemental data.
Supplemental Figures 1–5 are available at www.jneurosci.org as supplemental material.
Results
We used the intact hippocampal preparation (Khalilov et al., 1997) to preserve the neuronal network and avoid potential alterations in chloride transport and intracellular chloride concentration related to neuronal injury and axotomy (van den Pol et al., 1996; Nabekura et al., 2002) that may occur during slice preparation.
Distribution of resting [Cl−]i and GABA action in the intact neonatal hippocampus
High-resolution two-photon fluorescence chloride imaging was performed in the intact hippocampus in vitro of the neonatal (P5–P7) transgenic CLM-1 mice expressing Clomeleon (Kuner and Augustine, 2000; Berglund et al., 2008; Glykys et al., 2009). The ratio of fluorescence resonance energy transfer-dependent emission of the chloride-sensitive YFP to the chloride-insensitive CFP was used to measure [Cl−]i in CA3 pyramidal cells and interneurons (see Materials and Methods) (Fig. 1 a,b). Under control conditions, the resting [Cl−]i in the individual neurons of the intact hippocampal preparations (n = 10 hippocampi) varied from 1 to 40 mm (Fig. 1) depending on cell size, type (data not shown), and postnatal age (Tyzio et al., 2007; Glykys et al., 2009). Gaussian fits revealed a wide range of the resting intracellular chloride (Fig. 1 b), suggesting heterogeneous populations of neurons, some with an E Cl below the resting membrane potential, and others whose E Cl is positive to resting membrane potential (Tyzio et al., 2003). This wide range of neuronal Cl− is consistent with previously published data (Tyzio et al., 2007; Berglund et al., 2008; Glykys et al., 2009) and implies that both hyperpolarizing as well as depolarizing GABAA receptor (GABAA-R)-mediated signaling occurs in large populations of developing neurons, via inwardly and outwardly directed Cl− fluxes.
Figure 1.

Distribution of resting [Cl−]i and GABA action in the intact neonatal hippocampus. a, Two-photon fluorescence confocal scanning images of the CA3 pyramidal cell layer in the intact hippocampus in vitro of the neonatal (P7) transgenic CLM-1 mouse expressing Clomeleon. Top, Overlay of confocal multiple planes (150 samples; 2 μm step) of the Cl−-sensitive YFP signals of the CA3 neurons. Inset shows the photograph of the intact hippocampus. Bottom, Pseudocolored regions of interest from neurons according to [Cl−]i. b, Distribution of resting [Cl−]i (bin size, 2.5 mm) from 81 individual neurons shown in a. A Gaussian fit yielded a peak at 17.4 ± 0.6 mm and width of 14.6 ± 2.3 mm. Multipeak fit yielded three peaks at 6.5 ± 0.6, 15.9 ± 1, and 25.3 ± 2.1 mm. c–e, Effect of isoguvacine (ISO) on MUA in the intact neonatal (P5–P7) hippocampal preparations. c, Extracellular recording of MUA in the CA3 pyramidal cell layer and corresponding frequency of MUA. Bath application of isoguvacine (10 μm) caused a transient decrease in the frequency of MUA; peak function fit is indicated by red line. d, e, Individual hippocampus responses to isoguvacine and corresponding mean frequency of MUA (*p = 0.00002, two sample t test). Decrease in MUA frequency indicates that the net effect of GABAA-R activation was inhibitory.
The net effect of GABAA-R activation on the frequency of spontaneous action potentials was measured in intact hippocampi prepared from neonatal (P5–P7) CLM-1 mice (Fig. 1 c–e). Non-invasive extracellular recordings of MUA were performed in the CA3 pyramidal cell layer (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Bath application of the selective agonist of GABAA-R isoguvacine (10 μm for 1 min) transiently reduced spontaneous neuronal firing rate by 91.5 ± 3.1% in 12 recordings from six intact hippocampi (Fig. 1 c–e), indicating that the net effect of GABAA-R activation in the intact hippocampal network was inhibitory. In contrast, in the neonatal hippocampal and neocortical slice preparations, a more uniformly depolarizing Cl− equilibrium potential (Glykys et al., 2009) and a consequent excitatory action of GABAA-R activation (Dzhala et al., 2005) leads to a net increase in spontaneous firing rates of neurons and an increased frequency of spontaneous population bursts in response to GABAA-R agonists (Tyzio et al., 2007; Dzhala et al., 2008) .
Progressive [Cl−]i accumulation and change in the action of GABA during recurrent seizures
Positive (excitatory) shifts in the reversal potential for GABAA-receptor-mediated responses were observed in immature neurons after prolonged seizure activity (Khalilov et al., 2003). We considered the possibility that neuronal chloride accumulation and a consequent excitatory shift in GABA action occur and persist during recurrent seizure activity. Simultaneous high-resolution two-photon fluorescence chloride imaging and extracellular field potential recordings were performed in intact hippocampi prepared from neonatal (P5–P7) CLM-1 mice (Kuner and Augustine, 2000; Berglund et al., 2008). Persistent bath application of low-Mg2+ ACSF induced recurrent interictal and ictal-like (seizure) epileptiform discharges (Fig. 2 a) (for details, see Dzhala et al., 2008), which gradually increased in frequency (Quilichini et al., 2002, 2003; Dzhala et al., 2008) and power (Dzhala et al., 2008) (Fig. 2 e–g) (see Fig. 5 e,f). Starting from onset, the mean interseizure interval significantly decreased by 39% from 515.4 ± 29.7 to 315.7 ± 35.6 s (n = 8, p = 0.0008) (see Fig. 5 e). Over a 1 h period of low-Mg2+ ACSF application, the normalized mean power of recurrent ictal-like events increased by 61 ± 24% (p = 0.039) (see Fig. 5 f). These data are consistent with the hypothesis that neonatal seizures increase the probability of subsequent seizures.
Figure 2.
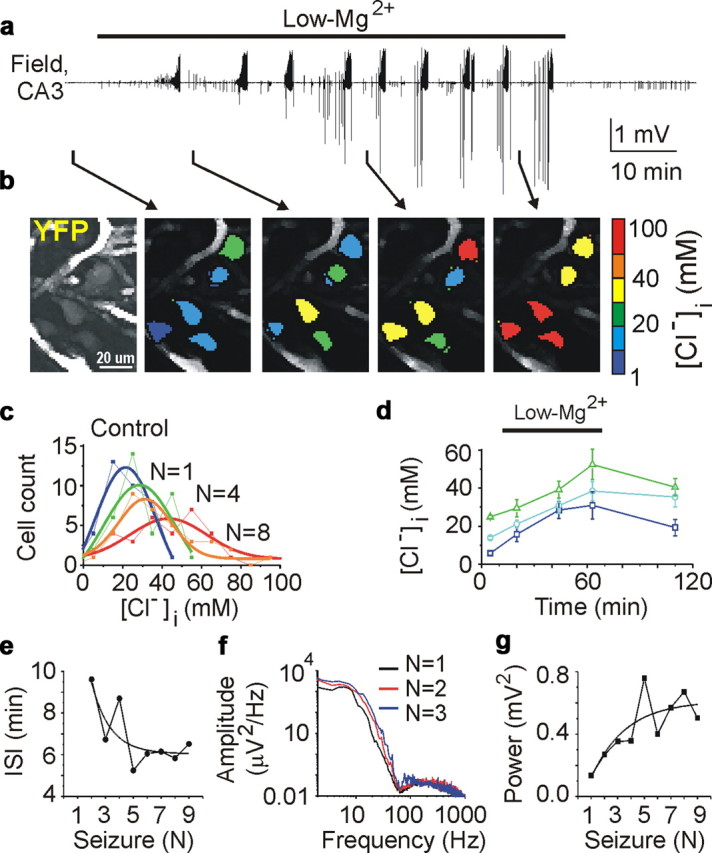
Neonatal seizure-induced chloride accumulation and facilitation of recurrent seizures. a, Extracellular field potential recordings in the CA3 pyramidal cell layer in the intact hippocampus in vitro of the neonatal (P5) CLM-1 mouse. Recurrent seizures were induced by low-Mg2+ ACSF and stopped by perfusion of control ACSF. b, Effect of recurrent seizures on [Cl−]i. Example of two-photon confocal imaging of YFP (left) of CA3 neurons yielded pseudocolor map of [Cl−]i before seizures, after the first, fourth, and eighth recurrent seizures. c, Distribution of [Cl−]i (bin size, 10 mm) in 36 identified neurons before seizures (dark blue), after one (green), four (orange), and eight (red) recurrent seizures. d, Corresponding changes of mean [Cl−]i in subpopulation of neurons with low (0–10 mm), medium (10–20 mm), and high (20–30 mm) range of control resting chloride. e–g, Seizure induced facilitation of recurrent seizures. e, Interseizure intervals (ISI) decreased as a function of the number of recurrent seizures. f, Power spectra of three consecutive seizures in frequency band from 1 to 1000 Hz. g, Corresponding power of recurrent seizures as a function of the number of seizures.
Figure 5.

The role of NKCC1 in seizure-induced chloride accumulation and facilitation of recurrent seizures. a, Top, Low-Mg2+ ACSF induced recurrent seizures in control and in the presence of NKCC1 blocker bumetanide (10 μm). Extracellular field potential recordings in the CA3 pyramidal cell layer in the intact hippocampus in vitro of neonatal (P5) CLM-1 mouse. Bottom, Example of two-photon confocal imaging of YFP (left) of CA3 neurons yielded pseudocolor map of [Cl−]i before seizures, after two seizures, and after nine seizures in the presence of bumetanide. b, Distribution of [Cl−]i (bin size, 10 mm) in 48 identified neurons before seizures (black), after two seizures in control (low-Mg) solution (red), and after nine seizures in the presence of bumetanide (Bum; green). Gaussian fits yielded mean ± width values of 22.1 ± 19 mm in control, 29.8 ± 18.4 mm after n = 2 seizures, and 44.4 ± 24.1 mm after n = 9 seizures. c, Seizure-induced changes of mean [Cl−]i in subpopulation of neurons with low (0–10 mm), medium (10–20 mm), and high (20–30 mm) range of control resting chloride. Bumetanide prevented seizure-induced neuronal chloride accumulation. d, Effects of recurrent seizures on intracellular chloride accumulation in control low-Mg2+ ACSF and in the presence of bumetanide. Solid lines represent linear regression [Control; low-Mg2+ ACSF (black): r = 0.97 ± 1.66; p = 0.03; bumetanide (green): r = −0.43 ± 2.6; p = 0.57]. In low-Mg2+ ACSF, [Cl−]i progressively increases as a function of recurrent seizures. Bumetanide prevents progressive chloride accumulation induced by recurrent seizures. e, f, Mean interseizure intervals (e) and seizure power (f) as a function of the number of recurrent seizures. e, Solid lines represent linier regression fit of the mean interseizure intervals (ISIs) in control (black; r = −0.82 ± 1.37; p = 0.02) and in the presence of bumetanide (green: r = −0.55 ± 0.7; p = 0.2). f, Corresponding linear regression fit of normalized mean power in control (black; r = 0.95 ± 0.07; p = 0.0003; n = 8) and in the presence of bumetanide (green; r = 0.08 ± 0.08; p = 0.85; n = 8). Bumetanide prevents a progressive increase of seizure power and frequency.
The [Cl−]i in CA3 pyramidal cells was measured between recurrent seizures as a function of the quantity (n) of preceding seizures (Fig. 2 a–d; Table 2). After one to two seizures, 35% of CA3 neurons accumulated chloride >10 mm above the resting [Cl−]i. After seven to eight seizures, 71% of CA3 neurons accumulated chloride >10 mm above the resting [Cl−]i. These changes were of sufficient magnitude to shift the E Cl above the predicted resting membrane potential, implying a shift in GABAergic signaling from inhibitory to excitatory. Depolarized values of E Cl in a subpopulation of CA3 neurons persisted for a long time (at least 1–2 h) after cessation of recurrent seizures by perfusion of control ACSF (data not shown) and may contribute to the generation of subsequent spontaneous seizures (Khalilov et al., 2003; Nardou et al., 2009). Application of low-Mg2+ ACSF in the presence of the sodium channel blocker TTX (1 μm) did not alter intracellular chloride concentration (n = 3; data not shown), suggesting that changes in [Cl−]i were caused by seizure activity. Thus, recurrent seizures caused a progressive intracellular chloride accumulation as a function of the quantity of preceding seizures.
Table 2.
[Cl−]iaccumulation during low-Mg2+ induced recurrent seizures
| Seizures | Control |
+ Bumetanide |
||||||
|---|---|---|---|---|---|---|---|---|
| Δ[Cl−]i(mm) | n (cells) | IHF | p | Δ[Cl−]i(mm) | n (cells) | IHF | p | |
| 1–2 | 8.1 ± 1.2 | 187 | 4 | <0.05 | 6.8 ± 0.8 | 185 | 4 | <0.05 |
| 3–4 | 11.4 ± 1.2 | 187 | 4 | <0.05 | 10.4 ± 1 | 185 | 4 | <0.05 |
| 5–6 | 17.2 ± 2.7 | 187 | 4 | <0.05 | 7.5 ± 0.75 | 185 | 4 | <0.05 |
| 7–8 | 28 ± 1.8 | 187 | 4 | <0.05 | 5.2 ± 0.85 | 185 | 4 | <0.05 |
IHF, Number of hippocampi. p values correspond to one-way ANOVA, followed by Tukey's means comparison.
To test whether the ictal increase in [Cl−]i altered the effect of GABAA-R activation, we tested whether the action of the GABAA-R agonist isoguvacine on neuronal activity (Fig. 1 c–e) is altered by seizure activity. We tested whether isoguvacine increased or decreased multiunit activity, which allowed us to separate the effects of alterations in E GABA (Dzhala and Staley, 2003; Ben-Ari et al., 2007) from changes in GABA release (Hirsch et al., 1999) and postsynaptic GABAA-R conductance (Naylor et al., 2005; Goodkin et al., 2008). If GABA-mediated excitation progressively increases during the time course of spontaneous epileptiform activity, then the GABAA receptor agonist should increase the neuronal firing rate and the frequency of epileptiform discharges proportionally to the number and duration of preceding seizures. Conversely, if GABAA-R function is inhibitory but limited by postsynaptic reductions in GABA conductance (Naylor et al., 2005; Goodkin et al., 2008), we would see a reduction but not a reversal in the inhibitory effects of exogenous GABAA-R agonists. Finally if GABA effects were limited by presynaptic reduction in GABA release (Hirsch et al., 1999), we would see no net change in the effects of exogenous GABAA-R agonists.
Interictal and ictal-like epileptiform discharges were induced by persistent bath application of low-Mg2+ ACSF in intact hippocampi prepared from neonatal (P5–P7) CLM-1 mice. Figure 3 illustrates the typical effects of isoguvacine (10 μm for 1 min) on MUA and epileptiform discharges. Application of isoguvacine after the first seizure, shortly after recovery from post-ictal depression, invariably reduced MUA frequency (n = 6). After two to three seizures, isoguvacine transiently increased the frequency of MUA, in association with a transient increase of the frequency of interictal epileptiform discharges (IEDs). The transient excitatory action of isoguvacine was followed by a suppression of interictal and multiple-unit activity (Fig. 3 a–c), consistent with activity-dependent reduction in [Cl−]i (Brumback and Staley, 2008). In three of six experiments, application of isoguvacine failed to block interictal activity. After four to six recurrent seizures, isoguvacine transiently increased the frequency of MUA and IEDs and often induced ictal-like epileptiform discharges (six of nine applications of isoguvacine). However, this does not exclude a coincidence of the effect of isoguvacine with recurrent seizure activity (data not shown).
Figure 3.

Seizure-dependent changes in the effects of the GABAA agonist isoguvacine (ISO) on MUA and epileptiform discharges. a, b, Extracellular field potential recordings in the CA3 pyramidal cell layer in the intact hippocampus in vitro of the neonatal (P5) CLM-1 mouse. Recurrent seizures were induced by low-Mg2+ ACSF. Application of isoguvacine (ISO; 10 μm for 1 min) after n = 1 seizure decreased neuronal firing. Similar applications of isoguvacine after n = 2 and n = 3 seizures transiently increased neuronal firing rate in association with a transient increase of the frequency of IEDs. c, Corresponding normalized frequency of MUA. Excitatory action of isoguvacine increased as a function of recurrent seizures. Red lines represent adjacent averaging of 20 data points. Vertical dashed lines on b and c delineate isoguvacine application.
Thus, neonatal seizures alter polarity of the GABAA-R-mediated signaling and invert the net effect of GABAA-R activation from inhibition to transient excitation. Positive shifts in E Cl and corresponding changes in GABA action may contribute to facilitation of recurrent seizures and resistance of recurrent seizures to GABAergic anticonvulsants.
Efficacy of phenobarbital depends on the quantity of seizure activity
The ictal shifts in [Cl−]i and E GABA can initiate a positive feedback cycle in neonatal seizures (Staley and Smith, 2001), and a depolarized value of E GABA may contribute to the reduced efficacy of GABA-enhancing anticonvulsants (Dzhala et al., 2005, 2008). We therefore tested the effects of phenobarbital on recurrent seizure activity as a function of the number of preceding seizures. Extracellular field potential recordings were performed in the CA3 pyramidal cell layer in the neonatal rat (P3–P6) whole-hippocampal preparations in vitro. Bath application of low-Mg2+ ACSF induced recurrent ictal-like (seizure) epileptiform activity (Fig. 4 a,d) (for details, see Dzhala et al., 2008).
Figure 4.

Seizure-dependent changes in the effects of phenobarbital on neonatal seizures. a, Low-Mg2+ ACSF-induced recurrent seizures in the intact hippocampus in vitro. b, Application of phenobarbital (100 μm) in low-Mg2+ ACSF after one ictal-like event (n = 1) abolished seizures in the intact neonatal hippocampus. c, Application of phenobarbital (100 μm) after five ictal-like events (n = 5) failed to abolish recurrent ictal-like activity in low-Mg2+ ACSF. a–c, Extracellular field potential recordings were performed in the CA3 pyramidal cell layer in the intact hippocampus in vitro of neonatal (P5–P6) rats. d, Frequency of recurrent seizures as a function of preceding seizures (n) in control low-Mg2+ ACSF and after application of phenobarbital. Phenobarbital applied after n = 1 and n = 3 seizures significantly reduced the mean frequency of recurrent seizures (*p < 0.05). Phenobarbital applied after more than n = 5 seizures failed to reduce the frequency of recurrent seizures. e, Fraction of hippocampi in low-Mg2+ ACSF in which seizures were abolished by 100 μm phenobarbital, plotted as a function of the number of seizures before phenobarbital application. Efficacy of phenobarbital in neonatal seizures decreased as a function of quantity of previous seizure activity (n).
The anticonvulsant efficacy of phenobarbital, measured in terms of reduction in the frequency and power of recurrent seizures, was evaluated as a function of the quantity (n) of preceding seizures (Fig. 4 b–e). A high concentration of phenobarbital (100 μm) was bath applied for 60 min. The mean frequency of seizures in the presence of phenobarbital after one preceding seizure (n = 8) was significantly lower than in control low-Mg2+ ACSF (Fig. 4 d). After seven preceding seizures, the mean frequency of recurrent seizures in the presence of phenobarbital was 3.3 ± 0.6 seizures per hour (n = 7), not significantly different from control (p > 0.05) (Fig. 4 d). Application of phenobarbital after one or two seizures abolished recurrent seizures in four of eight experiments (50%) (Fig. 4 e). After three to five preceding seizures, phenobarbital abolished recurrent seizures in two of seven experiments (28.6%), and after six to eight seizures, in 1 of 11 experiments (9%). Phenobarbital applied after one seizure depressed the power of extracellular field potential activity by 81.8 ± 8.4% (n = 8, p = 0.0036) of preceding control power, but phenobarbital applied after seven preceding seizures decreased power by only 45 ± 15.3% (n = 7, p = 0.027) of preceding power.
Thus, neonatal seizures progressively reduce the anticonvulsant efficacy of phenobarbital. The anticonvulsant efficacy of phenobarbital was inversely proportional to the quantity of preceding seizures (Fig. 4).
NKCC1 cotransporter contributes to seizure-induced chloride accumulation and the facilitation of recurrent seizures
Immature neurons express high levels of the NKCC1 (Delpire, 2000; Wang et al., 2002; Dzhala et al., 2005) electroneutral cotransporter that imports Na+, K+, and Cl−. We therefore assessed the contribution of NKCC1 cotransporter to ictal [Cl−]i accumulation by perfusing the NKCC1 antagonist bumetanide (10 μm) after the first or second seizure induced by continuous superfusion of low-Mg2+ ACSF in the intact hippocampus in vitro of the neonatal (P5–P6) CLM-1 mice (Fig. 5; Table 2). Figure 5 d illustrates the mean effects of recurrent seizures on intracellular chloride accumulation in control low-Mg2+ + ACSF (mean ± SE; 187 cells from n = 4 intact hippocampi) and in the presence of bumetanide (185 cells from n = 4 hippocampi). Notably, bumetanide not only reduced the mean ictal increase in [Cl−]i (Fig. 5 b–d; Table 2) but also reduced the fraction of cells that accumulated chloride >10 mm above baseline chloride level in control low-Mg2+ ACSF. In the presence of bumetanide, the fraction of cells that accumulated chloride after seven to eight recurrent seizures >10 mm above the baseline resting chloride level was 31% (vs 71% in control).
Additionally, bumetanide prevented the progressive increase in the power and frequency of recurrent seizures induced by low-Mg2+ ACSF (Fig. 5 e,f) (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material). Over a 1 h period of simultaneous low-Mg2+ ACSF and bumetanide superfusion, the mean interseizure interval was not significantly different (495 ± 59 vs 417.4 ± 33.3 s; n = 8, p = 0.36) (Fig. 5 e). The mean power of ictal-like events was also not significantly different (105 ± 22% of control, p = 0.8) (Fig. 5 f).
We tested also whether NKCC1 block prevented excitatory shift in the action of GABA induced by recurrent seizures (Fig. 3). Figure 6 illustrates the typical effects of isoguvacine (10 μm for 1 min) on MUA and epileptiform discharges in the presence of bumetanide. Application of isoguvacine after the first seizure (n = 1), shortly after recovery from post-ictal depression, transiently reduced MUA frequency by 87 ± 4% (eight recordings in n = 4 hippocampi). Inhibitory action of isoguvacine persisted also after two to three as well as after four to five recurrent seizures (Fig. 6). After two to three seizures, isoguvacine application did not induce epileptiform discharges and transiently reduced the frequency of MUA by 83.5 ± 4% (eight recordings in n = 4 hippocampi). After four to six recurrent seizures, isoguvacine transiently reduced the frequency of MUA by 74.6 ± 6.2% (eight recordings in n = 4 hippocampi). In one of four experiments, recurrent seizures (n = 6) were followed by persistent interictal-like epileptiform discharges. Application of isoguvacine transiently abolished these epileptiform discharges (data not shown).
Figure 6.

The role of NKCC1 in seizure-induced changes in the action of GABA. a, b, Extracellular field potential recordings in the CA3 pyramidal cell layer in the intact hippocampus in vitro of the neonatal (P5) mouse. Recurrent seizures were induced by low-Mg2+ ACSF in the presence of NKCC1 blocker bumetanide (10 μm). Applications of isoguvacine (ISO; 10 μm for 1 min) after seizure and recovery from post-ictal depression transiently decreased neuronal firing rate. c, Corresponding frequency of MUA. Vertical dashed lines delineate isoguvacine application. Red lines represent adjacent averaging of 20 data points. Bumetanide prevented seizure-induced changes in the effects of the GABAA-R agonist isoguvacine on MUA and epileptiform discharges (Fig. 3).
Thus, the diuretic bumetanide prevented or reduced seizure-induced progressive [Cl−]i accumulation in large subpopulations of neurons (Fig. 5 c; Table 2), prevented changes in the action of GABA (Fig. 6), and reduced seizure-dependent facilitation of recurrent seizures (Fig. 5 e,f) (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material).
Potential mechanism of increased NKCC1 activity during recurrent seizures
NKCC1 is an electroneutral bidirectional transporter that can perform net Na+, K+, and 2Cl− ion influx or efflux, depending on the concentration gradients of the transported ions (Payne, 1997; Gamba, 2005; Brumback and Staley, 2008). The activation of NKCC1 requires phosphorylation of Thr-212 and Thr-217 in the cytoplasmic N terminus (Darman and Forbush, 2002; Flemmer et al., 2002; Payne et al., 2003). We monitored the phosphorylation of NKCC1 at these sites with anti-R5, an antibody that specifically recognizes phosphorylation of Thr-212 and Thr-217 in vitro and in vivo (Darman and Forbush, 2002; Flemmer et al., 2002; Gimenez and Forbush, 2003). Western blotting detected phospho-NKCC1 in both control and seizure conditions (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). These findings indicate that NKCC1 is present, phosphorylated, and presumably active in hippocampi from neonatal mice with recurrent seizures, but they do not provide evidence that increased seizure activity is associated with increased phosphorylation of NKCC1 at its known regulatory sites.
Recurrent seizures in neonatal and adult cortical structures have long been known to cause acute, activity-dependent alterations in neuronal ion concentrations (Moody et al., 1974; Heinemann et al., 1977; Khalilov et al., 1999b; Xiong and Stringer, 2000). In the intact neonatal hippocampal formation, recurrent ictal-like epileptiform discharges increase extracellular concentration of K+ ([K+]o) to 12.5 mm (Khalilov et al., 1999b). Ictal accumulation of extracellular potassium will increase the [Cl−]i at which NKCC1 transport reaches equilibrium (Brumback and Staley, 2008). We therefore assessed the contribution of NKCC1-mediated cotransport to intracellular chloride accumulation as a function of extracellular concentration of K+ (Fig. 7 a–e). In the presence of the sodium channel blocker TTX (1 μm), increasing [K+]o from 3.5 (control) to 8.5 mm resulted in a corresponding increase in [Cl−]i from 19.9 ± 0.8 to 26.9 ± 1.2 mm (p < 0.05 vs baseline [Cl−]i, ANOVA followed by Tukey's means comparison test; n = 138 cells in 4 hippocampi) (Fig. 7 e). Increasing [K+]o to 13.5 mm increased [Cl−]i to 45.6 ± 2 mm (p < 0.05). In the presence of bumetanide (10 μm), increasing [K+]o from 3.5 to 8.5 mm resulted in a corresponding increase in [Cl−]i from 17.4 ± 0.9 to 18.4 ± 1 mm; p = 0.43, n = 132 cells in 4 hippocampi (Fig. 7 e). Increasing [K+]o to 13.5 mm increased [Cl−]i to 21.8 ± 1 mm (p = 0.02). However, this increase in the presence of bumetanide was 23.8 mm lower than in control conditions (21.8 ± 1 vs 45.6 ± 2 mm; p < 0.05) (Fig. 7 e). Thus, seizure-induced [K+]o elevation is sufficient to increase [Cl−]i accumulation via NKCC1 cotransport, resulting in alterations of E GABA during recurrent seizures (Figs. 2, 3, 5, 6).
Figure 7.

Concentration gradient of potassium regulates NKCC1 activity. a–e, Changes in intracellular chloride distribution as a function of [K+]o in the presence of the sodium channel blocker TTX (1 μm). a, c, Examples of two-photon confocal imaging of YFP (left) of CA3 neurons yielded pseudocolor map of [Cl−]i at different [K+]o (3.5, 8.5, and 13.5 mm) in control (a) and in the presence of bumetanide (c). b, d, Corresponding distributions of [Cl−]i (bin size, 5 mm) in control (b; n = 30 identified neurons) and in the presence of bumetanide (d; n = 40 identified neurons). Data were fitted with a Gaussian function. e, Mean [Cl−]i as a function of [K+]o in control and in the presence of bumetanide.
NMDA receptor, NKCC1 activity, and inverted action of GABA
The NKCC1 phosphorylation data and [K+]o data suggest that at least some aspects of ictal changes in [Cl−]i may be independent of calcium-dependent signal transduction mechanisms. Conversely, epileptogenic transformation of the intact hippocampal neuronal network required voltage-dependent activation of NMDA receptors (NMDA-Rs) and long-term alterations in GABAergic synapses, which became excitatory because of a positive shift in the chloride reversal potential (Khalilov et al., 2003). We therefore determined the contribution of NMDA-R to seizure-induced changes in the net effect of GABAA receptor activation and corresponding alterations of recurrent seizures (Figs. 2, 3, 5). Because seizures are NMDA-R dependent in the low-Mg2+ model, we used the kainic acid model of epileptiform activities in the intact neonatal hippocampus in vitro of the neonatal (P5–P6) mice (Prince et al., 1973; Fisher et al., 1976; Khalilov et al., 1999b).
In control ACSF, transient application of kainic acid (1 μm for 4–5 min) progressively increased the frequency of MUA and induced interictal and ictal-like epileptiform discharges, followed by a post-ictal depression (eight recordings in n = 4 hippocampi) (Fig. 8 a). Repeated applications of kainic acid (five applications at 15 min intervals) induced inter-ictal and ictal-like epileptiform discharges that progressively increased in amplitude and power (Fig. 8 d). After five applications of kainic acid, spontaneous recurrent epileptiform discharges were generated in three of four intact hippocampi. Brief applications of isoguvacine (10 μm for 1 min) transiently increased the frequency of spontaneous epileptiform discharges by 225 ± 154% (p = 0.04; six applications in n = 3 hippocampi) (Fig. 8 a,e), suggesting seizure-induced changes in the action of GABA and contribution of the excitatory action of GABA to generation of spontaneous recurrent epileptiform discharges.
Figure 8.

Seizure-induced changes in the action of GABA are NMDA receptor independent. a, Repetitive applications of kainic acid (KA; 1 μm for 5 min; open bars) induced ictal-like epileptiform discharges (seizures) followed by spontaneous IEDs. Brief applications of isoguvacine (ISO; 10 μm for 1 min; filled bars) transiently increased the frequency of IEDs. b, Perfusion of the noncompetitive NMDA receptor antagonist MK801 (20 μm) depressed KA-induced seizures. Applications of isoguvacine increased the frequency of spontaneous IEDs. c, Simultaneous perfusion of MK801 and bumetanide (10 μm) strongly depressed kainic acid-induced seizures and prevented excitatory shift in the action of GABA. Applications of isoguvacine reduced the frequency of MUA and/or IEDs. a–c, Extracellular field potential recordings in the CA3 pyramidal cell layer in the intact hippocampus of neonatal (P5–P6) mice. d, Normalized power of KA-induced seizures (mean ± SE) in control (squares), in the presence of MK801 (circles), and both MK801 and bumetanide (Bum; triangles). e, Effects of isoguvacine on the frequency of spontaneous IEDs.
We determined whether the noncompetitive NMDA-R antagonist MK801 [(+)- 5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] prevented seizure-induced changes in the net effect of GABAA-R activation from inhibition to excitation assayed by the frequency of spontaneous action potentials and/or interictal epileptiform discharges (Fig. 8 b,e). In the presence of MK801 (20 μm), transient applications of kainic acid induced interictal and ictal-like epileptiform discharges that progressively decreased in amplitude and power (six recordings in n = 3 hippocampi) (Fig. 8 d). Persistent perfusion of MK801 did not prevent generation of spontaneous epileptiform discharges induced by repeated applications of kainic acid. Brief applications of isoguvacine (10 μm for 1 min) in the presence of MK801 transiently increased the frequency of spontaneous epileptiform discharges by 368 ± 129% (p = 0.015; six applications in n = 3 hippocampi) (Fig. 8 b,e) before depressing them. These effects are similar to what was seen in control conditions (Fig. 2), suggesting that NMDA-Rs did not contribute to seizure-induced chloride accumulation and the corresponding changes in the action of GABA.
As in control conditions, the NKCC1 antagonist bumetanide (10 μm) prevented the seizure-induced excitatory shift in the action of GABA (n = 3) (Fig. 8 c,e) in the presence of MK801 (20 μm). In the presence of MK801 and bumetanide, repeated applications of kainic acid (five applications for 5 min each; 15 min intervals) induced interictal and ictal-like epileptiform discharges that progressively decreased in power and amplitude (Fig. 8 c,d). Spontaneous epileptiform discharges after five applications of kainic acid were recorded in one of three experiments (33.3%). Brief applications of isoguvacine (10 μm for 1 min) abolished spontaneous epileptiform discharges (two recordings in n = 1 hippocampus) and/or reduced the frequency of MUA (six recordings in n = 3 hippocampi). Thus, seizure-induced NKCC1-dependent chloride accumulation and the excitatory shift in the action of GABA was NMDA-receptor independent.
Long-term pro-convulsive effect of GABAA receptor antagonist
The pro-convulsive effect of complete blockade of GABAA-R indicates that, at P4–P6, GABAA receptors subserve a significant inhibitory function (Khalilov et al., 1999a; Wells et al., 2000). In those studies, the GABAA-R antagonists bicuculline and picrotoxin induced glutamate-receptor-mediated interictal and ictal-like activities in the intact hippocampus. We determined the long-term effect of GABAA-R antagonist SR 95531 (gabazine) on spontaneous neuronal activity. We performed non-invasive extracellular field potential recordings of synchronous population activity and MUA in the CA3 pyramidal cell layer in the intact hippocampal preparations from P4–P6 rats (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In control ACSF, bath application of SR 95531 (10 μm for 3–4 min) progressively increased the frequency of MUA by 44 ± 13.2% from 13.5 ± 1.7 to 19.3 ± 2.8 spikes/s (p = 0.004; n = 14 recordings in 8 hippocampi) (data not shown) and rapidly, within 2–3 min, induced interictal and ictal-like epileptiform discharges, followed by a post-ictal depression with reduced neuronal activity (Fig. 9 a). After removal of the drug and shortly after post-ictal depression, recurrent interictal and tonic–clonic epileptiform discharges occurred in all intact hippocampi for an extended period of 30–60 min (mean, 42.8 ± 2.8 min for n = 16 hippocampi at P4–P5). Spontaneous epileptiform discharges, in some experiments, persisted for several hours in drug-free ACSF (data not shown), suggesting that their persistence was not related to residual antagonist but rather attributable to activity-dependent long-term changes in synaptic plasticity (Schneiderman et al., 1994; Bains et al., 1999; Abegg et al., 2004; Arnold et al., 2005). We considered the possibility that spontaneous epileptiform discharges induced by GABAA-R blockade were as effective as low-Mg2+-induced activity in inducing the shift in neuronal [Cl−]i and the net action of GABAA-R activation.
Figure 9.
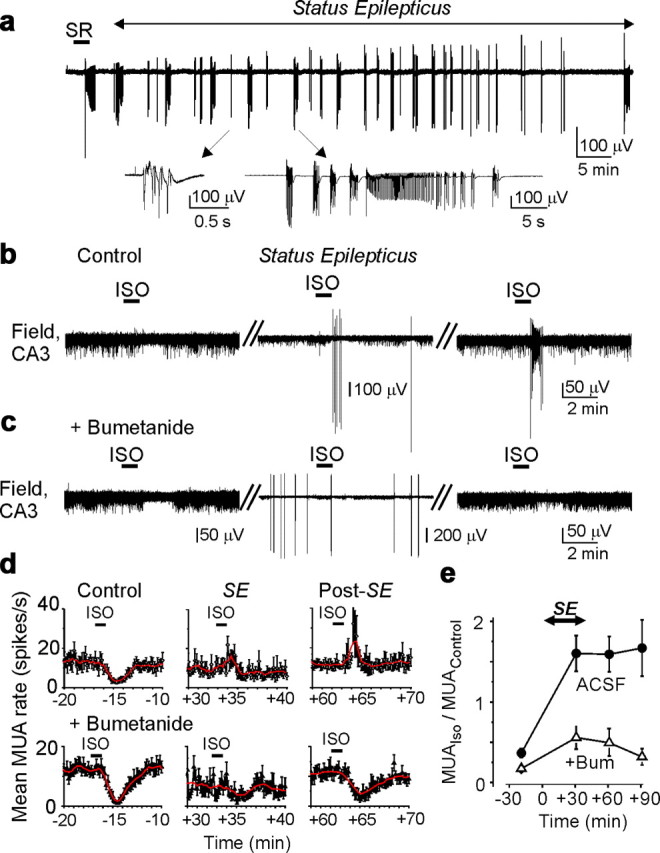
Recurrent seizures invert the effects of GABAA receptors via NKCC1-dependent mechanism. a, Spontaneous seizures (Status Epilepticus) were induced by brief bath application of the GABAA receptor antagonist SR 95531 (SR; 10 μm). In the bottom, spontaneous epileptiform discharges are shown on an expanded timescale. b, c, Effects of the GABAA-R agonist isoguvacine (ISO) on MUA and spontaneous epileptiform activity. In control ACSF, application isoguvacine (10 μm) decreased the frequency of MUA. At 30–40 min after onset of spontaneous seizures, isoguvacine increased the frequency of MUA and interictal epileptiform discharges. Excitatory effects of isoguvacine on population activity persisted at least 1–2 h after termination of seizures. c, Bumetanide (10 μm) prevented the excitatory action of isoguvacine on the MUA and interictal epileptiform discharges. a–c, Extracellular recordings of MUA and population activity were performed in the CA3 pyramidal cell layer of intact neonatal (P4–P5) hippocampal preparations in vitro. d, e, Mean frequency of MUA in control ACSF and in the presence of bumetanide (BUM; n = 6) before, during, and after spontaneous seizures. d, Time course of isoguvacine action on MUA frequency. Bumetanide (10 μm) prevented the seizure-induced reversal of the effects of GABAA-R activation.
Recurrent seizures cause progressive NKCC1-dependent change in the action of GABA
In control conditions, brief bath application of isoguvacine (10 μm for 1 min) transiently reduced the frequency of MUA by 63.4 ± 5.7% (p = 0.0009; n = 6) (Fig. 9 b,d,e). Spontaneous interictal and ictal-like epileptiform discharges were then induced by transient application of the GABAA-R antagonist SR 95531 (10 μm for 3 min) (Fig. 9 a). At 30 min after onset of spontaneous epileptiform discharges, bath application of isoguvacine transiently increased the frequency of multiple-unit activity frequency by 60.3 ± 22.1% (p = 0.007; n = 6), in association with a transient increase of the frequency of IEDs (Fig. 9 b,d). Subsequently, MUA frequency decreased, consistent with activity-dependent reduction in [Cl−]i (Brumback and Staley, 2008). At 60–70 min after onset of spontaneous seizures, isoguvacine transiently increased the frequency of MUA by 59.3 ± 22% (p = 0.015), in association with transiently induced IEDs, followed by a decrease as seen at 30 min. The biphasic action of isoguvacine persisted at least for 1–2 h after termination of spontaneous epileptiform discharges.
In the presence of bumetanide, application of isoguvacine (10 μm for 1 min) transiently reduced the frequency of MUA by 82.7 ± 4.2% (p = 0.0004; n = 6) (Fig. 9 c–e). At 30 min after onset of spontaneous epileptiform discharges induced by SR 95531, isoguvacine reduced MUA frequency by 44.3 ± 14.4% (p = 0.015; n = 6) and did not induce epileptiform discharges or increase the frequency of epileptiform discharges (Fig. 9 c,d). At 60–70 min after onset of spontaneous epileptiform discharges, isoguvacine invariably diminished MUA frequency by 50.7 ± 16.3% (p = 0.002; n = 6). Thus, the diuretic bumetanide prevents or reduces seizure-induced changes in the action of GABA during spontaneous seizures and after termination of seizures. In summary, although these data do not exclude presynaptic or postsynaptic alterations in GABA conductance, they demonstrate a clear reversal of the effect of GABAA-R activation that is NKCC1 dependent, and independent of the mechanism of seizure induction. These data support the idea that NKCC1 activity underlies the post-ictal increase in [Cl−]i (Figs. 2, 5) and the excitatory effects of GABA.
NKCC1-dependent changes contribute to the time course of spontaneous seizures
To determine whether the seizure-induced, NKCC1-dependent changes in GABAA-R function contribute to the increased probability of subsequent seizures (Dzhala and Staley, 2003; Khazipov et al., 2004; Dzhala et al., 2005, 2008), we assessed the effects of NKCC1 cotransporter blocker bumetanide on the time course of spontaneous seizures assayed by the quantity, duration, and power of spontaneous epileptiform activity in the P3–P5 intact hippocampal preparations (supplemental Fig. 5a–e, available at www.jneurosci.org as supplemental material). In control ACSF, brief application of SR 95531 (10 μm) induced spontaneous seizures in 16 of 18 preparations. The duration of this status epilepticus was 41.7 ± 2.5 min (n = 16). In the presence of bumetanide, similarly brief application of SR 95531 induced a shorter period of spontaneous interictal and ictal-like discharges (31.3 ± 2.2 min; p = 0.004) (supplemental Fig. 5c, available at www.jneurosci.org as supplemental material) in 10 of 15 preparations. The number of spontaneous ictal-like episodes induced by GABAA-R block was not significantly different: 2.1 ± 0.3 seizures in SR 95531 and 1.8 ± 0.3 seizures in the presence of SR 95531 + bumetanide (p = 0.7) (supplemental Fig. 5c, available at www.jneurosci.org as supplemental material). We also assessed the power of SR 95531-induced epileptiform activity in control ACSF and in the presence of bumetanide (supplemental Fig. 5d,e, available at www.jneurosci.org as supplemental material). The effect of bumetanide on EEG power was relatively low shortly after washout of GABAA-R antagonists (∼10%) but gradually increased to ∼45% by the end of the seizure activity induced by transient exposure to SR 95531. Although these data support a progressive, seizure-induced increase in [Cl−]i and excitatory shift in GABAA-R function, the alternative possibility was that SR 95531 was incompletely washed out early, in which case the impact of bumetanide on NKCC1 would initially be minimized as a result of reduced GABAA-receptor-mediated chloride conductance. This was unlikely because we had already seen a stable impact of GABAA-R agonist application (Fig. 9 d) at different time points after washout of SR 95531 during and after spontaneous seizures. We conclude that neonatal seizures increase the probability of recurrent neonatal seizures via NKCC1-dependent chloride accumulation and consequent alteration in GABA actions.
Neonatal seizures change the polarity of synaptic GABAA responses
To further characterize the mechanisms of seizure-induced alterations in [Cl−]i, GABA actions, and seizure activity, we assessed the effects of endogenously released GABA on neuronal firing rate and epileptiform activity and as a function of the number of seizures.
The net effect of GABAA-R activation may be inhibitory even when E GABA is positive to resting membrane potential. Large GABAA-R-mediated anion conductance effectively reduces the input membrane resistance and thereby shunts excitatory glutamatergic inputs, resulting in suppression of neuronal activity (Qian and Sejnowski, 1990; Staley and Mody, 1992; Khalilov et al., 1999a). We determined whether the inhibitory operation of GABAA-R in the intact hippocampal network was mediated by synaptic inhibitory mechanism and whether neonatal seizures alter this inhibition. Pharmacological protocols were used to determine the contributions of intrinsic and synaptic activities to the generation of neuronal discharges and estimate the degree to which endogenous synaptic activity altered spontaneous neuronal firing rate in population of neurons (Cohen and Miles, 2000; Dzhala and Staley, 2003). In control conditions, bath application of the AMPA and NMDA receptor antagonists kynurenic acid (2 mm) and the metabotropic GABAB receptor antagonist CGP 55845 (1 μm) significantly reduced neuronal firing rate by 32.5%, from 12.3 ± 1.3 to 8.3 ± 0.8 spikes/s (n = 14; p = 0.0002, paired t test) (Fig. 10 a). Subsequent application of the selective, competitive GABAA-R antagonist SR 95531 (10 μm), in the presence of KYNA and CGP 55845, increased neuronal firing rate in 10 of 14 recordings and did not significantly change neuronal firing rate in the remaining four recordings (Fig. 10 a). The average mean frequency of neuronal discharges under these conditions increased by 21% from 8.3 ± 0.8 to 10.04 ± 0.9 spikes/s (p = 0.009). These findings are consistent with the pro-convulsive effect of GABAA-R antagonists in the intact hippocampal preparation (Khalilov et al., 1999a) and indicate a net inhibitory action of endogenously released GABA in the intact CA3 hippocampal network at this early postnatal age (P3–P5).
Figure 10.
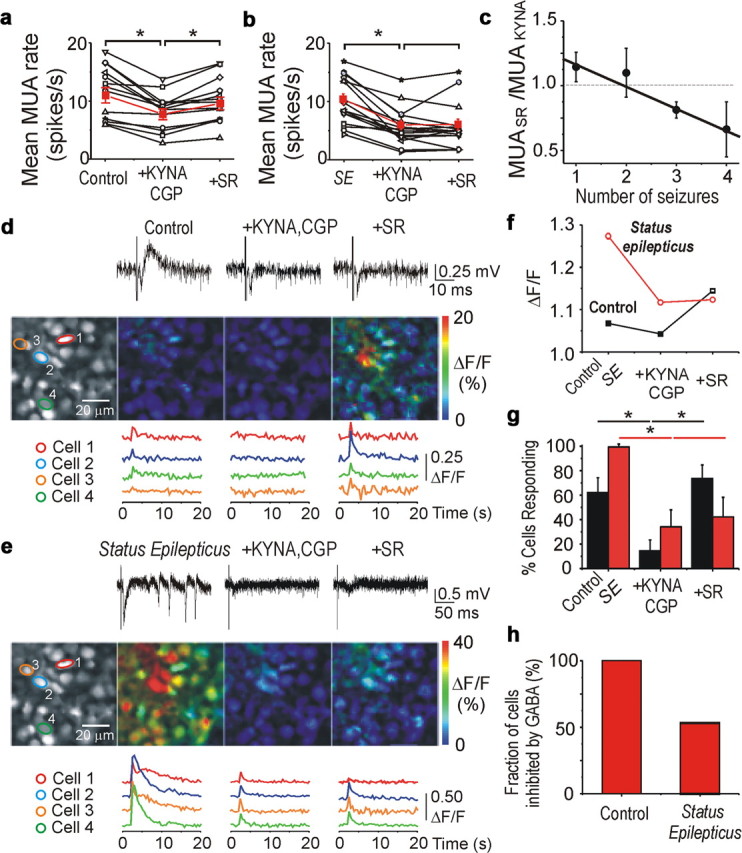
Neonatal seizures alter the effect of synaptic activation of GABAA receptors. a–c, Effects on spontaneous neuronal firing rate of suppressing synaptic excitation and inhibition in control and during seizures. In control (a), KYNA (2 mm) coapplied with CGP 55845 (CGP; 1 μm) reduced the mean frequency of MUA in the CA3 pyramidal cell layer in the intact neonatal hippocampi at P4–P5. Subsequent application of the GABAA-R antagonist SR 95531 (SR), in the presence of KYNA and CGP 55845, increased MUA frequency, demonstrating the net inhibitory effect of GABAA-R when activated by endogenously released GABA. b, Spontaneous seizures were induced by brief (2–3 min) application of SR 95531. After 30 min of spontaneous seizure activity after washout of SR 95531, application of KYNA and CGP 55845 stopped recurrent seizures and reduced MUA frequency. Subsequent application of SR 95531 did not change the mean frequency of MUA. Red plots represent the averaged mean frequency of MUA. SE, Status epilepticus. c, Mean ratio of MUA frequency [SR 95531/(KYNA + CGP 55845)] reversed as a function of the number of seizures. Solid line represents corresponding linear regression fit (r = −0.98; p = 0.02). d, e, Synchronous extracellular field potentials and two-photon fluorescence [Ca2+]i imaging in the CA3 pyramidal cell layer at P5 in the intact hippocampal preparation. Neurons were labeled by the calcium indicator dye OGB. Field EPSPs and intracellular calcium signals were induced by electrical stimulation in the stratum radiatum proximal of the CA3 area. In control (d), bath application of KYNA (2 mm) and CGP 55845 (1 μm) depressed calcium signals (ΔF/F) in the majority of CA3 neurons. Subsequent application of SR 95531 (10 μm), in the presence of KYNA and CGP 55845, increased ΔF/F in the majority of the cells. The increase in Ca2+ in the presence of ionotropic glutamate and GABA receptor antagonists likely represents metabotropic receptor activation and direct electrical stimulation. e, After spontaneous seizures, the effect of SR 95531 on ΔF/F was reversed in ∼50% of neurons. f, g, Mean population response (ΔF/F) and fraction of cells with a stimulus-induced increase in calcium responses during applications of drugs in control and after spontaneous seizures. *p < 0.05. h, After recurrent seizure activity, nearly half of neurons lose their inhibitory response to endogenous GABA or switch their inhibitory response from inhibition to excitation.
To determine whether the action of endogenously released GABA is altered by previous seizure activity, we measured the impact of GABAA-R block on the rate of spontaneous action potentials as a function of the number of preceding seizures in the CA3 pyramidal cell layer of the intact neonatal (P3–P5) hippocampus. Spontaneous seizures were induced by brief application of SR 95531 (10 μm for 3–4 min). At 30–40 min after onset of spontaneous epileptiform discharges, bath application of KYNA (2 mm for 10 min) in combination with CGP 55845 (1 μm) abolished epileptiform discharges and reduced the neuronal firing rate by 50%, from 10.3 ± 2.3 to 5.3 ± 1.8 spikes/s (n = 14; p = 0.002) (Fig. 10 b). Subsequent application of the GABAA-R antagonist SR 95531, in the presence of KYNA and CGP 55845, induced three types of responses on MUA frequency: (1) significantly increased MUA frequency in 3 of 14 total recordings, (2) unchanged MUA frequency in six recordings, and (3) significantly decreased MUA frequency in the remaining five recordings (Fig. 10 b). The absence of an effect of SR 95531 on action potential frequency in the majority of recordings indicates that the net effect of GABA is no longer inhibitory after neonatal seizures. We also assessed the effect of endogenous GABA on neuronal firing rates as a function of the number of spontaneous seizures (Fig. 10 c). We found a linear correlation between the number of spontaneous seizures and the action of GABA on neuronal firing rates. After one to two spontaneous seizures, the GABAA-R antagonist SR 95531, when AMPA and NMDA receptors were blocked, did not significantly change the neuronal firing rate (112 ± 9.9%; p = 0.38). After three to four spontaneous seizures, SR 95531 diminished neuronal firing rate to 76 ± 11% (p = 0.017), indicating that endogenous GABA was excitatory in a large population of neurons. Thus, spontaneous epileptiform discharges progressively reduce the rate of GABAA-R-mediated inhibition. This could be attributable to a number of factors, including long-term potentiation of glutamatergic synapses between pyramidal cells induced by epileptiform activity (Ben-Ari and Gho, 1988; Bains et al., 1999), as evidenced by the increased impact of KYNA on MUA during status epilepticus. However, glutamatergic mechanisms do not explain the alterations in MUA observed in the presence of KYNA. The loss of inhibitory effect of endogenously released GABA could arise from (1) a positive shift in the chloride equilibrium potential in a large population of cells (Khalilov et al., 2003), (2) a presynaptic reduction in GABA release (Hirsch et al., 1999), or (3) a reduction in the postsynaptic GABAA conductance (Naylor et al., 2005). However, other than a long-term shift in E GABA, none of these effects explain the post-ictal reduction in MUA frequency when GABAA-Rs were blocked.
Cell-specific actions of GABA on intracellular Ca2+ influx
The heterogeneity of neuronal [Cl−]i (Fig. 1) and the post-ictal effects of endogenously released GABA in the preceding experiments raise the possibility that subpopulations of neurons may have distinct responses to GABA, either excitatory or inhibitory, and the MUA assay was measuring the network-wide average of those distinct responses. To analyze the response to GABA at the level of individual neurons, the action of GABA was assayed by changes in intracellular calcium (Stosiek et al., 2003). Large populations of CA3 neurons in the intact neonatal (P4–P5) hippocampus were loaded with the calcium indicator dye OGB (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Synchronous postsynaptic field potential responses and intracellular Ca2+ signals were induced by electrical stimulation in the stratum radiatum of CA3 area (Fig. 10 d,e).
Under control conditions, electrical stimulation induced field EPSP and synchronous Ca2+ transients in hundreds of CA3 neurons (Fig. 10 d). On average, excitatory cellular responses (transient increases in [Ca2+]i measured as ΔF/F) were detected in 62.2 ± 12.0% of neurons (383 of 616 cells; n = 3 recordings) with an average ΔF/F of 6.8 ± 0.1% (Fig. 10 f,g). We determined the degree to which synaptic excitation and inhibition contribute to these responses. Bath application of KYNA (2 mm), in combination with CGP 55845 (1 μm) depressed the evoked field EPSPs and intracellular Ca2+ signals. Under these conditions, increases in [Ca2+]i in response to electrical stimulation were detected in only 14.5% of neurons (91 of 624 cells). In those cells that show increased [Ca2+]i, the average peak signal intensity was a ΔF/F of 4.3 ± 0.2%. Subsequent application of the GABAA-R antagonist SR 95531 (10 μm), in the presence of KYNA and CGP 55845, elicited an increase in the response to electrical stimulation measured by intracellular Ca2+ signals. Under these conditions, fluorescence calcium imaging revealed that both the percentage of responding cells (73.5 ± 12.0%) and the response amplitude (ΔF/F = 14.5 ± 0.4%) were significantly higher than under the previous conditions (p < 0.01, ANOVA with post hoc correction) (Fig. 10 f,g). These evoked increases in calcium in the presence of glutamate and GABA antagonists were presumably attributable to direct electrical stimulation of dendrites. After washing out of drugs, transient application of SR 95531 (10 μm for 5 min) induced sustained ictal-like epileptiform activity, followed by recurrent interictal and ictal-like epileptiform discharges. Under these conditions, 30–40 min after onset of spontaneous seizures, electrical stimulation induced epileptiform discharges and corresponding Ca2+ signals in 99.4% of all cells (Fig. 10 e–g) (820 of 825 cells; n = 3 recordings). These responses were, on average, significantly amplified compared with control (ΔF/F = 27.4 ± 0.6%; p < 0.01 ANOVA with post hoc). In line with previous data, bath application of KYNA (2 mm) and CGP 55845 (1 μm) abolished spontaneous and evoked epileptiform discharges and significantly decreased the percentage of responding cells (34.2%) and the corresponding response amplitude (ΔF/F = 11.7 ± 0.5%) (Fig. 10 f,g). Subsequent application of SR 95531 (10 μm), in the presence of KYNA and CGP 55845, failed to increase either cellular response probability (42.2%, 436 of 1032 cells) or response amplitude (ΔF/F = 12.4 ± 0.5%, 436 cells), indicating altered synaptic operation of the GABAA receptors. This is in contrast to the response elicited in control conditions, before seizure activity, in which block of GABAA-R caused a dramatic rise of cellular response probability and ΔF/F. Our data strongly suggest that neonatal seizures reduce the net inhibitory effect of endogenously released GABA.
The results described above do not differentiate whether GABA effects are homogenously reduced after seizures, as could occur from either a shift in E GABA or a reduction in the postsynaptic GABA conductance response, or whether GABA became excitatory in only a subpopulation of neurons. We therefore coregistered our images from the control and after-seizure activity datasets and automatically detected those cells that were clearly identifiable under both conditions. We then developed a simple measure to determine whether any of those cells switched from being inhibited by GABA in control conditions to being non-inhibited or excited by GABA post-ictally (Fig. 10 h). We determined in which cells the stimulus-induced calcium transient was increased after GABAA-R blockade under control conditions and then, after spontaneous seizures, measured the fraction of those cells that still had an increase in stimulus-induced calcium transient after GABAA-R blockade. We found that cellular responses were not uniform: not all cells had a loss of GABA-mediated inhibition after spontaneous seizures. Blockade of GABAA-R under these conditions still enhanced the Ca2+ signal in 224 of 423 cells (p < 0.001, Mann–Whitney test), indicating that these cells were still inhibited by endogenously released GABA. The remaining 199 cells (∼43%) switched post-ictally from being inhibited by endogenously released GABA to being excited or not inhibited by GABA [χ2 test, 3 (inhibitory, neutral, excitatory) × 2 (before seizures, after seizures), p < 0.001]. Cells in this category demonstrated after block of GABAA-Rs either no stimulus-induced change in the calcium transients (157 cells, ∼34%; p > 0.05, Mann–Whitney test) or significantly reduced calcium transients (42 cells, ∼9%; p < 0.05, Mann–Whitney test). Thus, after spontaneous seizure activity, nearly half of all neurons undergo a transformation that causes them to lose their inhibitory response or switch their inhibitory response to endogenous GABA from inhibition to excitation.
Discussion
Our results demonstrate that NKCC1 activity strongly contributes to the intracellular chloride accumulation induced by recurrent seizures (Figs. 2, 5; Table 2). Activity-dependent shifts in [K+]o (Moody et al., 1974; Heinemann et al., 1977), which alter the [Cl−]i at which NKCC1 comes to equilibrium, are sufficient to cause these short-term changes in [Cl−]i (Fig. 7), although other mechanisms may also be important. As a consequence, there is a sustained switch in the net effect of GABAA-R activation from inhibition to excitation (Figs. 3, 6, 9). Shifts in E Cl and GABA action contribute to facilitation of recurrent seizures and additional chloride accumulation. Under these conditions, recurrent seizures respond less well to GABAergic anticonvulsants and better to the NKCC1 antagonist bumetanide, particularly when administered in combination with a positive allosteric GABA receptor modulator such as phenobarbital (Dzhala et al., 2008). Our data provide mechanistic insights into GABAergic excitation during neonatal seizures (Derchansky et al., 2008).
We demonstrated previously the anticonvulsant effects of bumetanide in a model of neonatal seizures in vitro (Dzhala et al., 2005, 2008). The reduced efficacy of bumetanide in other in vitro models of neonatal seizures (Kilb et al., 2007) likely reflects differences in the number of seizures before administration of bumetanide because, as predicted by the ictal, NKCC1-mediated increase in [Cl−]i (Fig. 5) and as illustrated in supplemental Figure 3 (available at www.jneurosci.org as supplemental material), the anticonvulsant efficacy of bumetanide increases as a function of the number of seizures experienced by the preparation. Experimental data regarding the efficacy of GABA-enhancing anticonvulsants in neonatal seizures are controversial (Table 1). Some of this controversy may arise as a consequence of the heterogeneous values of E GABA in the developing cortical networks (Figs. 1, 2, 5, 7), such that the net actions of GABAergic agents in a particular slice preparation might be similarly varied. In addition, the ictal time course of changes in neuronal Cl− are important experimental variables. In vivo models of neonatal seizures, primarily induced by GABA antagonists, demonstrate efficacy of GABAergic anticonvulsants administered before or coincident with convulsants (Velísek et al., 1995; Haugvicova et al., 1999; Isaev et al., 2007). As demonstrated in Figures 1 –4, administration of phenobarbital before or just after the first seizure, when [Cl−]i is relatively low and the net effect of GABA is still inhibitory, may prevent or reduce the positive feedback cycle of seizure-dependent disinhibition and subsequent disinhibition-induced seizures that limit the subsequent efficacy of GABAergic anticonvulsants.
Several mechanisms may be involved in activation of NKCC1 during recurrent seizure activity. NKCC1 activity is thought to be increased by phosphorylation (Darman and Forbush, 2002) and decreased by protein phosphatase 1-mediated dephosphorylation (Gagnon et al., 2007). NKCC1 phosphorylation corresponded to an increase in intracellular chloride in an in vitro model of ischemia (Pond et al., 2006). Our data (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) provide no indication that NKCC1 phosphorylation is increased by seizure activity but do not address total NKCC1 protein levels nor membrane trafficking. However, under conditions studied so far, activity-dependent increases in NKCC1-mediated Cl− accumulation do not involve changes in NKCC1 maximum velocity or affinity for chloride but rather changes in the transmembrane ion gradients of the cotransported cations (Brumback and Staley, 2008), i.e., they do not involve alteration in total NKCC1 protein, NKCC1 membrane trafficking, or changes in NKCC1 phosphorylation (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
[K+]o increases during seizures (Prince et al., 1973; Fisher et al., 1976; Khalilov et al., 1999b), and elevated [K+]o markedly alters the driving force for transmembrane Cl− cotransport (Thompson and Gähwiler, 1989; Staley and Proctor, 1999; Blaesse et al., 2009). This shift in free energy would be expected to lead to a corresponding increase in the [Cl−]i at which NKCC1 reached equilibrium (Brumback and Staley, 2008) and would thus drive Cl− influx via NKCC1 (Sun and Murali, 1998). Our experiments with increased [K+]o strongly support this mechanism (Fig. 7) but do not exclude the possibility of additional mechanisms of modulation of NKCC1 and chloride accumulation.
Finally, paroxysmal depolarizing shifts in the membrane potential during seizures will induce Cl− accumulation through open GABAA-Rs; this phenomenon has been frequently exploited experimentally to load neurons with Cl− (Staley and Proctor, 1999; Brumback and Staley, 2008). Such a mechanism may contribute to the small NKCC1-independent ictal increase in [Cl−]i (Fig. 5 B).
All differential interference contrast patch clampers select neurons to patch based on visual criteria related to cell turgor. Given the strong relationship between the activities of cation-chloride cotransporters and cell volume (Kahle et al., 2008), turgor is likely to be strongly correlated with neuronal chloride concentration. Patchable cells represent a very small subset of all neurons imaged with Clomeleon. Our results using Clomeleon extend the spectrum of actions of GABA during postnatal development (Ben-Ari et al., 2007) by demonstrating the wide range of [Cl−]i and GABA effects in individual neurons in the developing hippocampus. Our data also underscore the importance of the history of activity of the preparation on neuronal [Cl−]i and the consequent effects of GABAA receptor activation. Our findings are consistent with previous observations of shifts in E GABA as a consequence of trains of action potentials (Woodin et al., 2003; Brumback and Staley, 2008), seizures (Khalilov et al., 2003), trauma (van den Pol et al., 1996; Nabekura et al., 2002), and hypoxia (Pond et al., 2006). These long-term, experience-dependent shifts in E GABA may explain the incongruent results that have been obtained in developing preparations (Table 1).
The opposite effects of the GABAA-R activation in the intact hippocampal preparations versus the hippocampal slices probably arise from acute traumatic neuronal damage during slicing that may cause an increase in intracellular Cl− concentration and consequent inversion of GABA action (van den Pol et al., 1996; Nabekura et al., 2002). Our results support reports of a predominantly inhibitory hyperpolarizing effect of GABAA receptors in the neonatal hippocampus in vivo (Isaev et al., 2007). Additionally, perinatal hippocampal slice studies of the effects of GABAA-R activation on [Ca2+]i in CA3 neurons estimate that 50% of P5 neurons are not excited by GABA (Tyzio et al., 2006), in agreement with Figure 10.
The biphasic effects of the GABAA-R agonist isoguvacine on network action potential frequency (Figs. 3, 8, 9) likely represents activity-dependent shift in E GABA. When NKCC1 is active and E GABA is positive to membrane potential, low initial concentrations of isoguvacine activate GABAA receptors and depolarize neurons. Subsequently, GABA channels are opened so persistently that the high fluxes of chloride overwhelm NKCC1-mediated chloride transport (Dzhala and Staley, 2003; Brumback and Staley, 2008). The clinical correlate of this experimental finding is that very high concentrations of GABAergic anticonvulsants are effective treatments of neonatal seizures (Mizrahi and Kellaway, 1987), presumably via the same mechanism: overwhelming NKCC1-mediated Cl− transport with chloride flux though persistently opened GABAA-R-operated ion channels. The consequent passive transmembrane distribution of Cl−, together with the low membrane resistivity induced by persistent GABAA-R activation, permits effective shunting inhibition. The potential disadvantage of using high doses of GABAergic anticonvulsants to treat neonatal seizures is the level of GABA-mediated suppression of brainstem and spinal circuits that have already undergone the developmental switch from NKCC1-mediated neuronal Cl− import to KCC2-mediated neuronal Cl− export (Stein et al., 2004; Glykys et al., 2009), as well as increased anticonvulsant-induced neuronal apoptosis (Ikonomidou, 2009). Although there are also potentially deleterious effects of NKCC1 inhibition (Ge et al., 2006; Cancedda et al., 2007; Wang et al., 2008), these are evident after much more prolonged inhibition than would be necessary to control most neonatal seizures (Painter et al., 1999).
Recurrent seizures in neonatal and adult cortical structures cause activity-dependent alterations in ion concentrations (Fisher et al., 1976; Cohen et al., 2002; Khalilov et al., 2003; Galanopoulou, 2008; Li et al., 2008). Here we demonstrate that changes in ion transport persist beyond the end of seizure activity to contribute to recurrence of seizures. These findings are of particular interest in understanding the natural history of neonatal seizures. Prospective EEG studies in which clinically silent electrographic seizures were untreated (Connell et al., 1989) support the clinical impression that seizures attributable to brain injury begin hours after the insult and continue for ∼72 h (Volpe, 2001). The NKCC1-mediated delayed increase in neuronal chloride concentration after hypoxic/aglycemic injury (Pond et al., 2006) together with the observation that increases in NKCC1 transcription persist for 72 h after injury in vivo (Dai et al., 2005) support the hypothesis that increases in NKCC1 activity and consequent compromise of GABA-mediated inhibition contribute significantly to the time course of neonatal seizures after hypoxic–ischemic brain injury (Cowan et al., 2003; Tekgul et al., 2006). Induction of NKCC1 by hypoxic injury may explain why rapid EEG diagnosis and treatment of neonatal seizures has not had a bigger impact on the efficacy of phenobarbital therapy (Painter et al., 1999; Bartha et al., 2007), as might be predicted from Figure 2. By the time the first seizure has occurred, NKCC1 activity may already be increased as a result of the previous hypoxia so that early treatment with phenobarbital is still too late to rescue GABAA-R-mediated inhibition. An open question is whether prophylactic treatment with bumetanide would reduce the incidence or severity of neonatal seizures.
Footnotes
This work was supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke Grant NS 40109-06 (K.J.S.), NIH Grants EB000768 (B.J.B.) and NS580752 (K.V.K.), an American Epilepsy Society grant (J.C.G.), and a Hearst Foundation grant (V.I.D.).
References
- Abegg MH, Savic N, Ehrengruber MU, McKinney RA, Gähwiler BH. Epileptiform activity in rat hippocampus strengthens excitatory synapses. J Physiol. 2004;554:439–448. doi: 10.1113/jphysiol.2003.052662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold FJ, Hofmann F, Bengtson CP, Wittmann M, Vanhoutte P, Bading H. Microelectrode array recordings of cultured hippocampal networks reveal a simple model for transcription and protein synthesis-dependent plasticity. J Physiol. 2005;564:3–19. doi: 10.1113/jphysiol.2004.077446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains JS, Longacher JM, Staley KJ. Reciprocal interactions between CA3 network activity and strength of recurrent collateral synapses. Nat Neurosci. 1999;2:720–726. doi: 10.1038/11184. [DOI] [PubMed] [Google Scholar]
- Bartha AI, Shen J, Katz KH, Mischel RE, Yap KR, Ivacko JA, Andrews EM, Ferriero DM, Ment LR, Silverstein FS. Neonatal seizures: multicenter variability in current treatment practices. Pediatr Neurol. 2007;37:85–90. doi: 10.1016/j.pediatrneurol.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gho M. Long-lasting modification of the synaptic properties of rat CA3 hippocampal neurones induced by kainic acid. J Physiol. 1988;404:365–384. doi: 10.1113/jphysiol.1988.sp017294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Berglund K, Schleich W, Wang H, Feng G, Hall WC, Kuner T, Augustine GJ. Imaging synaptic inhibition throughout the brain via genetically targeted Clomeleon. Brain Cell Biol. 2008;36:101–118. doi: 10.1007/s11068-008-9031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Brumback AC, Staley KJ. Thermodynamic regulation of NKCC1-mediated Cl− cotransport underlies plasticity of GABAA signaling in neonatal neurons. J Neurosci. 2008;28:1301–1312. doi: 10.1523/JNEUROSCI.3378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo . J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo KB, Barr P. Drug treatment of neonatal seizures by neonatologists and paediatric neurologists. J Paediatr Child Health. 2005;41:313–316. doi: 10.1111/j.1440-1754.2005.00638.x. [DOI] [PubMed] [Google Scholar]
- Cohen I, Miles R. Contributions of intrinsic and synaptic activities to the generation of neuronal discharges in in vitro hippocampus. J Physiol. 2000;524:485–502. doi: 10.1111/j.1469-7793.2000.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Connell J, Oozeer R, de Vries L, Dubowitz LM, Dubowitz V. Clinical and EEG response to anticonvulsants in neonatal seizures. Arch Dis Child. 1989;64:459–464. doi: 10.1136/adc.64.4_spec_no.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, Bydder GM, Meiners LC, Dubowitz LM, de Vries LS. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361:736–742. doi: 10.1016/S0140-6736(03)12658-X. [DOI] [PubMed] [Google Scholar]
- Dai Y, Tang J, Zhang JH. Role of Cl− in cerebral vascular tone and expression of Na+-K+-2Cl− co-transporter after neonatal hypoxia-ischemia. Can J Physiol Pharmacol. 2005;83:767–773. doi: 10.1139/y05-076. [DOI] [PubMed] [Google Scholar]
- Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem. 2002;277:37542–37550. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- Delpire E. Cation-chloride cotransporters in neuronal communication. News Physiol Sci. 2000;15:309–312. doi: 10.1152/physiologyonline.2000.15.6.309. [DOI] [PubMed] [Google Scholar]
- Derchansky M, Jahromi SS, Mamani M, Shin DS, Sik A, Carlen PL. Transition to seizures in the isolated immature mouse hippocampus: a switch from dominant phasic inhibition to dominant phasic excitation. J Physiol. 2008;586:477–494. doi: 10.1113/jphysiol.2007.143065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duebel J, Haverkamp S, Schleich W, Feng G, Augustine GJ, Kuner T, Euler T. Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron. 2006;49:81–94. doi: 10.1016/j.neuron.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Staley KJ. Excitatory actions of endogenously released GABA contribute to initiation of ictal epileptiform activity in the developing hippocampus. J Neurosci. 2003;23:1840–1846. doi: 10.1523/JNEUROSCI.23-05-01840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–235. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Pedley TA, Moody WJ, Jr, Prince DA. The role of extracellular potassium in hippocampal epilepsy. Arch Neurol. 1976;33:76–83. doi: 10.1001/archneur.1976.00500020004002. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Woodin MA. Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr Opin Neurobiol. 2007;17:81–86. doi: 10.1016/j.conb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem. 2002;277:37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, England R, Diehl L, Delpire E. Apoptosis-associated tyrosine kinase scaffolding of protein phosphatase 1 and SPAK reveals a novel pathway for Na-K-2C1 cotransporter regulation. Am J Physiol Cell Physiol. 2007;292:C1809–C1815. doi: 10.1152/ajpcell.00580.2006. [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS. Dissociated gender-specific effects of recurrent seizures on GABA signaling in CA1 pyramidal neurons: role of GABAA receptors. J Neurosci. 2008;28:1557–1567. doi: 10.1523/JNEUROSCI.5180-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giménez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem. 2003;278:26946–26951. doi: 10.1074/jbc.M303435200. [DOI] [PubMed] [Google Scholar]
- Glykys J, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–672. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannaert P, Alvarez-Guerra M, Pirot D, Nazaret C, Garay RP. Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:193–199. doi: 10.1007/s00210-001-0521-y. [DOI] [PubMed] [Google Scholar]
- Haugvicová R, Kubová H, Mares P. Antipentylenetetrazol action of clobazam in developing rats. Physiol Res. 1999;48:501–507. [PubMed] [Google Scholar]
- Heinemann U, Lux HD, Gutnick MJ. Extracellular free calcium and potassium during paroxsmal activity in the cerebral cortex of the cat. Exp Brain Res. 1977;27:237–243. doi: 10.1007/BF00235500. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Agassandian C, Merchán-Pérez A, Ben-Ari Y, DeFelipe J, Esclapez M, Bernard C. Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nat Neurosci. 1999;2:499–500. doi: 10.1038/9142. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C. Triggers of apoptosis in the immature brain. Brain Dev. 2009;31:488–492. doi: 10.1016/j.braindev.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Isaev D, Isaeva E, Khazipov R, Holmes GL. Shunting and hyperpolarizing GABAergic inhibition in the high-potassium model of ictogenesis in the developing rat hippocampus. Hippocampus. 2007;17:210–219. doi: 10.1002/hipo.20259. [DOI] [PubMed] [Google Scholar]
- Isenring P, Jacoby SC, Payne JA, Forbush B., 3rd Comparison of Na-K-Cl cotransporters. NKCC1, NKCC2, and the HEK cell Na-L-Cl cotransporter. J Biol Chem. 1998;273:11295–11301. doi: 10.1074/jbc.273.18.11295. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4:490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Esclapez M, Medina I, Aggoun D, Lamsa K, Leinekugel X, Khazipov R, Ben-Ari Y. A novel in vitro preparation: the intact hippocampal formation. Neuron. 1997;19:743–749. doi: 10.1016/s0896-6273(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Dzhala V, Ben-Ari Y, Khazipov R. Dual role of GABA in the neonatal rat hippocampus. Dev Neurosci. 1999a;21:310–319. doi: 10.1159/000017380. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Dzhala V, Medina I, Leinekugel X, Melyan Z, Lamsa K, Khazipov R, Ben-Ari Y. Maturation of kainate-induced epileptiform activities in interconnected intact neonatal limbic structures in vitro. Eur J Neurosci. 1999b;11:3468–3480. doi: 10.1046/j.1460-9568.1999.00768.x. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Holmes GL, Ben-Ari Y. In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat Neurosci. 2003;6:1079–1085. doi: 10.1038/nn1125. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci. 2004;19:590–600. doi: 10.1111/j.0953-816x.2003.03152.x. [DOI] [PubMed] [Google Scholar]
- Kilb W, Sinning A, Luhmann HJ. Model-specific effects of bumetanide on epileptiform activity in the in vitro intact hippocampus of the newborn mouse. Neuropharmacology. 2007;53:524–533. doi: 10.1016/j.neuropharm.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Li X, Zhou J, Chen Z, Chen S, Zhu F, Zhou L. Long-term expressional changes of Na+-K+-Cl− co-transporter 1 (NKCC1) and K+-Cl− co-transporter 2 (KCC2) in CA1 region of hippocampus following lithium-pilocarpine induced status epilepticus (PISE) Brain Res. 2008;1221:141–146. doi: 10.1016/j.brainres.2008.04.047. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Shin D, Sankar R. Bumetanide inhibits rapid kindling in neonatal rats. Epilepsia. 2009;50:2117–2122. doi: 10.1111/j.1528-1167.2009.02048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi EM, Kellaway P. Characterization and classification of neonatal seizures. Neurology. 1987;37:1837–1844. doi: 10.1212/wnl.37.12.1837. [DOI] [PubMed] [Google Scholar]
- Moody WJ, Futamachi KJ, Prince DA. Extracellular potassium activity during epileptogenesis. Exp Neurol. 1974;42:248–263. doi: 10.1016/0014-4886(74)90023-5. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardou R, Ben-Ari Y, Khalilov I. Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J Neurophysiol. 2009;101:2878–2888. doi: 10.1152/jn.90761.2008. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MJ, Scher MS, Stein AD, Armatti S, Wang Z, Gardiner JC, Paneth N, Minnigh B, Alvin J. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485–489. doi: 10.1056/NEJM199908123410704. [DOI] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;273:C1516–C1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD. The chloride transporter Na+-K+-Cl− cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci. 2006;26:1396–1406. doi: 10.1523/JNEUROSCI.1421-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA, Lux HD, Neher E. Measurement of extracellular potassium activity in cat cortex. Brain Res. 1973;50:489–495. doi: 10.1016/0006-8993(73)90758-0. [DOI] [PubMed] [Google Scholar]
- Qian N, Sejnowski TJ. When is an inhibitory synapse effective? Proc Natl Acad Sci U S A. 1990;87:8145–8149. doi: 10.1073/pnas.87.20.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quilichini PP, Diabira D, Chiron C, Ben-Ari Y, Gozlan H. Persistent epileptiform activity induced by low Mg2+ in intact immature brain structures. Eur J Neurosci. 2002;16:850–860. doi: 10.1046/j.1460-9568.2002.02143.x. [DOI] [PubMed] [Google Scholar]
- Quilichini PP, Diabira D, Chiron C, Milh M, Ben-Ari Y, Gozlan H. Effects of antiepileptic drugs on refractory seizures in the intact immature corticohippocampal formation in vitro. Epilepsia. 2003;44:1365–1374. doi: 10.1046/j.1528-1157.2003.19503.x. [DOI] [PubMed] [Google Scholar]
- Raol YH, Lapides DA, Keating JG, Brooks-Kayal AR, Cooper EC. A KCNQ channel opener for experimental neonatal seizures and status epilepticus. Ann Neurol. 2009;65:326–336. doi: 10.1002/ana.21593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie JM, Boylan GB. Neonatal seizures and their treatment. Curr Opin Neurol. 2003;16:177–181. doi: 10.1097/01.wco.0000063768.15877.23. [DOI] [PubMed] [Google Scholar]
- Rheims S, Minlebaev M, Ivanov A, Represa A, Khazipov R, Holmes GL, Ben-Ari Y, Zilberter Y. Excitatory GABA in rodent developing neocortex in vitro. J Neurophysiol. 2008;100:609–619. doi: 10.1152/jn.90402.2008. [DOI] [PubMed] [Google Scholar]
- Schneiderman JH, Sterling CA, Luo R. Hippocampal plasticity following epileptiform bursting produced by GABAA antagonists. Neuroscience. 1994;59:259–273. doi: 10.1016/0306-4522(94)90594-0. [DOI] [PubMed] [Google Scholar]
- Sipilä ST, Schuchmann S, Voipio J, Yamada J, Kaila K. The cation-chloride cotransporter NKCC1 promotes sharp waves in the neonatal rat hippocampus. J Physiol. 2006;573:765–773. doi: 10.1113/jphysiol.2006.107086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley K, Smith R. A new form of feedback at the GABAA receptor. Nat Neurosci. 2001;4:674–676. doi: 10.1038/89439. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Mody I. Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J Neurophysiol. 1992;68:197–212. doi: 10.1152/jn.1992.68.1.197. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and. J Physiol. 1999;519:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Murali SG. Stimulation of Na+-K+-2Cl− cotransporter in neuronal cells by excitatory neurotransmitter glutamate. Am J Physiol. 1998;275:C772–C779. doi: 10.1152/ajpcell.1998.275.3.C772. [DOI] [PubMed] [Google Scholar]
- Tekgul H, Gauvreau K, Soul J, Murphy L, Robertson R, Stewart J, Volpe J, Bourgeois B, du Plessis AJ. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics. 2006;117:1270–1280. doi: 10.1542/peds.2005-1178. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl− in hippocampal CA3 neurons. J Neurophysiol. 1989;61:512–523. doi: 10.1152/jn.1989.61.3.512. [DOI] [PubMed] [Google Scholar]
- Twyman RE, Rogers CJ, Macdonald RL. Differential regulation of gamma-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann Neurol. 1989;25:213–220. doi: 10.1002/ana.410250302. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Ivanov A, Bernard C, Holmes GL, Ben-Ari Y, Khazipov R. Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J Neurophysiol. 2003;90:2964–2972. doi: 10.1152/jn.00172.2003. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hübner CA, Represa A, Ben-Ari Y, Khazipov R. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 2006;314:1788–1792. doi: 10.1126/science.1133212. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, Khazipov R. Timing of the developmental switch in GABAA mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia. 2007;48(Suppl 5):96–105. doi: 10.1111/j.1528-1167.2007.01295.x. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velísek L, Velísková J, Ptachewich Y, Ortíz J, Shinnar S, Moshé SL. Age-dependent effects of gamma-aminobutyric acid agents on flurothyl seizures. Epilepsia. 1995;36:636–643. doi: 10.1111/j.1528-1157.1995.tb01039.x. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Philadelphia: Saunders; 2001. Neurology of the newborn. [Google Scholar]
- Wang C, Shimizu-Okabe C, Watanabe K, Okabe A, Matsuzaki H, Ogawa T, Mori N, Fukuda A, Sato K. Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Brain Res Dev Brain Res. 2002;139:59–66. doi: 10.1016/s0165-3806(02)00536-9. [DOI] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR, Ben-Ari Y. GABA regulates stem cell proliferation before nervous system formation. Epilepsy Curr. 2008;8:137–139. doi: 10.1111/j.1535-7511.2008.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JE, Porter JT, Agmon A. GABAergic inhibition suppresses paroxysmal network activity in the neonatal rodent hippocampus and neocortex. J Neurosci. 2000;20:8822–8830. doi: 10.1523/JNEUROSCI.20-23-08822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM. Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron. 2003;39:807–820. doi: 10.1016/s0896-6273(03)00507-5. [DOI] [PubMed] [Google Scholar]
- Xiong ZQ, Stringer JL. Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol. 2000;83:1443–1451. doi: 10.1152/jn.2000.83.3.1443. [DOI] [PubMed] [Google Scholar]
- Xiong ZQ, Saggau P, Stringer JL. Activity-dependent intracellular acidification correlates with the duration of seizure activity. J Neurosci. 2000;20:1290–1296. doi: 10.1523/JNEUROSCI.20-04-01290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]