

Abstract
Signal transducer and activator of transcription (STAT) proteins are well known for their essential roles in transmitting cytokine-mediated signals and specifying T helper (TH) cell differentiation; however, recent technological advances have led to a new level of understanding of transcription factor action. This work has revealed that STAT proteins have broad and complex roles in gene regulation and epigenetic control, including important roles as functional repressors. However, the challenge remains how to link signal transduction, nucleosome biology and gene regulation. The relevance of tackling this problem is highlighted by genome-wide association studies that link cytokine signalling and STATs to a variety of autoimmune or immune deficiency disorders. Defining exactly how extrinsic signals control the specification and plasticity of TH cells will provide important insights and perhaps therapeutic opportunities in these diseases.
CD4+ T cells are essential for host defence, as exemplified by the effects of depletion of CD4+ T cells associated with HIV/AIDS. This loss of T cells leads to profound impairment of the immune response and a range of opportunistic infections. Conversely, CD4+ T cells are also fundamental drivers of autoimmunity when a loss of tolerance occurs. CD4+ T cells mainly direct immune responses through the cytokines they produce, and our understanding of the range of cytokines produced by CD4+ T cells has become considerably expanded than before1. In addition to T helper 1 (TH1) and TH2 cells, which produce interferon-γ (IFNγ) and interleukin-4 (IL-4), respectively, new subsets of T cells continue to be recognized. These include regulatory T (TReg) cells2, which comprise both natural and induced TReg cells, IL-17-producing TH17 cells2–4, IL-9-producing TH9 cells5–7 and a subset of IL-22-producing TH cells7, 8. IL-21-producing follicular helper T (TFH) cells provide help to B cells, but their identity as a distinct lineage and relationship to other CD4+ T cell subsets remain a source of some controversy9, 10.
What is clear is that the cytokine milieu is crucial for CD4+ T cell differentiation. Signal transducer and activator of transcription (STAT) family proteins have essential roles in transmitting many cytokine-mediated signals and thereby have similarly crucial roles in TH cell differentiation1, 11. The first STAT proteins (STAT1 and STAT2) were discovered as a mediators of interferon (IFN) action to induce de novo gene transcription12, 13; the essential, non-redundant functions of the seven members of the STAT family have been extensively defined by generating individual gene knockout mice and by careful analysis of the effects on gene expression14–18. One of the challenges to interpreting such gene expression data is to distinguish direct actions of STATs on individual genes from secondary, indirect effects of STAT deficiency. Recent technologies have enabled investigators to construct a genome-wide view of transcription factor binding to distinguish direct from indirect effects.
In this Review, we discuss the impact of next generation sequencing19, 20, and illustrate how this technology has allowed us to begin to construct a quantitative map of not only of genome-wide transcription factor binding, but also of their effect on genome-wide epigenetic changes. Specifically, we review the genome-wide STAT binding studies that have been reported so far. We discuss the relationship between STAT binding and local epigenetic patterns and how STAT proteins can integrate extrinsic signals to influence epigenetic changes associated with T cell lineage commitment. Finally, we review emerging new information regarding mutations and polymorphisms of STAT proteins that are associated with human immune disorders.
Genome-wide views offered by next generation sequencing
STATs are DNA-binding transcription factors that induce the transcription of their target genes by recognizing specific DNA consensus sequences. Analysis of direct STAT binding to DNA was initially analyzed by electrophoretic mobility shift assay (EMSA). Later, analysis of binding to specific genes was carried out by chromatin immunoprecipitation followed by the detection of precipitated DNA by PCR using primers against pre-selected regions. This type of targeted analysis inevitably limited its application to a small subset of genes and regions. However with the arrival of next generation sequencing methods, an unbiased genome-wide view of protein–DNA association has become a reality (Box 1 and Fig. 1), allowing us to catalogue the entire range of STAT target genes on a genome-wide scale. Equally important has been the capability to map histone epigenetic marks throughout the entire genome to gain insight into how chromatin accessibility relates to STAT binding and ultimately to transcriptional regulation.
Text box 1. Genome-wide chromatin immunoprecipitation (ChIP) to study protein–DNA interactions.
Chromatin immunoprecipitation (ChIP) has been used to profile DNA–protein interactions. By choosing appropriate antibodies specific for the protein or epigenetic modification of interest, both transcription factor binding and histone epigenetic marks can now be profiled on a genome-wide scale. After protein-associating DNA fragments are enriched and purified through immunoprecipitation, the DNA fragments can be measured and mapped to reference genomes by either hybridization (ChIP -on chip) or high-throughput next generation sequencing (ChIP-seq).
ChIP -on chip, which is based on microarray hybridization technology133, 134, has the intrinsic limitation that only pre-selected regions of the genome are included in the arrays, such as proximal gene promoter regions. Also, array-based methods are restricted by the variation and limitation implicit in nucleotide hybridization. By contrast, chromatin immunoprecipitation coupled with next generation sequencing methods (ChIP-seq) covers the entire genome without any preconceived bias19, 20, 135. Because the DNA fragments of interest are sequenced directly instead of being hybridized to microarray chips, ChIP-seq provides higher resolution, greater genomic coverage, fewer artifacts and a larger dynamic range of signal strength than ChIP-on chip. Although the relatively short reads (35–75 bp) generated by various next generation sequencing platforms could pose technical difficulties for certain other applications such as RNA-seq, the technology is well-suited for a ChIP-seq approach 136.
In addition to mapping trascription factor binding and histone epigenetic marks, Chip-seq has also been applied to map the binding of CCCTC-binding factor (CTCF), which regulates chromatin architecture, and p300, which marks enhancer elements27. This next generation sequencing platform is also used to define nucleosome positioning and accessibility by coupling with micorococcal nuclease digestion (MNase-seq) 137 or with the detection of DNAse hypersensitivity sites (DNase-seq)138, 139. Next generation sequencing can also be used to generate comprehensive methylome mapping 140, which will provide yet more insights on stable and heritable aspect of epigenome. Next generation sequencing has also been used to profile various types of RNA, such as microRNAs (miRNAs)141, 142, long noncoding intervening RNAs143 and enhancer RNAs144.
Figure 1. Experimental flow of ChIP-seq analysis.
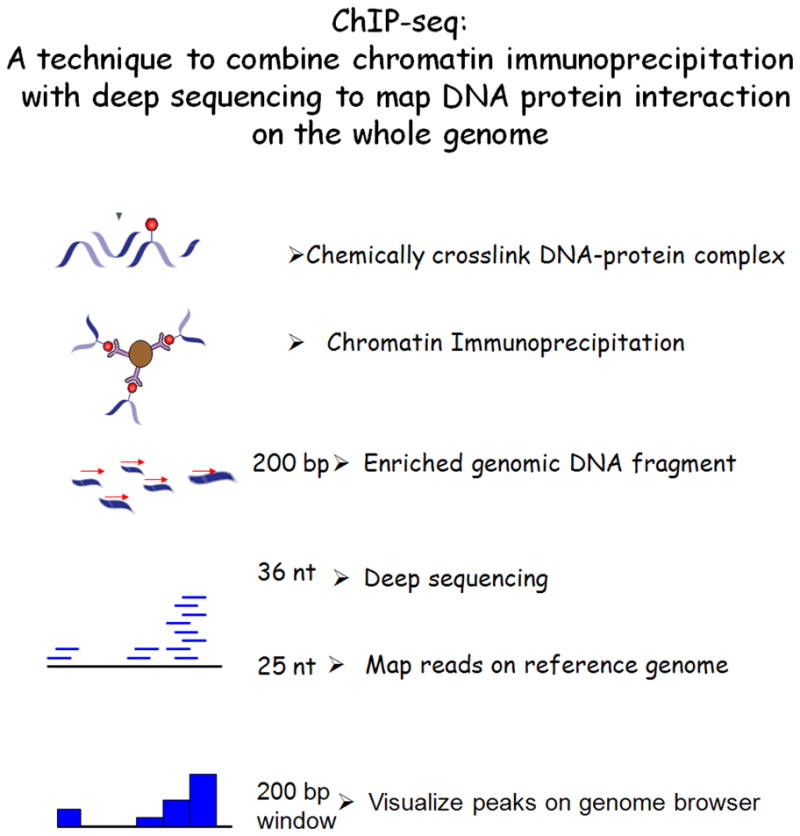
A technique to combine chromatin immunoprecipitation with next generation sequencing to map DNA–protein interactions across the whole genome is shown. Chemically cross-linked DNA-protein complexes are immunoprecipitated and the protein-bound DNA fragments are isolated. The crosslinks are reversed, and the purified DNA was used to generate a library for sequencing. Automated reactions yield 36 nt long sequence reads of over 20 million per sample (Illumina GA platform). The sequence reads are aligned onto the reference genome and the distribution of protein-DNA interaction sites are visualized as “peaks” on the genome browser.
Evolving views of epigenome
It has become clear since the original discovery of the STATs that in addition to transcription factor binding, a crucial part of gene regulation is epigenetic regulation and that the modifications comprised by this term are highly dynamic21. It is beyond the scope of the present Review to comprehensively discuss this incredibly dynamic field; but briefly, factors that influence the accessibility of chromatin for active transcription include DNA methylation, ATP-dependent nucleosome remodeling and a large number of post translational histone modifications. Together, these modifications, in addition to classic transcription factors, have major effects on gene expression 22–24. Early comprehensive genome-wide histone-modification maps generated by ChIP-seq suggested novel functions for histone modifications and showed the importance of combinatorial patterns of modifications 25, 26. Although acetylation is always associated with active chromatin regions, the functional significance of histone methylation is more complex with respect to gene expression. For example, histone 3 lysine 4 (H3K4), H3K36 and H3K79 trimethylation are associated with active genes (‘permissive’ marks), whereas H3K27, H3K9 and H4K20 di/trimethylation are linked to gene silencing (‘repressive’ marks). Importantly, in contrast to classic views of the epigenome, it is now clear that some of these modifications can occur rapidly in response to exogenous signals27–29. Therefore, the nucleosome is increasingly viewed as a nuclear sensor that responds to various signals from the cellular environment.
Cytokine signaling and the T cell epigenome
The ability to measure genome-wide changes in histone modifications by ChIP-seq provided an opportunity to ask a simple but crucial question about T cell biology, namely whether the observed epigenetic modifications in T cells are more consistent with a model of stable terminal differentiation of CD4+ T cells or intrinsic flexibility in T cell responses 6, 7. The stability of various T cell subsets continues to be intensively debated, with remarkable examples of T cell plasticity appearing in the literature30, 31 Rubtsov, #320, measuring the epigenetic landscape of TH cells has proven to be illuminating.
Genome-wide H3K4 (permissive) and H3K27 (repressive) trimethylation maps in naïve CD4+ T cells and fully polarized TH1, TH2, TH17, induced TReg and natural TReg cells have now been obtained 32. The data show that the histone modifications of genes encoding TH cell subset signature cytokines are consistent with the signature of terminal commitment, such that permissive marks on a particular cytokine gene are selectively present in the relevant lineage that expresses that cytokine and repressive marks are present in other CD4+ T cell lineages that do not express the cytokine. However, genes encoding “master regulator” transcription factors, such as Tbx21 for TH1 cells and Gata3 for TH2 cells, were found to have “bivalent poised domains”, meaning that both permissive and repressive histone marks are present on these genes in the lineages of alternative fates32. Originally identified in stem cells, bivalent domains seem to allow for flexibility in expression once the cells receive signals for a differentiation 33, 34. Bivalent domains were also present on genes encoding other key transcription factors including Runx3, Bcl6 and Blimp1 32,. Thus, the answer to the question initially posed is that epigenetic analysis provided evidences for both terminal commitment (e.g. cytokine genes) and flexible plasticity (e.g. master regulators genes) of T helper cells depending which genes are examined. As such, the extent to which T cells subsets really behave as “lineages” or behave as flexible populations will continue be the focus of ongoing research and controversy.
Although many interesting observations have arisen from this genome-wide epigenetic profiling, many other questions remain. First, the cell preparation used for these studies was generated in vitro and characterized at a single time point. The dynamic nature of chromatin remodeling/modification over the course of T cell differentiation is yet to be fully elucidated by genome-wide assays. It will be interesting to know whether the bivalent marks noted on the master transcriptional regulators are already in place in the very early phases of TH cell differentiation to guide the transcriptional programme or if these marks gradually evolve over time. Equally, it will be crucial to determine how the recruitment of STAT proteins affects the deposition/removal of epigenetic marks, and how all the aspects of nucleosome remodeling are acquired over time. We also do not yet know the degree of similarity between in vitro-generated cells and bona fide TH cells that arise in vivo during the course of infection or autoimmunity in mice and in humans.
Profiling of genome-wide STAT binding
Initial work using Chip-seq to map STAT1 binding sites in the genome revealed more than 11,000 sites in unstimulated HeLa cells and 40,000 sites after IFN-γ stimulation 35. However, it was not clear from these data whether STAT1 is an important initiator of gene regulation in all cases of binding or whether STAT1 has a major role in creating the local epigenetic patterns around these binding sites. Subsequent work showed that for most genes, deposition of the local histone modification has preceded the recruitment of ligand-induced STAT1 binding36. While this study was an important breakthrough, it will be important to link STAT1 action in various primary cells and to link transcriptional and epigenetic changes using STAT1-deficient cells. (This point is discussed further with additional ChIP-seq data for STAT binding below). Since these initial reports, all STATs, with the exception of STAT2, have been profiled by ChIP-seq and the original datasets are publically available through Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) as shown in Table 1. These datasets will serve as enormous resources to promote further genomic research in the scientific community, and deposition of original datasets to publicly accessible domains such as GEO will be key. On the other hand, it will be important to keep in mind the degree of compatibility among different datasets, which have been generated by different sequencing platforms and by different investigators under different experimental conditions could impose certain limitation over comparable analysis. As the field matures, we await a better ways to “quality control” these sequencing dataset to allow broader across-the-board analyses including the utilization of appropriate reference controls to score ChIP-seq peaks37. Nevertheless, from the STAT binding data listed in Table 1, we can start to address the unique as well as shared functions of individual STAT proteins in directing epigenetic modifications and gene expression in T cells. For the sake of briefness, we will only discuss genome wide STAT data in Table 1 that are derived from T cells with genome scale analysis provided.
Table 1.
Summary of ChIP-seq data reported for STAT family proteins
| Stimuli | Cell-type | Species | GEO Accession number | Reference | |
|---|---|---|---|---|---|
| STAT1 | IFNg | HeLa | human | GSE15353 | 35 |
| IFNg | HeLa | human | GSE12783 | 37, 145 | |
| STAT2 | N/A | ||||
| STAT3 | IL-6 | TH17 polarized T cells | mouse | GSE21670 | 76 |
| IL-21 | CD4+ T cells | mouse | GSE19198 | 89 | |
| LIF? | ESC cells | mouse | GSE11431 | 146 | |
| STAT4 | IL-12 | TH1 polarized T cell | mouse | GSE22105 | 42 |
| STAT5(a,b) | IL-2? | CD4+ T cell | mouse | GSE12346 | 68 |
| STAT6 | IL-4 | TH2 polarized T cell | mouse | GSE22105 | 42 |
| IL-4 | TH2 polarized T cell | human | GSE18017 | 59 | |
IFN, interferon; IL, interleukin; TH, T helper
All of the original data listed in the table are accessible through Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) by the corresponding GEO accession number.
STAT4 in TH1 cells
Landscape of STAT4 targets
Unlike other STATs, the expression of which is found in a wide range of cell types, the expression of STAT4 is relatively restricted to immune cells and testis. Accordingly its nonredundant functions are manifested mainly in immune cells, resulting in a very discrete phenotype of STAT4 deficiency involving decreased IFNγ production38. STAT4 is activated mainly by IL-12, IL-23 and type I IFNs, and it functions predominantly in promoting TH1 cell differentiation. STAT4 is also the major regulator of Ifng expression in innate immune cells such as NK cells39, 40.
Before the advent of genomic approaches, only a limited number of direct STAT4 target genes had been identified (such as Ifng, Il18r1, Hlx, Map3k8 and Furin). The first effort to increase our knowledge of STAT4 targets was through the use of ChIP-on-chip technology, which showed that STAT4 bound the promoters of many previously unidentified targets such as Gadd45g, Lcp2 and Myd88 41. The expanded list of STAT4 target genes also showed that all genomic STAT4-binding events are not equal and that in some cases the binding of STAT4 to a target gene was not translated into a change in gene expression as a result of IL-12 stimulation. Binding per se was not the only determinant of STAT4-dependent gene programming during TH1 cell differentiation.
Whereas the analysis provided by ChIP-on-chip technology is limited to predefined regions of the genome, ChIP-seq data generate an unbiased genome-wide map of where STATs bind. Using ChIP-seq 42, STAT4 was found to have 10,000 binding sites in in vitro-differentiated murine TH1 cells, 40% of which were localized to the promoters or gene bodies of approximately 4,000 annotated genes. 60% of the STAT4-binding sites occurred in intergenic regions, where some of the distal enhancer elements are thought to reside away from annotated genes. In sharp contrast to the implications of the STAT1 data described above, comparative epigenomic analysis of wild-type versus STAT4-deficient TH1 cells provided evidence that of the ~4,000 genes bound by STAT4, nearly 1,000 of these genes showed STAT4-dependent alterations in epigenetic modifications. Of these 1,000 genes, 200 genes showed highly STAT4-dependent gene expression as determined by microarray analysis of wild-type versus STAT4-deficient cells. These genes therefore represent a core subset of direct STAT4 targets, which are highly dependent on STAT4 for promoting gene expression and the local epigenetic signature. Importantly their dependence on STAT4 cannot be compensated for by other STAT proteins or transcription factors. The subset not only included signature TH1 cell genes such as Ifng and Tbox21 but also others including Il18rb, Icos, Lilrb4 and Nkg7. This implies a potential role for these genes in maintaining the phenotype of fully polarized TH1 cells, which may be of interest to examine in the future.
The analysis of STAT4 target genes also showed that some cytokine genes previously considered to define other TH cell lineages were STAT4 targets in TH1 cells. Initially denoted as a TH2 cell gene in mice, subsequent work has shown that IL-10 is expressed by multiple T cell subsets43. It was interesting to note that the Il10 gene was bound and positively regulated by STAT4 in TH1 cells, but also by STAT6 in TH2 cells. Initially noted to be a product of T cells following TCR stimulation, IL-21 was later reported to be produced by TH17 cells in a STAT3-dependent manner 44–47. More recently IL-21 has also been reported as a lineage-defining cytokine for TFH cells 10. However, Chip-seq data indicated that STAT4 can bind and regulate the Il21 gene, which is consistent with the recent finding that IL-12 (acting via STAT4) can induce the expression of IL-21 in human T cells 48. Thus, IL-10 and IL-21 are two examples of genes that can be regulated by multiple STATs.
STAT4 as a transcriptional repressor
Generally, transcription factors that drive lineage commitment positively regulate the expression of phenotype-defining genes, but they can also repress the expression of genes associated with alternative fates. Although STATs were originally discovered as activators of gene transcription, a point that has been well confirmed by genome-wide analysis, there have been indications that STATs can also function as functional repressors. Microarray data provided evidence of genes whose expression was increased when a given STAT was deleted 49, 50, but there were few examples of genes for which STATs seemed to function as direct transcriptional repressors 51, 52. In this regard, several possible ways for a STAT protein to cause gene silencing were reported that include recruitment of DNMT1 and HDAC153 or direct interaction with HP1 for heterochromatin formation54. In T cells, STAT4-dependent repressive histone marks were identified on several TH2 cell-expressed genes, including STAT6 target genes, which are actively repressed by STAT4 in TH1 cells. Although the total number of such genes is small (around 40 genes), the data clearly point to a role for STAT4 as a transcriptional repressor as well as its more widely recognized role as a transcriptional activator (Figure 2). It is not yet clear how a transcription factor can drive expression of one gene and repress the expression of another gene in the same cell, but it will be informative to analyze the associating factors/proteins that are locally recruited to genes bound and repressed by STAT proteins. The successful identification of specific chromatin modifications associated with STAT binding represents different way of utilizing the genomic approach, aside from obtaining a list of target genes. The genomic approach that integrates different types of readouts enables us to “examine the forest” rather than just “finding the trees” on the genome. Not to say that the trees are not interesting, but the big picture is important as well.
Figure 2. Distinctive epigenetic patterns are formed by STAT proteins in differentiated T helper effector cells.

A key role of signal transducer and activator of transcription (STAT) proteins includes shaping epigenetic patterns on target gene loci to maintain cell lineage specificity. Five distinct epigenetic patterns were found to be STAT4 dependent in T helper 1 (TH1) cells that included both permissive chromatin signatures (high histone 3 lysine 4 trimethylation (H3K4me3) marks, high H3K36me3 marks or low H3K27me3 marks) and repressive chromatin signatures (high H3K27me3 or low H3K36me3). Permissive chromatin signatures are found on TH1 cell-expressed genes, whereas repressed chromatin signatures are found on TH2 cell-expressed genes in TH1 cells. The figure was reproduced from online version of Wei et al, Immunity 201042.
STAT6 in TH2 cells
STAT6 as a driver of TH2 cell differentiation
TH2 cell differentiation is induced by IL-4, and the importance of STAT6 for this process has been well established in mice 55–58. The actions of STAT6 and its downstream targets in pathological TH2 cell responses such as asthma and allergy are also of great interest, given the public health impact of these diseases. Consequently, both mouse 16, 18 and human 59 systems have been studied in terms of STAT6 functions. In human cells, STAT6 mediates the expression of more than 80% of IL-4-regulated genes, a higher proportion than was reported in previous studies using mouse cells 16. The function and cellular distribution of identified STAT6 targets are broad, reflecting the fundamental role of STAT6 in regulating multiple aspects of activities in cells.
Genome-wide kinetic profiling of STAT6-dependent gene expression and analysis of the STAT6-dependent gene network in humans59 confirm that STAT6 is a major, direct contributor to the transcriptional profile associated with the TH2 cell phenotype. The findings also show that IL-4-induced regulation of gene transcription in human cells is highly dynamic; only a subset of the genes that were differentially regulated within the first few hours after IL-4 treatment remained differentially expressed at later time points up to 72 hrs. These findings indicate that in addition to genes that provide the appropriate ‘switch signal’ for TH2 cell differentiation at early times, factors other than STAT6 are required for the transition along the developmental pathway and maintenance of the acquired phenotype. Through genome-wide differential gene expression analysis using small interfering RNA (siRNA), 453 genes were identified as being regulated by STAT6 in human cells59. Of these, only 6% of genes had been previously identified as STAT6 targets, including genes such as GATA3, SOCS1 and IL24. The new target genes suggest new functions and processes that are mediated by STAT6 signalling. In general, the findings underscore the importance of using genome-wide approaches to further explore whether there are species-specific roles of STAT proteins in humans and mice.
The early signalling network: connection to different TH cell fates
By gene network analysis, the IL-4- and STAT6-regulated transcription factors were found to form a compact core interaction network of signalling (Figure 3). These data underscore the importance of combinatorial signalling pathways that function together to determine TH cell commitment and fate. Of the newly identified direct STAT6 targets, three transcription factors that form hubs in the regulatory network — RUNX1, EPAS1 and BATF — are of particular interest. RUNX proteins have a central role in regulating TH cell differentiation in general 60, but RUNX1 preferentially inhibits TH2 cell differentiation by downregulating GATA3 expression61 and binds to the IL4 silencer region62. In addition, RUNX1 can form a complex with FOXP3 or RORC, and is necessary for TReg and TH17 cell function, respectively 63, 64. Interestingly, EPAS1 binds to the RUNX1 promoter, potentially amplifying the effect of STAT6 on RUNX1 expression 65. BATF, which is also directly regulated by STAT6, regulates both TH17 and TH2 cell differentiation 66, 67. The connection between Th2 differentiation program and programs for other Th cell subsets can be further examined through the network of key transcription factors. As shown in Fig. 3, it is notable that within STAT6-mediated Th2 differentiation program, a close connection between STAT6 and other STAT family proteins is evident through only a few intermediate molecules. This in turn underscores the importance of understanding cooperative and counter-operative interactions between the STATs as well as between downstream transcription factors in directing TH cell fate. For example, a comparison of ChIP-seq data for STAT659 and STAT5A 68 showed that they have overlapping targets and indicated that these two STATs might have cooperative roles 59. This is consistent with the known contribution of STAT5 to STAT6-independent TH2 cell commitment68–70.
Figure 3. The STAT6 signalling network identified during the initial TH2 cell differentiation stage.

Interleukin-4 (IL-4)- and signal transducer and activator of transcription 6 (STAT6)-regulated transcription factors form a core network of interacting nodes. Genes shown in red color boxes are upregulated and those in green color boxes are downregulated by STAT6 in transcriptomics studies. STAT6-mediated regulation of genes detected by ChIP-seq is marked with red arrows (solid line for direct regulation, dashed line for indirect regulation). Furthermore, known direct interactions between the putative downstream transcriptional regulators of STAT6 in humans were added to the figure. Blue lines correspond to protein-protein interactions, and black lines correspond to other types of interaction or regulation. The networks were generated through the use of Ingenuity Pathways
Analysis (Ingenuity Systems, www.ingenuity.com) with some modifications based on published reports. The figure was modified from Elo et al. Immunity 201059 with the permission from Immunity.
A stabilizer of the TH2 cell phenotype
The crucial role of STAT6 in the initiation stage of TH2 cell differentiation is evident, but STAT6 also contributes to the maintenance of the TH2 cell phenotype in differentiated cells, as supported by combinatorial genome-wide analysis of STAT6 binding, STAT6-induced epigenetic patterns and gene expression 42. Similar to STAT4, STAT6 is responsible for the maintenance of distinct epigenetic patterns on selected target genes. STAT6 predominantly functions as a transcriptional activator, but as is the case with STAT4, it also functions as a functional repressor for a subset of genes. In terms of activating target genes, STAT6 more frequently opposes the deposition of repressive epigenetic marks than it promotes permissive epigenetic marks; in this regard, STAT6 seems to be subtly different from STAT4.
Of particular interest are a subset of genes that are bound by STAT4 in TH1 cells and by STAT6 in TH2 cells, for which the respective STAT has opposing effects on local epigenetic patterns. A notable example is the Il18r1-Il18rap locus42. That is, whereas one STAT promotes active marks (in this case STAT4), the other STAT (STAT6) promotes repressive marks on the same locus. This divergent action of STAT4 and STAT6 on the same genes provides a back-up for inducing gene expression in one lineage and repressing gene expression in the other lineage (Figure 2).
STAT3 and TH17 cell differentiation
TH cells that selectively produce IL-17 (known as TH17 cells) are one of the newest T cell subsets to be recognized. They have crucial roles in host defence against extracellular bacteria and fungi, but also in the pathogenesis of various autoimmune diseases 2, 3. Cytokines that promote IL-17 production include IL-1, transforming growth factor-β1 (TGFβ1), IL-6, IL-21 and IL-23. The latter three cytokines activate STAT3. Although STAT3 is activated by a large number of cytokines and has crucial functions in various tissues 71, 72, T cell-specific deletion of STAT3 mainly affects the expression of IL-17 and IL-21 73–75, and consequently results in decreased severity of several autoimmune disease models 73, 76–79. Conversely, the deletion of Socs3, which increases STAT3 activation, results in increased numbers of TH17 cells 74.
Landscape of STAT3 targets
ChIP-seq analysis of STAT3 binding in T cells, coupled with gene expression analysis, has confirmed that the Il17 and Il21 genes are direct targets of STAT3 46, 74, 76. Of note, STAT3 binds to multiple sites in the Il17 locus80, the most prominent of which are intergenic regions that coincide with conserved non-coding sequences (CNS) 81. These sites also bind p300 in a STAT3-dependent manner and so are probably enhancer elements (Golnaz Vahedi, Yuka Kanno, John O’Shea, unpublished observation). Furthermore, analogous to the role of STAT6, STAT3 directly binds to genes encoding multiple transcription factors that are crucial for programming TH17 cells. These include Rorc 82, Rora83, Ahr84, Batf 66, Irf4 85 and c-Maf 86. Other important direct targets of STAT3 that define the TH17 cell phenotype include Il23r and Il6ra 45, 76. Notably, the ability of STAT3 to positively regulate expression of these genes was associated with permissive H3K4me3 marks. The prominent role of STAT3 in this process led to a re-evaluation of the factors involved in specification of TH17 cells. While TGF-β signaling constitutes an important aspect of Th17 differentiation, an alternative mode of generating Th17 cells in the absence of TGF-β has also been recognized87. It was found that activation of STAT3 in conjunction with IL-1 were sufficient to promote expression of the IL-23 receptor87. Acquisition of the receptor allowed responsiveness to IL-23, which has a major role in driving pathogenic IL-17-dependent responses88. As a result, pathogenic TH17 cells were generated in the absence of TGF-β signalling87 via activation of STAT3 and other cooperating factors. In this regard, it is interesting that genome wide STAT3 binding sites and that of IRF4 site overlap significantly following IL21 stimulation89
In addition to the role of STAT3 in regulating the expression of TH17 cell-related cytokines and transcription factors, Chip-seq analysis also pointed to a role for STAT3 in regulating T cell proliferation and survival. This phenotype was not evident in the initial description of deletion of STAT3 in T cells90; however, Stat3−/− T cells have delayed proliferation and poor clonal expansion, particularly in the setting of inflammation76. In this regard, newly identified STAT3 target genes in T cells include the anti-apoptotic genes Bcl290 and ler3, and the proto-oncogenic transcription factors Fos, Jun, and Fosl2.
Complex roles of STAT3 in TReg cells
IL-6 also inhibits FOXP3 expression, an effect that depends on STAT3 91. Accordingly, deletion of STAT3 in T cells resulted in the clonal expansion of induced TReg cells in the setting of colitis, but not in the normal gut 76, consistent with the relief of IL-6-mediated inhibition of TReg cells. Curiously, when STAT3 was deleted only in the TReg cell population, the ability of TReg cells to constrain a pathogenic TH17 cell response was selectively impaired, whereas suppression of TH1 or TH2 cell responses remained intact 92. These data were interpreted to indicate that intrinsic activation of STAT3 in TReg cells endows these cells with the ability to specifically suppress TH17 cell responses. Gene expression analysis of STAT3-deficient TReg cells showed impaired expression of genes potentially contributing to the suppressor function of TReg cells (such as Prf1, Gzmb, Klrg1, Ccr6, Il1r1 or Il6ra). As Chip-seq datasets are generated, it will be of considerable interest to dissect how STAT3 controls the suppressor function of TReg cells that directed towards a specific TH cell subset.
STAT5 and TReg cell differentiation
Essential regulators of lymphoid development and peripheral tolerance
Stat5a and Stat5b genes are adjacent on the same chromosome in both mice and humans, have 96% similarity in sequence and have overlapping functions in diverse tissues93, 94. Similar to STAT3, germline deletion of Stat5a and Stat5b (collectively referred to as Stat5) is embryonic lethal 95, 96. The few mice that survive are extremely runted and anemic. Deletion of Stat5 has marked effects on all lymphoid lineages including T cells (thymic and peripheral), B cells and NK cells, pointing to a crucial role for STAT5 in lymphoid development. It should also be noted that the targeted disruption of the Stat5a or Stat5b genes individually yielded distinctive phenotypes, suggesting signaling mechanisms unique to each94.
Elucidation of STAT5 target genes in T cells68 and other cells has been accomplished, and these data helped to explain the role of STAT5 in TH2 cell differentiation by up-regulating expression of the IL-4 receptor68. However, there has been remarkably little analysis so far of the non-redundant roles of STAT5 in regulating the development and survival ofn T cells. Given the profound effects of STAT5 deficiency in T cells, more extensive analysis of the STAT5 ChIP-seq data sets is warranted.
The few peripheral T cells that develop in STAT5-deficient mice have an activated phenotype leading to the development of autoimmunity 97–99. One major factor underlying this autoimmune phenotype is the impaired TReg cell development in both thymus and periphery that results from deletion of Stat5 in CD4+ T cells. Indeed, it was recognized that STAT5 binds to the promoter and first intron of the Foxp3 gene, to activate transcription of this TReg cell master regulator95, 96,100. In addition, STAT5 influences the survival of TReg cells by regulating expression of the IL-2 receptor α-chain (Cd25) and the anti-apoptotic gene Bcl2. Although it is probable that STAT5 regulates many aspects of lymphoid survival, a direct comparison of STAT5 targets in TReg cells and conventional CD4+ T cells has not been carried out. Equally, a direct comparison of STAT5 and STAT3 targets in TReg cells would be particularly interesting.
Whereas IL-2-mediated activation of STAT5 is indispensable for the maintenance of TReg cells through upregulation of FOXP3 expression, other cytokines negatively influence FOXP3 expression. For example, IL-4-activated STAT6, IL-12-activated STAT4 and IL-6-activated STAT3 all decrease the expression of FOXP3 and affect chromatin modification at this locus 73, 95, 101, 102. However, the exact mechanisms by which STAT4 and STAT6 function to negatively regulate TReg cells have not been elucidated on a genome-wide scale.
Analogous to the relationship between STAT4 and STAT6 in determining TH1 versus TH2 cell differentiation, the relationship and balance between STAT5 and STAT3 seem to dictate the dichotomy of TReg cells and TH17 cells80. In addition to its role in positively regulating TReg cell function, STAT5 inhibits TH17 cell differentiation103. To address the potential mechanisms underlying this action, mapping of STAT5 targets in IL-2-activated TH17 cells was carried out by ChIP-seq. One important finding was the extensive overlap between STAT3 and STAT5 binding sites in the Il17 gene 80. It was found that STAT5 competes with STAT3 for binding to Il17 and inhibits the function of STAT3 in activating Il17 transcription. The opposing effects of STAT3 and STAT5 on Il17 transcription explain why IL-2 inhibits IL-17 production, although effects of signaling molecules other than STATs, which activated by these distinct cytokines, may also be contributors. In many other cases, STAT3 and STAT5 will work together to enhance gene expression, but given the example of Il17, the precedent is clear that these two highly related transcription factors can act in opposition. Exactly how these factors can accomplish global versus gene-specific effects warrants further investigation.
STATs and human disease
In addition to abundant data pointing to critical functions of STAT proteins in animal models, evidence of their importance in humans is quickly emerging from studies in primary immunodeficiency and in autoimmune diseases.
New insights into primary immunodeficiency disorders
Previous work has established that STAT5A/B mutations in humans are associated with impaired TReg cell function104 and that STAT1 mutations are associated with susceptibility to viral and mycobacterial infections105, 106. Furthermore, recent work has established that another classic primary immunodeficiency, Hyper-IgE or Job’s syndrome (HIES) is a result of dominant-negative mutations of STAT3 107, 108. This finding was interesting because germline deletion of Stat3 in mice is embryonic lethal. As a result, the restricted pathology seen in humans with STAT3 mutations was not anticipated; presumably, this is because the mutant allele interferes with, but does not totally abrogate, STAT3 function. An important aspect of this immunodeficiency disorder is absence of TH17 cells 109–112. A classic feature of HIES is infection without the typical signs of inflammation (i.e. redness and warmth) that results in “cold abscesses”. It is tempting to speculate that what might underlie this unique feature of HIES is the defect of TH17 cells to produce IL-17, which in turn results in the failure to recruit neutrophils to sites of infection. It remains to be elucidated how impaired function of STAT3 in other tissues contributes to the pathology seen in HIES in various tissues.
Genetic polymorphisms and human autoimmunity
Although various animal models have implicated STATs and altered cytokine signalling in autoimmunity, the issue always arises as to whether these models really mirror immunopathogenic mechanisms in humans. However, large scale genome-wide association studies (GWAS) have now provided evidence that various genes involved in cytokine signalling, and STATs in particular, are linked to the development of autoimmunity in humans (Table 2). For example, polymorphisms in STAT3 are linked to susceptibility to Crohn’s disease (a form of inflammatory bowel disease) and ankylosing spondylitis 113, 114. Equally compelling is the evidence that polymorphisms of IL23R and JAK2, both of which signal through STAT3, are linked to the same diseases 115–120, suggesting a profound involvement of the IL-23–STAT3 axis in the genesis of autoimmune diseases.
Table 2.
Genetic analysis of human autoimmune diseases showing linkages to JAK-STAT signalling pathway.
| disease | Immunological phenotype | gene | Mutation/linkage | referense |
|---|---|---|---|---|
| Hyper-IgE syndrome | Skin abscess, cystic lung infection, elevated serum IgE, impaired Th17 generation | STAT3 | Missensemutations leading to dominant negative STAT3 protein |
107 108 |
| Immune dysfunction/growth hormone insensitivity | Impaired Treg function | STAT5b | Missense mutation -A630P in SH2 domain leading to failure to respond to activation signal | 104 |
| Immune dysfunction | Susceptibility to infection | STAT1 | Missense mutation-L600P, 1757- 1758delAG, L706S leading to loss of function |
106 105 |
| Crohn’s disease (CD) | Overactive mucosal immune reaction of GI tract triggered by commensal intestinal bacteria | STAT3 JAK2 STAT4 |
11 previously reported loci + 21 additional loci linked to CD by meta-analysis A SNP (rs7574865) linked to early onset and colonic CD |
113 122 |
| Reumatoid arthritis Systemic lupus erythematosus |
Immune reaction against the lining of small joints Systemic immune reaction against own tissue and organs |
STAT4 | A SNP(re7574865)linked to both RA and SLE | 121 |
| Primary Sjögren’s Syndrome(pSS) | Inflammation of salivary and lacrimal glands leading to a dry mouth and dry eyes | STAT4 | A SNP(rs7574865) linked to pSS | 123 |
Components of the JAK–STAT signaling pathway have been identified as causal genes for autoimmune diseases and have also been implicated in genetic linkage studies as having statistically significant differences between patients and controls.
In multiple studies, a variant allele of STAT4 has been found to be associated with increased risk of developing systemic lupus erythematosus (SLE), rheumatoid arthritis, Sjogen’s syndrome and Crohn’s disease 121–123. The connection between STAT4 and SLE is perhaps unexpected insofar as this disease is not a prototypical TH1 cell-mediated disease. However, it is worth noting that STAT4 can be activated by type I IFNs124, and an important aspect of the pathogenesis of SLE is the “interferon signature” 125. Polymorphisms in TYK2, whose gene product is activated by IFNs, IL-12 and other cytokines, have also been reported to be associated with SLE 126, providing further evidence in support of the importance of STAT4 in the pathophysiology of this disorder. As the STAT4 polymorphisms do not fall within the coding region of the gene, they presumably influence the level of gene expression, but clearly much more work is required to confirm this hypothesis.
Concluding remarks and future directions
The powerful genome-wide approaches now available to researchers have enabled a comprehensive evaluation of the role of individual STAT proteins in specifying TH cell lineages and they provide a quantitative determination of the target genes that are mobilized during the process of TH cell differentiation. These findings have established that STATs have multiple roles during the initiation stage as well as the maintenance stage of a TH cell fate decision. For the latter case, a key role of STATs involves the induction and/or maintenance of epigenetic patterns on target loci. STAT proteins induce both permissive and repressive epigenetic patterns. Although a particular STAT can be assigned to each TH cell lineage as a dominant regulatory factor, it is clear that this is an overly simplistic view of defining TH cell lineages. Emerging evidence points to a functional network, with STATs working cooperatively and in opposition with other transcription factors to ensure the desired balance between different T cell fates, and in certain cases even to promote phenotypical plasticity.
Fortunately, we now have genome-wide approaches to define the breadth of transcription factor action. In addition, we also have the ability to carry out many chromatin-related assays on a genome-wide scale to examine the activity of genomic regions 127 through common chromatin signatures 128, and to determine the state of dynamic genomic organization (Figure 4). In particular, the extensive coverage of the genome afforded by next generation sequencing offers the possibility of exploring so called “gene desert” or intergenic regions for distal enhancers and other types of regulatory element21, 24, 129. This is an exciting opportunity to analyse previously unexplored regions of the genome, and in fact, recent reports have shown that the patterns of distal enhancers are quite unique and different between different cell types 130. The challenge now of course is to understand how “master regulators” of cell fate and other transcription factors, such as STATs, contribute to the activity of distal enhancers in a manner that creates cell identity21, 130. It is quite possible that some of the polymorphisms that have been linked to autoimmune diseases reside in enhancer regions of the genome 131 that are crucial for regulating tissue-specific patterns of gene expression from a distance.
Figure 4. Markers of genomic organization to define activities of chromosome regions.

Genomic organization encompassing the interferon-g (Ifng) locus in T helper (TH) cells. In TH1 cells, in which the Ifng locus is actively transcribed in a signal transducer and activator of transcription 4 (STAT4)-dependent manner, the promoter is marked by permissive histone 3 lysine 4 trimethylation (H3K4me3) and STAT4 binding, and the gene body is marked by permissive H3K4me3 and H3K36me3 marks. One of the distal enhancer elements (shaded in gray) is marked by H3K4me1 and STAT4 binding in TH1 cells and by repressive H3K27me3 in TH2 cells. Further 5′ upstream of the Ifng locus, an insulator site marked by CTCF binding is located and all permissive histone marks and DNase hypersensitivity sites are restricted beyond that point.
Components of the JAK–STAT signaling pathway have been identified as causal genes for autoimmune diseases and have also been implicated in genetic linkage studies as having statistically significant differences between patients and controls.
Equally, the entire notion of the epigenome is in the midst of a not so quiet revolution27, 132. Increasingly, “epigenetics” is being viewed as an extension of signal transduction. Nonetheless, it is certainly not clear how all of the components of epigenetic information are linked to each other, to signalling and to transcription factor binding. Indeed, we are really in our infancy of understanding how signal transduction and nucleosome biology relate. Because the STAT pathway provides a rapid means of transmitting signals from the extracellular environment to the nucleus, investigating how cytokines drive TH cell specification will provide remarkable opportunities to link signal transduction, transcription factor binding, nucleosome biology and gene expression. Hopefully, we will witness valuable new information about gene regulation coming from systematic database analysis of STAT-mediated cytokine signalling in differentiating T cells.
Glossary
- Next generation sequencing
High throughput sequencing methods that produce rapid, inexpensive, accurate sequencing data that can cover entire genomes. Based on different chemistries, several different platforms are available including: Illumina/GA, Roche/454, ABI/SOLID and Helicos/HeliScope
- Chromatin immunoprecipitation
A technique for the detection of proteins bound to specific regions of chromatin. These assays involve chemically crosslinking proteins bound to the DNA sequences, followed by immunoprecipitation with an antibody that is specific for the crosslinked protein
- Epigenome and Epigenetic regulation
These terms refer to the heritable, but potentially reversible, states of gene activity that are imposed by the structure of chromatin such as covalent modifications of DNA or of nucleosomal histones. The epigenome pertains to the aspects of heritable cellular phenotype that is not explained by DNA sequence
- ChIP-seq
A technique in which chromatin immunoprecipitation is followed by high throughput sequencing to generate maps the genome-wide distribution of protein-DNA interaction. This technique can be used to measure transcription factor binding or histone modifications
- Nucleosome
A nucleosome consists of histone protein core and a segment of DNA wrapped around it. It is the minimum unit to make up chromosome
- ChIP-on chip
The term refers to a technique that combines chromatin immunoprecipitation (“ChIP”) with microarray technology (“chip”) to investigate DNA-protein interaction in vivo on a genome-wide basis
- Enhancer element
A control element in DNA to which regulatory proteins bind and influence the rate of gene transcription of the associated gene(s). Enhancers function in an orientation- and position-independent manner so that they can function either from upstream or downstream of the associated gene, or in an intron
- Genome-wide association study (GWAS)
GWAS refers to a study in which genomewide genetic variation is linked to a particular phenotype, most often a clinical disorder by applying high-throughput genotyping techniques to profile single-nucleotide polymorphisms (SNPs) of controls vs. patient
References
- 1.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140:845–58. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 3.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 4.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Veldhoen M, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–6. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 6.Locksley RM. Nine lives: plasticity among T helper cell subsets. J Exp Med. 2009;206:1643–6. doi: 10.1084/jem.20091442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17–31. doi: 10.1007/s00281-009-0188-x. [DOI] [PubMed] [Google Scholar]
- 9.Yu D, Batten M, Mackay CR, King C. Lineage specification and heterogeneity of T follicular helper cells. Curr Opin Immunol. 2009;21:619–25. doi: 10.1016/j.coi.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 10.Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity. 2009;30:324–35. doi: 10.1016/j.immuni.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 12.Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993;261:1744–6. doi: 10.1126/science.7690989. One of the first reports that defined the importance of STAT1 in interferon signaling. [DOI] [PubMed] [Google Scholar]
- 13.Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–51. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 14.Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 15.Hoey T, et al. Distinct requirements for the naturally occurring splice forms Stat4alpha and Stat4beta in IL-12 responses. EMBO J. 2003;22:4237–48. doi: 10.1093/emboj/cdg393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, et al. Identification of novel IL-4/Stat6-regulated genes in T lymphocytes. J Immunol. 2003;171:3627–35. doi: 10.4049/jimmunol.171.7.3627. [DOI] [PubMed] [Google Scholar]
- 17.Lund RJ, Chen Z, Scheinin J, Lahesmaa R. Early target genes of IL-12 and STAT4 signaling in th cells. J Immunol. 2004;172:6775–82. doi: 10.4049/jimmunol.172.11.6775. [DOI] [PubMed] [Google Scholar]
- 18.Tuomela S, Rautajoki KJ, Moulder R, Nyman TA, Lahesmaa R. Identification of novel Stat6 regulated proteins in IL-4-treated mouse lymphocytes. Proteomics. 2009;9:1087–98. doi: 10.1002/pmic.200800161. [DOI] [PubMed] [Google Scholar]
- 19.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. Basic chemistries employed by next generation sequencing platforms on market are compared and discussed. [DOI] [PubMed] [Google Scholar]
- 20.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 21.Natoli G. Maintaining cell identity through global control of genomic organization. Immunity. 2010;33:12–24. doi: 10.1016/j.immuni.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 24.Heintzman ND, Ren B. Finding distal regulatory elements in the human genome. Curr Opin Genet Dev. 2009;19:541–9. doi: 10.1016/j.gde.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. One of the first epigenome mapping studies to report genome wide distribution of 20 histone modification marks. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohammad HP, Baylin SB. Linking cell signaling and the epigenetic machinery. Nat Biotechnol. 2010;28:1033–8. doi: 10.1038/nbt1010-1033. [DOI] [PubMed] [Google Scholar]
- 28.Liefke R, et al. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev. 2010;24:590–601. doi: 10.1101/gad.563210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, Edwards JP, Mosser DM. Dynamic and transient remodeling of the macrophage IL-10 promoter during transcription. J Immunol. 2006;177:1282–8. doi: 10.4049/jimmunol.177.2.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–7. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hegazy AN, et al. Interferons direct Th2 cell reprogramming to generate a stable GATA-3(+)T-bet(+) cell subset with combined Th2 and Th1 cell functions. Immunity. 2010;32:116–28. doi: 10.1016/j.immuni.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Wei G, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–67. doi: 10.1016/j.immuni.2008.12.009. The presence of bivalent epigenetic marks on transcription factors noted in this study helps explain emerging views of T helper cell plasticity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–81. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev. 2009;19:127–34. doi: 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–7. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 36.Robertson AG, et al. Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding. Genome Res. 2008;18:1906–17. doi: 10.1101/gr.078519.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Auerbach RK, et al. Mapping accessible chromatin regions using Sono-Seq. Proc Natl Acad Sci U S A. 2009;106:14926–31. doi: 10.1073/pnas.0905443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaplan MH. STAT4: a critical regulator of inflammation in vivo. Immunol Res. 2005;31:231–42. doi: 10.1385/IR:31:3:231. An excellent review that summarizes the function of STAT4 in immune responses. [DOI] [PubMed] [Google Scholar]
- 39.Uemura A, et al. Natural killer cell is a major producer of interferon gamma that is critical for the IL-12-induced anti-tumor effect in mice. Cancer Immunol Immunother. 2010;59:453–63. doi: 10.1007/s00262-009-0764-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyagi T, et al. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. 2007;204:2383–96. doi: 10.1084/jem.20070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Good SR, et al. Temporal induction pattern of STAT4 target genes defines potential for Th1 lineage-specific programming. J Immunol. 2009;183:3839–47. doi: 10.4049/jimmunol.0901411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei L, et al. Discrete Roles of STAT4 and STAT6 Transcription Factors in Tuning Epigenetic Modifications and Transcription during T Helper Cell Differentiation. Immunity. 2010;32:840–851. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–81. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 44.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–3. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 45.Zhou L, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 46.Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605–10. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spolski R, Leonard WJ. Interleukin-21: Basic Biology and Implications for Cancer and Autoimmunity. Annu Rev Immunol. 2007;26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316. [DOI] [PubMed] [Google Scholar]
- 48.Schmitt N, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity. 2009;31:158–69. doi: 10.1016/j.immuni.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gil MP, et al. Biologic consequences of Stat1-independent IFN signaling. Proc Natl Acad Sci U S A. 2001;98:6680–5. doi: 10.1073/pnas.111163898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramana CV, et al. Stat1-independent regulation of gene expression in response to IFN-gamma. Proc Natl Acad Sci U S A. 2001;98:6674–9. doi: 10.1073/pnas.111164198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walker SR, Nelson EA, Frank DA. STAT5 represses BCL6 expression by binding to a regulatory region frequently mutated in lymphomas. Oncogene. 2007;26:224–33. doi: 10.1038/sj.onc.1209775. [DOI] [PubMed] [Google Scholar]
- 52.Tran TH, et al. Prolactin inhibits BCL6 expression in breast cancer through a Stat5a-dependent mechanism. Cancer Res. 2010;70:1711–21. doi: 10.1158/0008-5472.CAN-09-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q, et al. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:6948–53. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi S, et al. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat Cell Biol. 2008;10:489–96. doi: 10.1038/ncb1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–9. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 56.Shimoda K, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–3. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 57.Takeda K, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–30. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 58.Zhu J, Guo L, Watson CJ, Hu-Li J, Paul WE. Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J Immunol. 2001;166:7276–81. doi: 10.4049/jimmunol.166.12.7276. [DOI] [PubMed] [Google Scholar]
- 59.Elo LL, et al. Genome-wide Profiling of Interleukin-4 and STAT6 Transcription Factor Regulation of Human Th2 Cell Programming. Immunity. 2010;32:852–862. doi: 10.1016/j.immuni.2010.06.011. The first report to present genome wide binding of STAT6 in human Th2 cells. [DOI] [PubMed] [Google Scholar]
- 60.Collins A, Littman DR, Taniuchi I. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat Rev Immunol. 2009;9:106–15. doi: 10.1038/nri2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Komine O, et al. The Runx1 transcription factor inhibits the differentiation of naive CD4+ T cells into the Th2 lineage by repressing GATA3 expression. J Exp Med. 2003;198:51–61. doi: 10.1084/jem.20021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naoe Y, et al. Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med. 2007;204:1749–55. doi: 10.1084/jem.20062456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitoh A, et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity. 2009;31:609–20. doi: 10.1016/j.immuni.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 64.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mole DR, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–75. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schraml BU, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–9. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Betz BC, et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med. 2010;207:933–42. doi: 10.1084/jem.20091548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liao W, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol. 2008;9:1288–96. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takatori H, et al. Stat5a inhibits IL-12-induced Th1 cell differentiation through the induction of suppressor of cytokine signaling 3 expression. J Immunol. 2005;174:4105–12. doi: 10.4049/jimmunol.174.7.4105. [DOI] [PubMed] [Google Scholar]
- 70.Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–48. doi: 10.1016/s1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
- 71.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 72.Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–11. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- 73.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–63. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 74.Chen Z, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–42. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mathur AN, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–7. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 76.Durant L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–15. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nishihara M, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol. 2007;19:695–702. doi: 10.1093/intimm/dxm045. [DOI] [PubMed] [Google Scholar]
- 78.Harris TJ, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–7. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 79.Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol. 2008;180:6070–6. doi: 10.4049/jimmunol.180.9.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang X, Ghoreschi K, O’Shea J J, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAt5. Nat Immunol. 2011 doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem. 2007;282:5969–72. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- 82.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 83.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Veldhoen M, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 85.Brustle A, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8:958–66. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- 86.Bauquet AT, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. 2009;10:167–75. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghoreschi K, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–71. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McGeachy MJ, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–24. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kwon H, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. 2009;31:941–52. doi: 10.1016/j.immuni.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takeda K, et al. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–60. [PubMed] [Google Scholar]
- 91.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 92.Chaudhry A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–91. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995;92:8831–5. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grimley PM, Dong F, Rui H. Stat5a and Stat5b: fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 1999;10:131–57. doi: 10.1016/s1359-6101(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 95.Yao Z, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–75. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–90. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 97.Nosaka T, et al. Defective lymphoid development in mice lacking Jak3. Science. 1995;270:800–2. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 98.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270:794–7. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 99.Antov A, Yang L, Vig M, Baltimore D, Van Parijs L. Essential role for STAT5 signaling in CD25+CD4+ regulatory T cell homeostasis and the maintenance of self-tolerance. J Immunol. 2003;171:3435–41. doi: 10.4049/jimmunol.171.7.3435. [DOI] [PubMed] [Google Scholar]
- 100.Zorn E, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–9. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Takaki H, et al. STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem. 2008;283:14955–62. doi: 10.1074/jbc.M801123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.O’Malley JT, et al. Signal transducer and activator of transcription 4 limits the development of adaptive regulatory T cells. Immunology. 2009;127:587–95. doi: 10.1111/j.1365-2567.2008.03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 104.Cohen AC, et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177:2770–4. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 105.Dupuis S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 106.Dupuis S, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–3. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 107.Holland SM, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. These two studies (107 and 108) were the first to describe unexpected dominant negative phenotype of missense mutations in STAT3 in patients. [DOI] [PubMed] [Google Scholar]
- 108.Minegishi Y, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 109.Milner JD, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van de Veerdonk FL, et al. Milder clinical hyperimmunoglobulin E syndrome phenotype is associated with partial interleukin-17 deficiency. Clin Exp Immunol. 2010;159:57–64. doi: 10.1111/j.1365-2249.2009.04043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ma CS, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–7. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Minegishi Y, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. 2009;206:1291–301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Danoy P, et al. Association of variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with Crohn’s disease. PLoS Genet. 2010;6:e1001195. doi: 10.1371/journal.pgen.1001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duerr RH, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cargill M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–90. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Burton PR, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–37. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Filer C, et al. Investigation of association of the IL12B and IL23R genes with psoriatic arthritis. Arthritis Rheum. 2008;58:3705–9. doi: 10.1002/art.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Remmers EF, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat Genet. 2010;42:698–702. doi: 10.1038/ng.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reveille JD, et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet. 2010;42:123–7. doi: 10.1038/ng.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Remmers EF, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–86. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Glas J, et al. Evidence for STAT4 as a common autoimmune gene: rs7574865 is associated with colonic Crohn’s disease and early disease onset. PLoS One. 2010;5:e10373. doi: 10.1371/journal.pone.0010373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Korman BD, et al. Variant form of STAT4 is associated with primary Sjogren’s syndrome. Genes Immun. 2008;9:267–70. doi: 10.1038/gene.2008.1. [DOI] [PubMed] [Google Scholar]
- 124.Cho SS, et al. Activation of STAT4 by IL-12 and IFN-alpha: evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol. 1996;157:4781–9. [PubMed] [Google Scholar]
- 125.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hellquist A, et al. Evidence for genetic association and interaction between the TYK2 and IRF5 genes in systemic lupus erythematosus. J Rheumatol. 2009;36:1631–8. doi: 10.3899/jrheum.081160. [DOI] [PubMed] [Google Scholar]
- 127.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. Basic concepts and application of next generation sequencing techniques are reviewed by experts in the field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hon G, Ren B, Wang W. ChromaSig: a probabilistic approach to finding common chromatin signatures in the human genome. PLoS Comput Biol. 2008;4:e1000201. doi: 10.1371/journal.pcbi.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. This study indicates that the location of tissue-specific distal enhancers can be accurately predicted by mapping genome-wide p300 binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ghisletti S, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–28. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 131.Kasowski M, et al. Variation in transcription factor binding among humans. Science. 2010;328:232–5. doi: 10.1126/science.1183621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–6. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Blat Y, Kleckner N. Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell. 1999;98:249–59. doi: 10.1016/s0092-8674(00)81019-3. [DOI] [PubMed] [Google Scholar]
- 134.Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–9. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 135.Gupta PK. Single-molecule DNA sequencing technologies for future genomics research. Trends Biotechnol. 2008;26:602–11. doi: 10.1016/j.tibtech.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 136.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schones DE, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–98. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Crawford GE, et al. Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites. Proc Natl Acad Sci U S A. 2004;101:992–7. doi: 10.1073/pnas.0307540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sabo PJ, et al. Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries. Proc Natl Acad Sci U S A. 2004;101:4537–42. doi: 10.1073/pnas.0400678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ji H, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kuchen S, et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010;32:828–39. doi: 10.1016/j.immuni.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guttman M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–7. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rozowsky J, et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol. 2009;27:66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–17. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]