

Abstract
Signal transducer and activator of transcription 3 (STAT3) is activated in human breast cancer and other malignancies. Mucin 1 (MUC1) is a heterodimeric cell surface glycoprotein that is overexpressed in human carcinomas and, like STAT3, promotes cell survival and induces transformation. Here, we showed that in breast cancer cells, the MUC1 carboxyl-terminal receptor subunit (MUC1-C) associated with the gp130–Janus-activated kinase 1 (JAK1)–STAT3 complex. The MUC1-C cytoplasmic domain interacted directly with JAK1 and STAT3, and MUC1-C was necessary for JAK1-mediated STAT3 activation. In turn, MUC1-C and activated STAT3 occupied the promoter of MUC1, and MUC1-C contributed to STAT3-mediated activation of MUC1 transcription. The MUC1-C inhibitor GO-201 blocked the MUC1-C interaction with STAT3, thereby decreasing MUC1-C and STAT3 occupancy on the MUC1 and STAT3 promoters and activation of STAT3 target genes, including MUC1 itself. These findings indicate that MUC1-C promotes STAT3 activation and that MUC1-C and STAT3 function in an autoinductive lopp that may play a role in cancer cell survival.
Keywords: MUC1, STAT3, JAK1, IL-6, transactivation, targeted therapy
Introduction
Members of the signal transducer and activator of transcription (STAT) family have been implicated in malignant transformation, as well as in tumor cell survival, invasion, and metastasis (1). The STAT3 transcription factor, which acts as an effector of the interleukin-6 (IL-6) inflammatory response (2), is activated by Janus-activated kinase (JAK)-1 phosphorylation of the IL-6 receptor and the subsequent recruitment and phosphorylation of STAT3 on a conserved tyrosine at position 705 (Tyr-705)(1). Phosphorylated STAT3 (p-STAT3) undergoes dimerization and translocates to the nucleus to activate the transcription of target genes encoding regulators of cell cycle progression (such as cyclin D1 and c-Myc) and inhibitors of apoptosis (such as survivin and Bcl-xL) (3,4). Constitutively activated STAT3 induces transformation (5) and STAT3 activation has been detected in various carcinomas and hematologic malignancies (6-8), consistent with its involvement in the transcription of genes that control cell proliferation and survival.
The unphosphorylated form of STAT3 has also been implicated in transcriptional activation and linked to oncogenesis (9,10). The gene encoding STAT3 is itself activated in response to IL-6-induced STAT3 phosphorylation (35) and the consequent increase in unphosphorylated STAT3 induces the transcription of additional genes, in part through STAT3 binding to the RelA subunit of the transcription factor nuclear factor κB (NF-κB) (10). Small molecule inhibitors of JAK-1 and STAT3 have anti-cancer activity in vitro and in animal models (11-14). Together, these findings support a role for STAT3 signaling in tumorigenesis.
The mucin family of glycoproteins function in protecting the apical surfaces of epithelial cells that interface with the external environment and line ducts (16). The mucin 1 (MUC1) transmembrane oncoprotein is overexpressed in various human carcinomas and certain hematologic malignancies (16). Indeed, MUC1 overexpression is sufficient to induce transformation (17,18). MUC1 is translated from a single transcript into a polypeptide that undergoes autocleavage into two subunits, which in turn form a heterodimer (16). MUC1 thus consists of an extracellular N-terminal mucin subunit (MUC1-N) that forms a complex with the transmembrane C-terminal subunit (MUC1-C) (19,20). MUC1-C contains a 58 amino acid extracellular domain that interacts with galectin-3 and thereby forms complexes with EGFR (21) and a cytoplasmic domain consisting of 72 amino acids including a Cys-Glu-Cys (CQC) motif necessary for MUC1-C oligomerization and function (22). The MUC1-C cytoplasmic domain (MUC1-CD) also contains sites that function as substrates for phosphorylation by EGFR, c-Met, c-Src, c-Abl, glycogen synthase kinase 3β, and protein kinase C (16). MUC1-CD binds directly to the Wnt effector β-catenin and contributes to activation of the Wnt pathway (16). c-Src phosphorylation of MUC1-CD increases the binding of MUC1-CD to β-catenin (16). MUC1-CD also interacts with the inhibitor of κB kinase (IKK) complex and RelA, and contributes to activation of the NF-κB pathway (23,24). MUC1-C thus has the potential for multiple functions in cell signaling and gene regulation as an adaptor or scaffold for interactions with client proteins that, in certain settings, are regulated by MUC1-CD phosphorylation (16).
With overexpression, MUC1-C accumulates in the cytoplasm and is targeted to the nucleus by a mechanism involving importin-α and Nup62 (22,25,26) and to the mitochondria by a mechanism involving the chaperones HSP70 and HSP90 (16,27,28). In the nucleus, MUC1-C interacts with various transcription factors, including p53, and binds to the promoters of their target genes (16,25,26). As a component of the mitochondrial outer membrane, MUC1-C attenuates loss of the transmembrane potential in response to genotoxic stress (16,27). Consistent with these findings, MUC1-C confers resistance to death in the cellular response to DNA damage, reactive oxygen species, hypoxia, or activation of the death receptor superfamily (16). Thus, like STAT3, MUC1-C has been linked to pathways that lead to malignant transformation and attenuation of cell death.
Here, we show that the MUC1-CD binds directly to JAK1 and STAT3 and that MUC1-C promotes JAK1-mediated phosphorylation of STAT3. Activated STAT3 induces expression of the MUC1 gene and, in an autoinductive loop, MUC1-C contributes to STAT3-mediated activation of MUC1 and other STAT3 target genes.
Results
MUC1-C associates with the gp130-JAK1-STAT3 complex
IL-6 and certain other inflammatory cytokines signal through a complex of the common receptor subunit gp130, JAK1, and STAT3. Immunoblot analyses of anti-MUC1-C precipitates (immunoprecipitates obtained with antibody directed against MUC1-C) from lysates of ZR-75-1 breast cancer cells, which overexpress endogenous MUC1, revealed that MUC1-C associates with gp130 (Fig. 1A).
Fig. 1. MUC1-C interacts with the gp130-JAK-STAT3 complex.
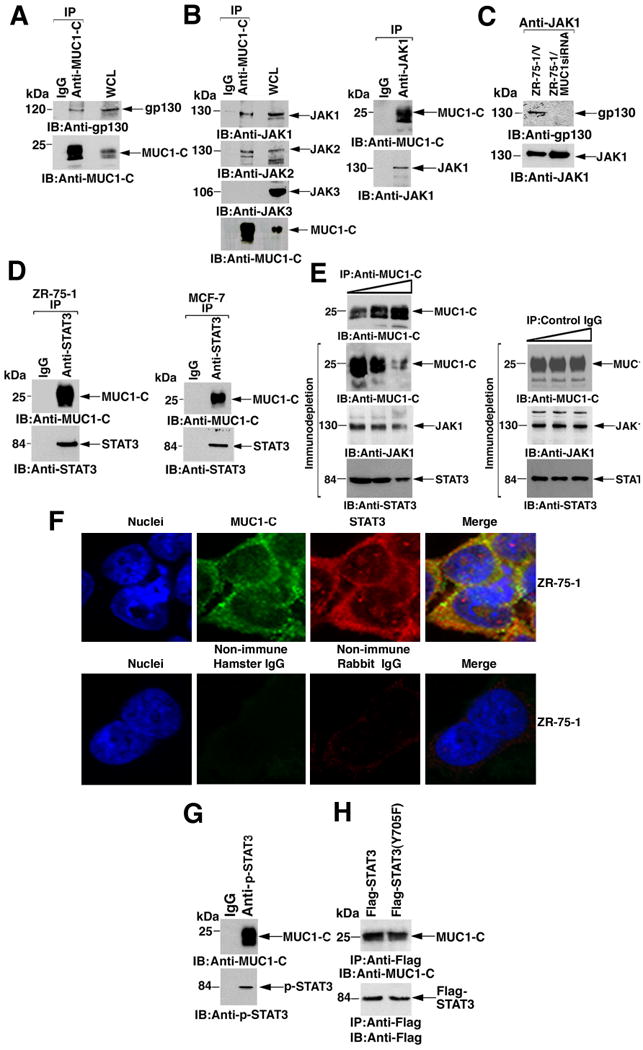
A and B. Lysates from ZR-75-1 cells were immunoprecipitated with control IgG or anti-MUC1-C (A and B, left). Lysate from ZR-75-1 cells immunoprecipitated with control IgG or anti-JAK1 (B, right). The precipitates were immunoblotted with the indicated antibodies. WCL: whole cell lysate. C. Lysates from ZR-75-1/vector and ZR-75-1/MUC1siRNA cells were immunoprecipitated with anti-JAK1 and the precipitates immunoblotted as indicated. D. Lysates from ZR-75-1 (left) and MCF-7 (right) cells were immunoprecipitated with control IgG or anti-STAT3 and the precipitates immunoblotted as indicated. E. ZR-75-1 cell lysates were immunoprecipitated with increasing amounts of anti-MUC1-C or control IgG. Immunoblot analysis of the anti-MUC1-C precipitates confirmed the precipitation of MUC1-C (left, upper panel). The anti-MUC1-C (left) and IgG (right) immunodepleted supernatants were immunoblotted with the indicated antibodies. Densitometric scanning of the signals demonstrated decreases (mean and range from two independent experiments) in JAK1 and STAT3 from lane 1 to lane 3 of 31.2% (range 27.4 to 35.0%) and 41.9% (range 40.2 to 43.5%), respectively. F. ZR-75-1 cells were fixed and stained with anti-MUC1-C (green) and anti-STAT3 (red) (upper panels). As controls, the cells were stained with non-immune hamster and rabbit IgGs (lower panels). Nuclei were stained with DAPI (blue). G. ZR-75-1 cell lysates were immunoprecipitated with a control IgG or anti-p-STAT3 and the precipitates immunoblotted as indicated. H. MUC1-null 293 cells were transfected to express MUC1 and Flag-STAT3 or Flag-STAT3(Y705F). Lysates prepared at 48 h after transfection were immunoprecipitated with anti-Flag. The precipitates were immunoblotted with anti-MUC1-C and anti-Flag.
Analysis of the anti-MUC1-C precipitates showed that MUC1-C also formed complexes with JAK1 and JAK2, but not with JAK3 (Fig. 1B, left). MUC1-C association with JAK1 was confirmed in experiments in which anti-JAK1 precipitates were immunoblotted with anti-MUC1-C (Fig. 1B, right), as was the association of JAK1 with gp130 (Fig. 1C). However, gp130-JAK1 complexes were not detectable in ZR-75-1 cells that had been stably silenced for MUC1 with a short interfering RNA directed against MUC1 (ZR-75-1/MUC1siRNA cells) (24) (Fig. 1C), indicating that MUC1-C contributes to formation of the gp130-JAK1 complex.
Analyses of anti-STAT3 precipitates from ZR-75-1 cells demonstrated that MUC1-C also associated with STAT3 (Fig. 1D, left). Similar results were obtained when MUC1-C-STAT3 co-immunoprecipitation studies were performed on MCF-7 breast cancer cells, which also overexpress endogenous MUC1 (Fig. 1D, right). Consistent with the MUC1-C-JAK1-STAT3 association, incubation of ZR-75-1 cell lysates with increasing amounts of anti-MUC1-C, but not a control IgG, led to immunodepletion of JAK1 and STAT3 (Fig. 1E). Confocal microscopic analysis of ZR-75-1 cells showed colocalization of MUC1-C and STAT3 at the cell membrane (Fig. 1F). Immunoprecipitation analyses demonstrated that p-STAT3 formed complexes with MUC1-C (Fig. 1G), indicating that MUC1-C associates with the activated form of STAT3. To determine whether association with MUC1-C depends on STAT3 phosphorylation, we coexpressed MUC1 with Flag-tagged wild-type STAT3 and a mutant form of STAT3 in which Tyr-705 is substituted with phenylalanine(Y705F). Coimmunoprecipitation studies showed that the MUC1-C-STAT3 interaction did not depend on the availability of Tyr-705 for phosphorylation (Fig. 1H).
MUC1-C interacts directly with JAK1 and STAT3
In vitro glutathione-S-transferase (GST) pull-down assays indicated that the purified MUC1-CD (Fig. 2A) bound the JAK1 kinase domain (Fig. 2B, left). In contrast, there was no detectable binding of the JAK2 kinase domain (JAK2-KD) to MUC1-CD (Fig. 2B, right). In the reciprocal experiment, GST-MUC1-CD bound to purified JAK1-KD (Fig. 2C). Experiments with truncated constructs revealed that this interaction was mediated by MUC1-CD(1-45) and not MUC1-CD(46-72) (Fig. 2C, left). Substitution of the cytosines in the MUC1-CD CQC motif (amino acids 1-3) with alanines abrogated MUC1-CD binding to JAK1-KD, whereas mutation of the serine-rich motif (SRM; SAGNGGSSLS, amino acids 50-59) to AAGNGGAAAA (mSRM) (24) had little effect (Fig. 2C, right). Incubation of ZR-75-1 cell lysates with GST or GST-MUC1-CD indicated that MUC1-CD is sufficient for binding with STAT3 (Fig. 2D). Binding studies performed with purified recombinant STAT3 revealed that GST-MUC1-CD, but not GST, bound directly to STAT3 (Fig. 2E, left). In contrast to its interaction with JAK1, MUC1-CD binding to STAT3 involved MUC1-CD(46-72), but not MUC1-CD(1-45) (Fig. 2E, left). Moreover, mutation of the MUC1-CD CQC motif to AQA had no apparent effect on binding to STAT3 (Fig. 2E, right), whereas mutation of the MUC1-CD SRM markedly decreased STAT3 binding (Fig. 2E, right).
Figure 2. MUC1-C binds directly to JAK1 and STAT3.
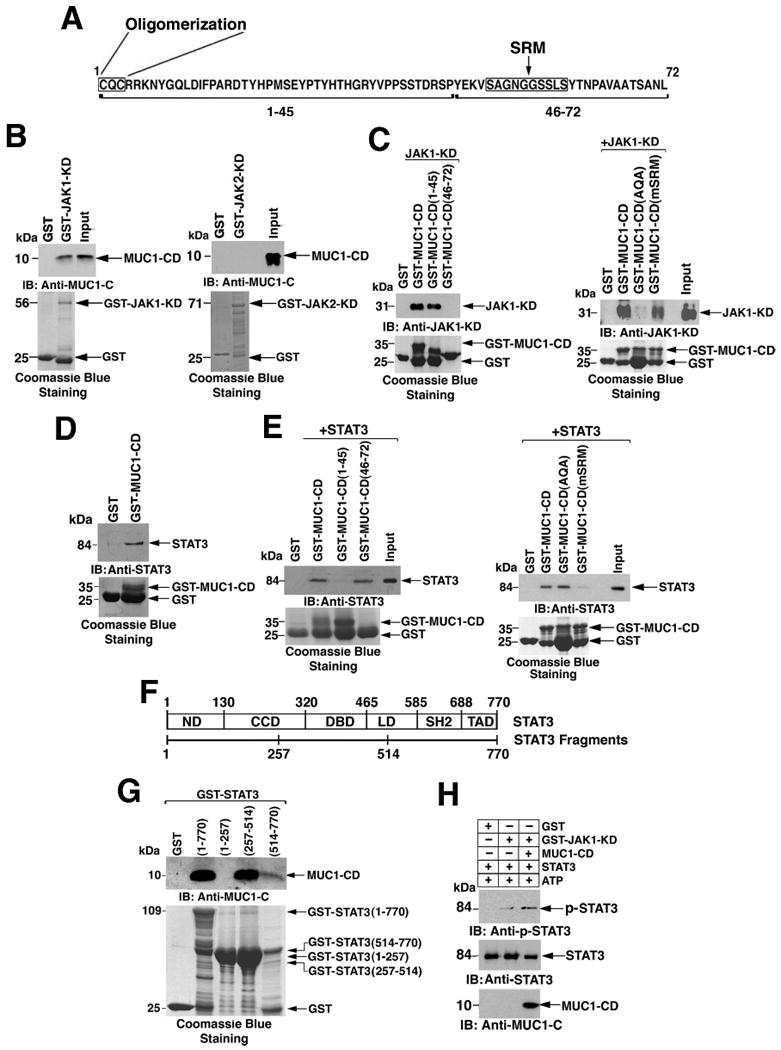
A. Amino acid sequence of the MUC1-CD with the oligomerization motif (CQC) and serine-rich motif (SRM) indicated. B. GST and GST-JAK1-KD (left) or GST-JAK2-KD (right) were incubated with purified MUC1-CD and the adsorbates immunoblotted with anti-MUC1-C. Loading of the GST, GST-JAK1-KD, and JAK2-KD proteins in the binding reactions was assessed by Coomassie blue staining. C. GST, GST-MUC1-CD, GST-MUC1-CD(1-45), GST-MUC1-CD(46-72) (left), GST-MUC1-CD(AQA), and GST-MUC1-CD(mSRM) (right) were incubated with purified JAK1-KD and the adsorbates immunoblotted with anti-JAK1. Input of the GST and GST-MUC1-CD fusion proteins was assessed by Coomassie blue staining. D. Lysates from ZR-75-1 cells were incubated with GST and GST-MUC1-CD bound to glutathione beads. The adsorbates were immunoblotted with anti-STAT3. Input of the GST and GST-MUC1-CD proteins was assessed by Coomassie blue staining. E. GST, GST-MUC1-CD, GST-MUC1-CD(1-45), and GST-MUC1-CD(46-72) (left), GST-MUC1-CD(AQA), and GST-MUC1-CD(mSRM) (right) were incubated with purified recombinant STAT3. The adsorbates were immunoblotted with anti-STAT3. Loading of the GST and GST-MUC1-CD fusion proteins was assessed by Coomassie blue staining. F. Structure of STAT3. ND, N-terminal domain; CCD, coiled coil domain; DBD, DNA binding domain; LD, linker domain; SH2, SH2 domain; TAD, transactivation domain. G. GST, GST-STAT3(full length; amino acids 1-770), GST-STAT3(1-257), GST-STAT3(257-514) and GST-STAT3(514-770) bound to glutathione beads were incubated with purified MUC1-CD. Adsorbates were immunoblotted with anti-MUC1-C. Loading of the GST and GST-STAT3 fusion proteins was assessed by Coomassie blue staining. H. GST and GST-JAK1-KD were incubated with STAT3 in kinase buffer containing ATP in the presence and absence of MUC1-CD. The reaction products were analyzed by immunoblotting with the indicated antibodies.
STAT3 has a dimerization domain at the N-terminus, a central DNA binding domain (DBD), and a C-terminal transactivation domain (Fig. 2F). Incubation of MUC1-CD with a construct consisting of GST and full-length STAT3 [GST-STAT3(1-770)] confirmed its direct interaction with STAT3 (Fig. 2G). To identify the responsible STAT3 region, we analyzed MUC1-CD binding to GST constructs containing STAT3(1-257), STAT3(257-514), or STAT3(514-770) deletion mutants. GST-STAT3(1-257) includes the N-terminus and part of the coiled-coil domain (Fig. 2F). GST-STAT3(257-514) includes the remainder of the coiled-coil domain and the DBD, and GST-STAT3(514-770) contains the C-terminal transactivation domain (Fig. 2F). The binding studies showed that MUC1-CD interacted predominantly with GST-STAT3(257-514) (Fig. 2G), indicating that MUC1-CD(46-72) binds to the STAT3 region that includes the DBD. Our data also indicate that different regions of the MUC1-CD form complexes with JAK1 and STAT3, suggesting that MUC1-C may facilitate the formation of JAK1-STAT3 complexes. Indeed, incubation of JAK1-KD and STAT3 in the absence or presence of MUC1-CD demonstrated that MUC1-CD promotes JAK1-KD-mediated phosphorylation of STAT3 (Fig. 2H).
MUC1-C promotes JAK1- but not JAK2-mediated STAT3 phosphorylation
To further assess the functional significance of the MUC1-C-JAK1-STAT3 interaction, we analyzed ZR-75-1 and MCF-7 cells that had been stably silenced for MUC1-C. ZR-75-1/MUC1siRNA cells showed a decrease in the abundance of p-STAT3 (Fig. 3A, left). Similar results were obtained with MUC1-C silencing in MCF-7 cells, indicating that MUC1-C promotes STAT3 phosphorylation (Fig. 3A, left). Consistent with these results, silencing MUC1-C was also associated with a decrease in accumulation of STAT3 in the nucleus (Fig. 3A, right). Primary breast cancer cells obtained from a malignant pleural effusion expressed MUC1-C and p-STAT3 (Fig. 3B) and transient silencing of MUC1-C was associated with decreased phosphorylation of STAT3 in these cells (Fig. 3B).
Figure 3. MUC1-C promotes JAK1 and STAT3 phosphorylation.
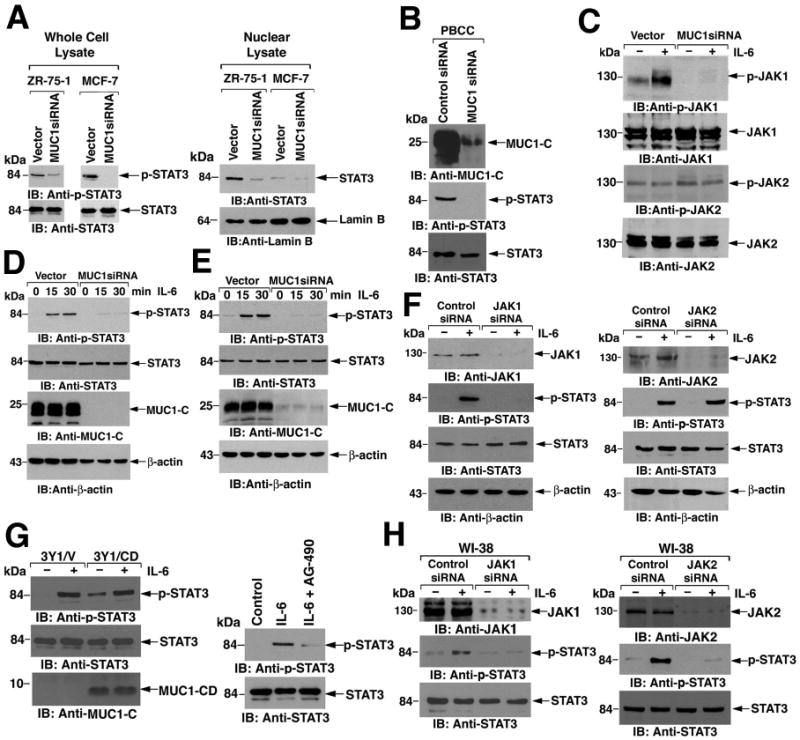
A. Total cell lysates (left) and nuclear lysates (right) from ZR-75-1 and MCF-7 cells stably transfected with empty vector or vector expressing MUC1siRNA were immunoblotted as indicated. Lamin B was included as a loading control. B. Primary breast cancer cells were infected with lentiviruses expressing control siRNA or MUC1siRNA (50) and lysates immunoblotted as indicated. C. ZR-75-1/vector and ZR-75-1/MUC1siRNA cells were left untreated or stimulated with IL-6 for 15 min. Lysates were immunoblotted with the indicated antibodies. D and E. The indicated ZR-75-1/vector, ZR-75-1/MUC1siRNA (D), MCF-7/vector, and MCF-7/MUC1siRNA (E) cells were serum starved for 18 h and then stimulated with IL-6 for the indicated times. Lysates were immunoblotted with the indicated antibodies. β-actin was included as a loading control. F. MCF-7 cells were transfected with control, JAK1 siRNA (left), or JAK2 siRNA (right) pools for 72 h and then stimulated with IL-6 for 15 min. Lysates were immunoblotted with the indicated antibodies. G. Rat 3Y1 fibroblasts stably expressing an empty vector or MUC1-CD were stimulated with IL-6 for 15 min (left). 3Y1 cells were treated with 50 μM AG-490 in serum free media for 16 h before stimulation with IL-6 for 15 min (right). Lysates were immunoblotted with the indicated antibodies. H. Human WI-38 fibroblasts were transfected with control, JAK1 (left) or JAK2 (right) siRNA pools for 72 h and then stimulated with IL-6 for 15 min. Lysates were immunoblotted with the indicated antibodies.
IL-6 induced JAK1 phosphorylation in serum-starved ZR-75-1 cells stably transfected with vector, but not in the ZR-75-1/MUC1siRNA cells (Fig. 3C). In contrast, silencing MUC1 had no effect on constitutive JAK2 phosphorylation (Fig. 3C). In addition, JAK2 phosphorylation was unchanged by IL-6 stimulation, consistent with involvement of JAK1 and not JAK2 in gp130 signaling (Fig. 3C). IL-6-dependent phosphorylation of STAT3 was also attenuated in ZR-75-1/MUC1siRNA cells (Fig. 3D). Similar results were obtained in MCF-7/MUC1siRNA cells (Fig. 3E), indicating that MUC1 knockdown affects activation of both JAK1 and STAT3. JAK1 knockdown in MCF-7 cells abrogated IL-6-induced STAT3 phosphorylation, consistent with its dependence on this kinase (Fig. 3F, left), whereas silencing JAK2 had no apparent effect on IL-6-induced STAT3 phosphorylation, indicating that MUC1-C regulates JAK1-mediated activation of STAT3 (Fig. 3F, right).
Rat 3Y1 fibroblasts lack Muc1 expression and thus represent a non-epithelial cell model to study JAK-mediated STAT3 activation in the absence of endogenous MUC1-C. IL-6-induced phosphorylation of STAT3 was detectable in 3Y1 cells stably transfected with empty vector (3Y1/vector cells) or with MUC1-CD (3Y1/MUC1-CD cells), (Fig. 3G, left). In addition, treatment of 3Y1/vector cells with the JAK2 inhibitor AG-490 (29) blocked STAT3 phosphorylation in response to IL-6 (Fig. 3G, right). Furthermore, we found that silencing JAK1 or JAK2 blocked IL-6-dependent STAT3 phosphorylation in human WI-38 fibroblasts, which are also null for MUC1 expression (fig. S1) (Figs. 3H, left and right). Thus, MUC1-C is dispensable for JAK1- and JAK2-mediated STAT3 activation in fibroblasts.
Activation of STAT3 in MCF-10A breast epithelial cells by IL-6, IL-10, and IL-22 depends on MUC1-C
To further assess the involvement of MUC1-C in the regulation of STAT3 activation, we studied MCF-10A epithelial cells, which are derived from normal breast tissue and express endogenous MUC1 in lower abundance than do ZR-75-1 and MCF-7 breast cancer cells (23). IL-6 stimulation of MCF-10A cells led to an increase in MUC1-C abundance (Fig. 4A). A more detailed time course of the effects of IL-6 stimulation on STAT3 abundance and phosphorylation demonstrated an increase in p-STAT3 at 15-30 min and then a decline of p-STAT3 to undetectable amounts at 24-48 h (Fig. 4B). In contrast to the biphasic increases in STAT3 at 15 to 30 min and at 24 h, the increase in MUC1-C abundance was detectable at 24-48 h (Fig. 4B). Compared to breast cancer cells, there was a low basal level of MUC1-C binding with STAT3 in MCF-10A cells (Fig. 4C). Stimulation with IL-6 for 24 h increased association of MUC1-C with STAT3 (Fig. 4C), and promoted nuclear accumulation of MUC1-C and STAT3 (Figs. 4D and E).
Figure 4. IL-6 induces MUC1-C-dependent STAT3 activation in MCF-10A cells.
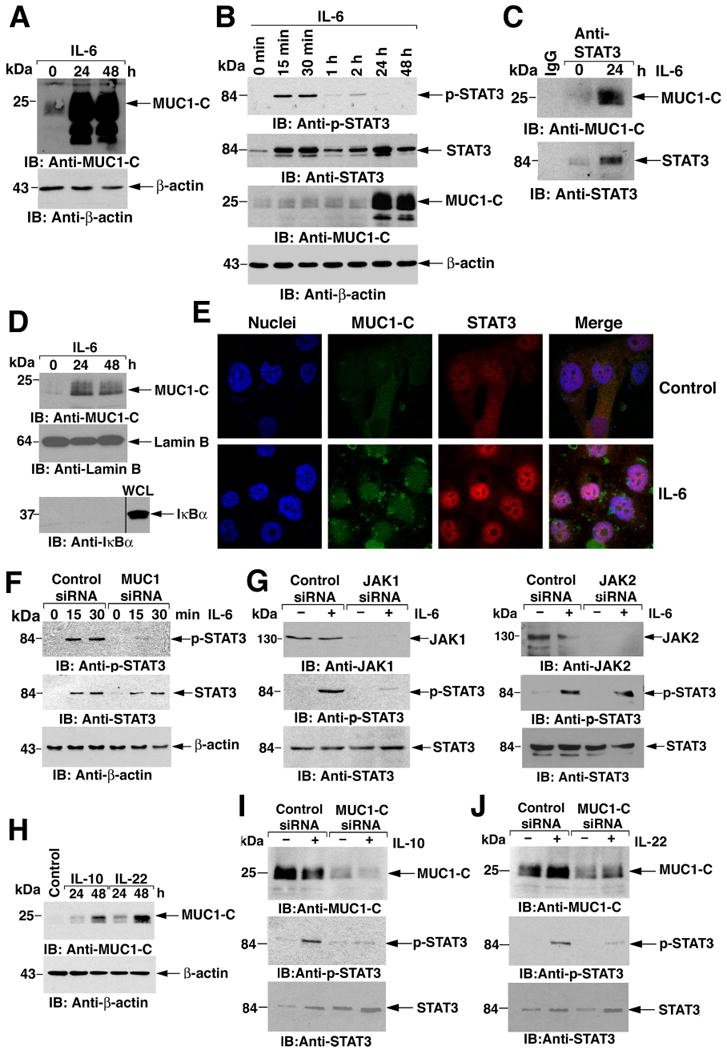
A and B. MCF-10A cells were stimulated with IL-6 for the indicated times. Lysates were immunoblotted with the indicated antibodies. As determined by densitometric scanning of the p-STAT3 and STAT3 signals, the fractional change (p-STAT3/STAT3; mean and range from two independent experiments) as compared to that prior to stimulation (0 min; assigned a value of 1) was 2.75 (range 1.4 to 4.1), 2.7 (range 1.4 to 4.0), 0.89 (range 0.85 to 0.94) and 1.4 (range 0.88 to 1.91) at 15 min, 30 min, 1 h and 2 h of IL-6 stimulation, respectively. C. MCF-10A cells were left untreated or stimulated with IL-6 for 24 h. Lysates were immunoprecipitated with a control IgG or anti-STAT3. The precipitates were immunoblotted with the indicated antibodies. D. Nuclear lysates from MCF-10A cells stimulated with IL-6 for 24 or 48 h were immunoblotted with the indicated antibodies. Whole cell lysate (WCL) was included as a control for detection of cytoplasmic IκBα. E. MCF-10A cells were left untreated (Control) or stimulated with IL-6 for 24 h, fixed and stained with anti-MUC1-C (green) and anti-STAT3 (red). Nuclei were stained with DAPI (blue). F. MCF-10A cells were transfected with control or MUC1 siRNA pools for 72 h and then stimulated with IL-6 for the indicated times. Lysates were immunoblotted with the indicated antibodies. G. MCF-10A cells were transfected with control, JAK1 siRNA (left), or JAK2 siRNA (right) pools for 72 h and then stimulated with IL-6 for 15 min. Lysates were immunoblotted with the indicated antibodies. H. MCF-10A cells were stimulated with IL-10 or IL-22 for the indicated times. Lysates were immunoblotted with the indicated antibodies. I and J. MCF-10A cells were transfected with control or MUC1 siRNA pools for 72 h and then stimulated with IL-10 (I) or IL-22 (J) for 15 min. Lysates were immunoblotted with the indicated antibodies.
Consistent with its role in STAT3 activation in breast cancer cells, silencing MUC1-C in MCF-10A cells abrogated IL-6-induced STAT3 phosphorylation (Fig. 4F). This implicates MUC1-C present under baseline conditions in STAT3 activation in the response to IL-6 and indicates that the increase in MUC1-C abundance, which occurs later (24-48 h), is not necessary for that response. Analyses of MCF-10A cells silenced for JAK1 expression confirmed that IL-6-induced activation of STAT3 depends on both MUC1-C and JAK1 (Fig. 4G, left), whereas JAK2 knockdown had no apparent effect on this response (Fig. 4G, right).
IL-10 and IL-22 signal through the IL-10 receptor family and JAK1 to activate the STAT3 pathway (30-32). As with IL-6, stimulation of MCF-10A cells with IL-10 and IL-22 increased MUC1-C abundance (Fig. 4H). Moreover, stimulation of MCF-10A cells with IL-10 increased STAT3 phosphorylation and MUC1-C knockdown attenuated this response (Fig. 4I). Phosphorylation of STAT3 in response to IL-22 also depended on MUC1-C (Fig. 4J). These findings indicate that, as found for gp130 signaling, MUC1-C promotes activation of STAT3 by the IL-10 receptor family through JAK1 and that IL-10 and IL-22 stimulate a late increase in MUC1-C abundance.
STAT3 and MUC1-C bind to the MUC1 promoter
To determine whether MUC1-C-STAT3 complexes activate MUC1 expression, we performed chromatin immunoprecipitation (ChIP) assays on a consensus STAT binding site (-503 to -495; TTCCGGGAA) in the MUC1 promoter (33) (Fig. 5A). Precipitation of chromatin from ZR-75-1 cells with antibody directed against STAT3 revealed association of STAT3 with the STAT binding region (SBR; -559 to −284), but not with a control region of the MUC1 gene (CR; +4596 to +4817) (Fig. 5B, left). Precipitation with anti-MUC1-C demonstrated that MUC1-C also occupies the STAT binding region (Fig. 5B, middle). Moreover, re-ChIP assays showed that MUC1-C occupies the MUC1 promoter with STAT3 (Fig. 5B, right). Occupancy of the MUC1 promoter by STAT3 and MUC1-C was confirmed by ChIP-quantitative PCR (qPCR) and re-ChIP-qPCR (Fig. 5C). ChIP analysis of the MUC1 promoter in MCF-7 cells confirmed that both STAT3 and MUC1-C occupy the STAT binding region and not the control region (figs. S2A and S2B) and that MUC1-C occupies the promoter with STAT3 (fig. S2C).
Figure 5. MUC1-C associates with the STAT3 transcription complex.
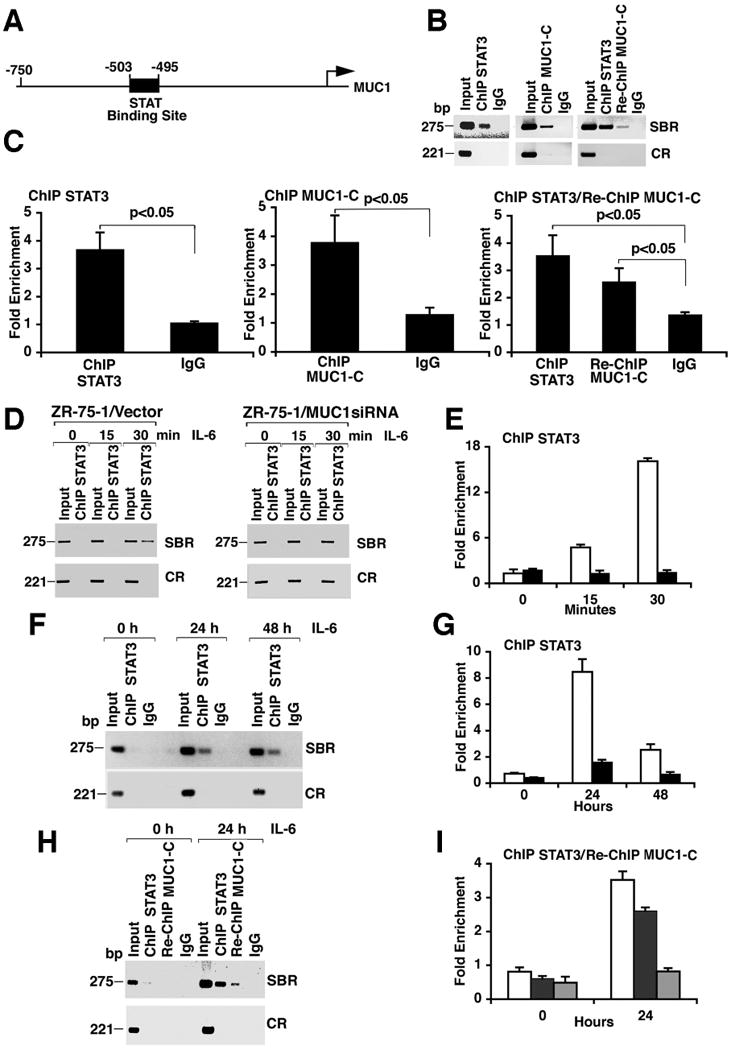
A. Schema of the MUC1 promoter region with location of the STAT binding site. B and C. Soluble chromatin from ZR-75-1 cells was immunoprecipitated with a control IgG, anti-STAT3 (left), or anti-MUC1-C (middle). Re-ChIP analysis, chromatin precipitated with anti-STAT3 was first released and then precipitated with anti-MUC1-C (right). The final DNA samples were amplified by PCR with pairs of primers flanking the STAT binding region (SBR; -559 to -284) or a control region (CR; +4596 to +4817) (B). The precipitated chromatin was also analyzed by qPCR (C). The results (mean±SD of three determinations) are expressed as the Fold Enrichment as compared to that obtained for the Control Region. D and E. ZR-75-1/vector and ZR-75-1/MUC1siRNA cells were serum starved overnight and then stimulated with IL-6 for the indicated times. Soluble chromatin was precipitated with anti-STAT3 and analyzed for MUC1 promoter SBR and CR sequences (D). The precipitated chromatin from ZR-75-1/vector (open bars) or ZR-75-1/MUC1siRNA (solid bars) cells was also analyzed by qPCR (E). F and G. Soluble chromatin from MCF-10A cells stimulated with IL-6 for the indicated times was precipitated with anti-STAT3 or a control IgG. The precipitates were analyzed for MUC1 promoter SBR or CR sequences (F). The anti-STAT3 (open bars) and IgG (solid bars) precipitated chromatin was also analyzed by qPCR (G). H and I. Soluble chromatin from control or IL-6-stimulated MCF-10A cells was precipitated with anti-STAT3 and analyzed for MUC1 promoter SBR or CR sequences (H). In the re-ChIP experiments, anti-STAT3 precipitates were released, reimmunoprecipitated with anti-MUC1-C and then analyzed for MUC1 promoter sequences (H). Precipitated chromatin was also analyzed by qPCR for the ChIP STAT3 (open bars), Re-ChIP MUC1-C (solid bars) and the IgG control (shaded bars) (I).
Polymerase chain reaction (PCR) (Fig. 5D) and qPCR (Fig. 5E) analyses of serum-starved ZR-75-1/vector and ZR-75-1/MUC1siRNA cells stimulated with IL-6 revealed that targeting of STAT3 to the MUC1 promoter depended on MUC1-C. Analysis of MCF-7/vector and MCF-7/MUC1siRNA cells confirmed that MUC1-C promoted STAT3 occupancy of the MUC1 promoter (figs. S2D and S2E). In MCF-10A cells, ChIP analysis of the MUC1 promoter showed that IL-6 stimulation for 24 and 48 h induced STAT3 occupancy of the STAT binding site (Figs. 5F and 5G), whereas STAT3 occupancy was not detectable at earlier times (fig. S3). Re-ChIP studies confirmed that MUC1-C is present with STAT3 on the MUC1 promoter following stimulation with IL-6 (Figs. 5H and 5I). These findings indicate that MUC1-C and STAT3 occupy the MUC1 promoter in ZR-75-1 and MCF-7 breast cancer cells and that IL-6 stimulates their association with the MUC1 promoter in MCF-10A cells.
MUC1-C promotes targeting of STAT3 to the MUC1 promoter
To determine whether STAT3 is responsible for the IL-6-induced increase in MUC1-C abundance, we silenced STAT3 in MCF-10A cells and then stimulated with IL-6 (Fig. 6A). We found that that IL-6 induced MUC1-C mRNA and protein expression by a STAT3-dependent mechanism (Fig. 6A and fig. S4). IL-6 increased luciferase activity in MCF-10A cells transfected with a plasmid encoding a MUC1 promoter-luciferase construct (pMUC1-Luc) (Fig. 6B), an effect that was attenuated by mutation of the STAT binding site (mSBS) Fig. 6B). Moreover--and consistent with involvement of STAT3--silencing STAT3 blocked IL-6-dependent activation of pMUC1-Luc (Fig. 6C).
Figure 6. MUC1-C promotes STAT3-mediated transcription.
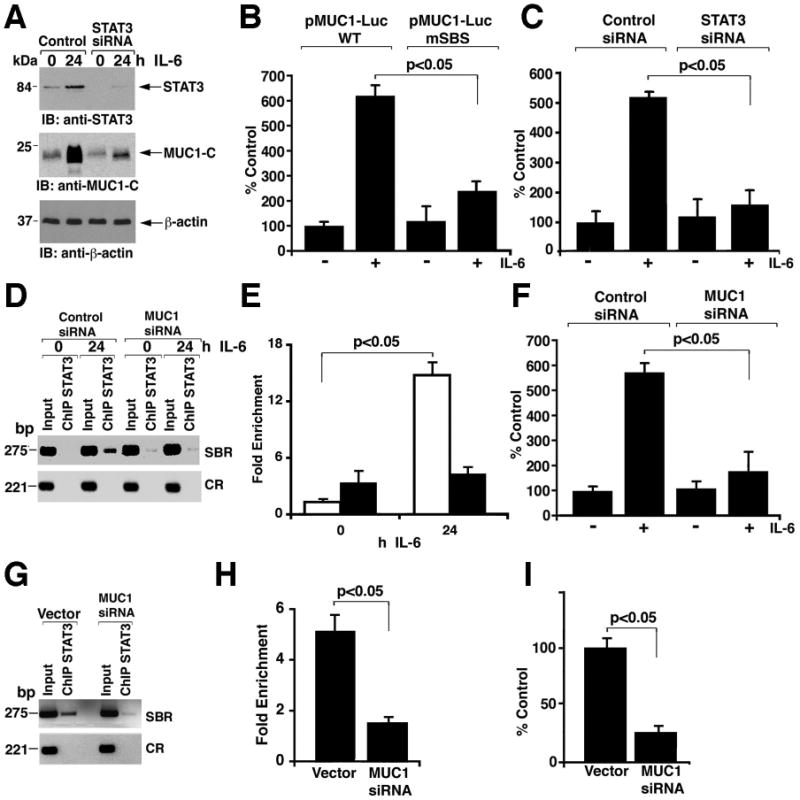
A. MCF-10A cells were transfected with control or STAT3 siRNA pools for 72 h. The transfected cells were then left untreated or stimulated with IL-6 for 24 h. Lysates were immunoblotted with the indicated antibodies. B. MCF-10A cells were transfected to express wild-type pMUC1-Luc, pMUC1-Luc mutated at the STAT binding site (mSBS), or Renilla-Luc. After 24 h, the cells were left untreated or stimulated with IL-6 for 24 h and then assayed for luciferase activity. The results are expressed as the percentage of control (mean±SD from three separate experiments) compared to that obtained with cells transfected with wild-type pMUC1-Luc and left untreated (assigned a value of 100%). C. MCF-10A cells were transfected with control or STAT3 siRNA pools for 72 h. The cells were then transfected with pMUC1-Luc or Renilla-Luc for 24 h and left untreated or stimulated with IL-6 for an additional 24 h before assaying for luciferase activity. The results are expressed as the percentage of control (mean±SD from three separate experiments) compared to that obtained with cells transfected with control siRNA and left untreated (assigned a value of 100%). D and E. MCF-10A cells were transfected with control siRNA or MUC1 siRNA pools for 72 h. The transfected cells were then left untreated or stimulated with IL-6 for 24 h. Soluble chromatin was precipitated with anti-STAT3 and analyzed for MUC1 promoter SBR and CR sequences (D). Precipitated chromatin from cells transfected with the control siRNA (open bars) or MUC1 siRNA (solid bars) was also analyzed by qPCR (E). F. MCF-10A cells were transfected with control or MUC1 siRNA pools for 72 h. The cells were then transfected with pMUC1-Luc or Renilla-Luc for 24 h and left untreated or stimulated with IL-6 for an additional 24 h before assaying for luciferase activity. The results are expressed as the percentage of control (mean±SD from three separate experiments) compared to that obtained with cells transfected with control siRNA and left untreated (assigned a value of 100%). G and H. Soluble chromatin from ZR-75-1/vector and ZR-75-1/MUC1siRNA cells was precipitated with anti-STAT3 and analyzed for MUC1 promoter SBR and CR sequences (G). The precipitated chromatin was also analyzed by qPCR (H). I. ZR-75-1/vector and ZR-75-1/MUC1siRNA cells were transfected to express pMUC1-Luc or Renilla-Luc. Luciferase activity was measured at 48 h after transfection. Results are expressed as the percentage of control (mean±SD from three separate experiments) compared to that obtained with ZR-75-1/vector cells (assigned a value of 100%).
To assess the effects of MUC1-C on the STAT3 transcription complex, we silenced MUC1 in MCF-10A cells and performed ChIP analyses of the MUC1 promoter. IL-6-induced targeting of STAT3 to the MUC1 promoter was attenuated by silencing MUC1 (Fig. 6D and 6E). Consistent with these results, silencing MUC1 attenuated IL-6-induced activation of the pMUC1-Luc reporter (Fig. 6F). Silencing MUC1 was also associated with decreased STAT3 occupancy of the MUC1 promoter and decreased activation of the pMUC1-Luc reporter (Fig. 6I) in ZR-75-1 cells (Figs. 6G and 6H). These findings indicate that MUC1 contributes to targeting of STAT3 to the MUC1 promoter and thereby to transcriptional activation of MUC1.
Inhibition of MUC1-C function blocks IL-6-induced targeting of STAT3 to the MUC1 promoter
To further assess the role of MUC1-C in the activation of STAT3, we performed studies with GO-201, a peptide that binds to the MUC1-C cytoplasmic domain and blocks its function (24,34). GO-201, but not the inactive control peptide CP-1 (24,34), inhibited the in vitro interaction between GST-STAT3 and MUC1-CD (Fig. 7A). GO-201, but not CP-1, also blocked the IL-6-dependent interaction between MUC1-C and STAT3 in MCF-10A cells (Fig. 7B). Furthermore, GO-201 attenuated the increase in MUC-1 abundance in response to IL-6- (Fig. 7C), and association of STAT3 and MUC1-C with the MUC1 promoter (fig. S5A).
Figure 7. Inhibition of MUC1-C decreases the abundance of STAT3 and the products of other STAT3 target genes.
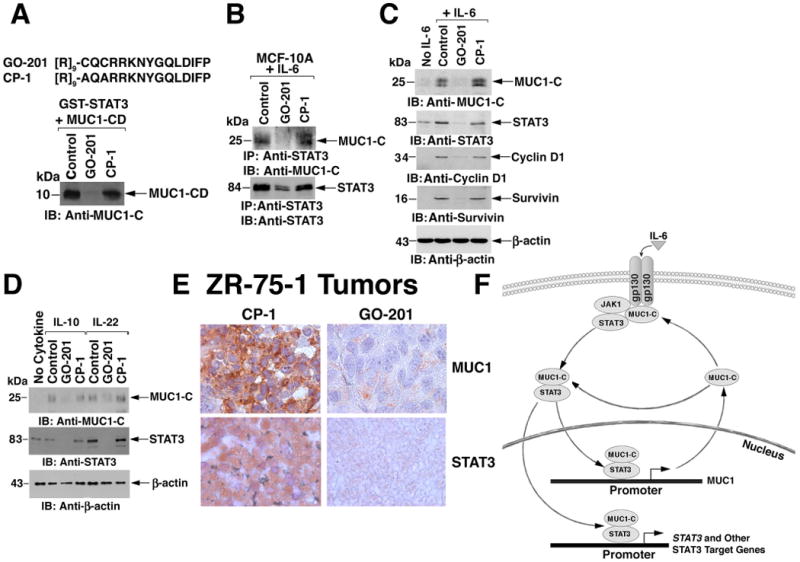
A. Amino acid sequences of GO-201 and CP-1 (upper panel). [R]9 denotes nine arginine residues. GST-STAT3 was incubated with purified MUC1-CD in the presence of a 20-fold excess of GO-201 or CP-1 for 2 h at room temperature. Adsorbates to glutathione beads were immunoblotted with anti-MUC1-C (lower panel). B. MCF-10A cells were stimulated with IL-6 in the absence (Control) or presence of 5 μM GO-201 or CP-1 added each 24 h for 72 h. Anti-STAT3 precipitates were immunoblotted with the indicated antibodies. C. MCF-10A cells were left untreated (No IL-6) or stimulated with IL-6 in the absence (Control) or presence of 5 μM GO-201 or CP-1 added each 24 h for 72 h. Lysates were immunoblotted with the indicated antibodies. D. MCF-10A cells were left untreated (No Cytokine) or stimulated with IL-10 or IL-22 in the absence (Control) or presence of 5 μM GO-201 or CP-1 added each 24 h for 72 h. Lysates were immunoblotted with the indicated antibodies. E. Female BALB/c nu/nu mice bearing subcutaneous ZR-75-1 breast tumor xenografts (∼150 mm3) were injected intraperitoneally with GO-201 or CP-1 at 30 mg/kg/day for 21 days. On day 28, tumors were harvested and fixed sections were analyzed for MUC1 and STAT3 expression by immunoperoxidase staining (reddish color). F. Proposed schema for a MUC1-C-STAT3 auto-inductive loop.
STAT3 activates STAT3 gene transcription by binding to a consensus site (-79 to -70; TTTCATGAA) in its own promoter (35). We performed ChIP analyses of MCF-10A cells stimulated with IL-6 and found that IL-6 induced occupancy of the STAT3 binding region (SBR; -315 to -70) of the STAT3 promoter, but not a control region (+5005 to +5347), by both STAT3 and MUC1-C (fig. S5B). Moreover, treatment with GO-201 (but not CP-1) attenuated IL-6-dependent association of STAT3 and MUC1-C with the STAT3 promoter (fig. S4B).
Cyclin D1 is another STAT3 target gene that has a consensus STAT3 binding site (-936 to -927; TTCCAGCAA) and cyclin D1 is required for STAT3-induced transformation (36). IL-6 stimulation of MCF-10A cells enhanced association of STAT3 and MUC1-C with the cyclin D1 promoter STAT binding region (SBR; -1045 to -839), a response that was attenuated by GO-201 but not CP-1 (fig. S5C). Consistent with these results, the IL-6-induced increase in abundance of STAT3 and cyclin D1 was blocked by GO-201 but not CP-1 (Fig. 7C). Similar results were obtained with survivin, which is encoded by another STAT3 target gene (Fig. 7C). GO-201 also attenuated expression of MUC1-C and STAT3 in the response of MCF-10A cells to IL-10 and IL-22 (Fig. 7D). EGFR has been associated with upregulation of the gp130/JAK/STAT3 pathway in breast cancer cells (37). However, in contrast to the effects of GO-201 on STAT3, it had no effect on EGFR phosphorylation (fig. S6).
To extend these findings with MUC1-C inhibition to an in vivo model, we treated established ZR-75-1 tumor xenografts in nude mice with GO-201 or CP-1. Tumor growth was inhibited by GO-201 but not CP-1 as previously reported (34). Treatment was continued for 21 days and tumors were harvested on day 28 for analysis of MUC1-C and STAT3 abundance. Consistent with the in vitro results, GO-201 but not CP-1 treatment was associated with decrease in the abundance of both MUC1-C and STAT3 (Fig. 7E). These findings are thus consistent with the proposed role of MUC1-C in promoting STAT3 signaling and thereby the activation of a MUC1-C-STAT3 auto-inductive loop (Fig. 7F).
Discussion
MUC1-C is necessary for STAT3 phosphorylation in breast cancer cells
Activation of STAT3 has been identified in various human carcinomas, including breast cancer, and some hematologic malignancies (6-8). MUC1 is overexpressed in breast and other carcinomas (16) and here, we show that the MUC1-C subunit constitutively associates with STAT3 in breast cancer cells and—to a lesser extent—in non-malignant breast epithelial cells. The interaction between MUC1-C and STAT3 was increased in IL-6-treated non-malignant MCF-10A breast epithelial cells, indicating that it may represent a physiologic response to inflammatory cytokines.
Silencing MUC1 in breast cancer cells was associated with a decrease in p-STAT3 abundance, supporting a functional role for the MUC1-C-STAT3 interaction. Silencing MUC1 also blocked IL-6-, IL-10-, and IL-22-induced increases in p-STAT3. MUC1-C interacted directly with JAK1 and activation of JAK1, but not that of JAK2, depended on MUC1-C in the response to IL-6. Genetic studies have demonstrated that different classes of cytokine receptors preferentially use one JAK or another, or a JAK combination (23). The IL-6, IL-10, and IL-22 receptors predominantly use JAK1, whereas cytokine receptors that stimulate hematopoietic cell growth predominantly use JAK2. Nonetheless, there is considerable variability in JAK usage that depends on specific receptors and cell context. Indeed, the present results support a model in which gp130 responses differentially depend on JAK1 in MUC1-C-expressing epithelial cells and both JAK1 and JAK2 in fibroblasts that lack MUC1.
Previous work has shown that the STAT3-interacting protein, StIP1, contributes to IL-6-induced STAT3 phosphorylation by acting as a scaffold for both JAK1 and STAT3 in mouse M1 myeloid leukemia cells (38). Here, we found that MUC1-CD interacted with both JAK1 and STAT3. Specifically, the N-terminal region of MUC1-CD (amino acids 1-45) bound directly to JAK1 and the C-terminal region of MUC1-CD (amino acids 46-72) formed direct complexes with STAT3, indicating that MUC1-C may function like StIP1 in providing a scaffold for JAK1 and STAT3. The interaction between MUC1-C and STAT3 did not depend on STAT3 phosphorylation. Moreover, MUC1-CD promoted JAK1-mediated phosphorylation of STAT3 in vitro, supporting our findings that MUC1-C contributes to JAK1 phosphorylation of STAT3 in breast cancer cells.
MUC1-C promotes STAT3-mediated transcription
The MUC1-C CD bound directly to the STAT3 region that includes the DBD. Few other proteins are known to interact with the STAT3 DBD (39). The C-terminal region of c-Jun binds to the STAT3 coiled-coil domain and DBD, and thereby contributes to cooperation between STAT3 and c-Jun in driving transcription (40). The STAT3 DBD is essential for mediating binding with NF-κB RelA (41). In addition, STAT3-mediated acetylation of NF-κB RelA, and thereby maintenance of NF-κB activity, requires the STAT3 DBD (42). Thus, binding of MUC1-C to the region of STAT3 that includes the DBD could affect STAT3 interactions with c-Jun or NF-κB RelA and thereby their regulation of gene transcription.
We found that MUC1-C associated with STAT3 on the STAT binding site in the MUC1 promoter. Occupancy of the MUC1 promoter STAT binding site by MUC1-C and STAT3 was detectable in breast cancer cells and inducible by IL-6 in MCF-10A breast epithelial cells. Other studies have shown that MUC1-C associates with STAT1 in the response to stimulation with interferon-γ (IFNγ) and that MUC1-C-STAT1 complexes occupy the STAT binding site in the MUC1 promoter (43). In contrast, STAT5 has not been detectable on the MUC1 promoter (33,43), indicating selectivity of such occupancy to STAT1 in the response to IFNγ and to STAT3 in the response to IL-6. We found that activation of the MUC1 promoter in the response of MCF-10A cells to IL-6 was mediated by STAT3. However, silencing MUC1 attenuated IL-6-induced targeting of STAT3 to the STAT binding site of the MUC1 promoter in MCF-10A cells, implicating MUC1-C in initiating STAT3 occupancy of that site or in delaying STAT3 latency. The demonstration that MUC1-C promotes STAT3-mediated activation of the MUC1 promoter supports the existence of an auto-inductive loop in which MUC1-C and STAT3 work cooperatively to activate MUC1 expression (Fig. 7F). Consistent with these observations, silencing MUC1 in breast cancer cells was associated with decreased STAT3 occupancy of the STAT binding site, and decreased activation of the MUC1 promoter.
Inhibition of MUC1-C function decreases signaling through the STAT3 pathway
Targeting of MUC1-C to the nucleus depends on its formation of oligomers through the CQC motif (22). The MUC1-C CQC motif is also necessary for its interaction with certain binding partners, such as NF-κB RelA (24), and for its ability to induce transformation (34,44). To determine whether disrupting MUC1-C function affects STAT3 signaling, we studied the effects of the MUC1-C inhibitor, GO-201, a mutant form of MUC1-C that binds to the MUC1-C CQC motif (34). Inhibition of MUC1-C with GO-201, and not the inactive control CP-1, blocked the interaction between MUC1-C and STAT3 in vitro and in cells. GO-201 also blocked IL-6-induced STAT3 phosphorylation and IL-6-induced targeting of MUC1-C and STAT3 to the MUC1 promoter, providing further support for a role of MUC1-C in promoting STAT3 occupancy of the STAT binding site. Targeting of both STAT3 and MUC1-C to the STAT3 and cyclin D1 promoters in the response to IL-6 was attenuated by GO-201-induced inhibition of MUC1-C function. Moreover, GO-201 blocked IL-6-induced increases in expression of MUC1-C, STAT3, cyclin D1 and survivin. These data provide further support for the existence of an auto-inductive loop in which MUC1-C and STAT3 function cooperatively to increase the expression of STAT3-dependent genes (Fig. 7F).
Silencing MUC1 in MCF-10A cells had little effect on STAT3 abundance, whereas treatment of MCF-10A cells with GO-201 was associated with decreased STAT3 abundance, indicating that MUC1-C inhibition by GO-201 was more effective than MUC1-C silencing at disrupting the MUC1-C-STAT3 auto-inductive loop. GO-201 contains the CQCRRKN sequence that is present in MUC1-CD, binds directly to the MUC1-CD CQC motif and thereby blocks the capacity for MUC1-C to form dimers and localize to the nucleus (34). In this context, GO-201 could act as a dominant-negative that disrupts activity of the wild-type protein and can thereby confer effects that differ from those obtained with gene silencing (45,46). GO-201 could also affect other signaling pathways that intersect with STAT3. Indeed, the present studies do not exclude the possibility that the inhibitory effects of GO-201 on NF-κB RelA activation (24) could contribute to disruption of the STAT3-NF-κB RelA interaction implicated in tumorigenesis (42).
Does the MUC1-C-STAT3 interaction contribute to a physiologic defense mechanism exploited by human tumors?
The epithelial cell barrier is exposed to various forms of stress, including inflammatory conditions associated with production of cytokines, such as IL-6. Epithelial cells thus need a robust defense mechanism to survive. Moreover, there is increasing evidence for the promotion of carcinomas by inflammatory signals from the surrounding microenvironment that activate STAT3 (47). MUC1 abundance increases in the response to the inflammatory cytokines TNFα and IFNγ (24,43). Here, we demonstrate that IL-6, IL-10, and IL-22 also increase MUC1 abundance. The available evidence indicates that MUC1 plays an important role in the inflammatory response in epithelial cells and that an aberrant increase in MUC1 abundance, as found in breast carcinomas, contributes to persistent activation of genes that confer inflammation (16,24,43).
The MUC1-CD confers resistance to induction of apoptosis and necrosis in the response to multiple insults, including genotoxic, oxidative, and hypoxic stress (16). Thus, overexpression of MUC1-C could promote transformation both by contributing to inflammatory signals and attenuating cell death (16). Our results and the prosurvival effects of MUC1-C could support a model in which MUC1-C promotes IL-6-mediated activation of the STAT3 pathway as a mechanism to protect against epithelial damage during an inflammatory response. In turn, STAT3 increases MUC1-C expression and MUC1-C contributes to STAT3-mediated transcription. In this model, MUC1-C could play a physiologic role in transiently promoting activation of STAT3-dependent genes and thereby growth and survival of the epithelial cell layer. Conversely, irreversible activation of a MUC1-C-STAT3 auto-inductive loop in breast cancer cells would contribute to a phenotype that is stably resistant to cell death. Therefore, a physiologic mechanism that protects epithelial cells during an inflammatory response may have been exploited by human breast carcinoma cells to survive under adverse conditions. Indeed, both MUC1 and STAT3 are upregulated in breast and other carcinomas.
Inhibitors of the STAT3 pathway have anti-cancer activity in animal models (11-14) and GO-201 treatment of nude mice bearing established breast tumor xenografts is associated with complete and prolonged regressions (34). Decreased abundance of both MUC1-C and STAT3 was observed in the GO-201-treated tumors, consistent with an important role for MUC1-C in activation of STAT3 signaling in these in vivo models. Therefore, MUC1-C and STAT3 pathway inhibitors could conceivably be more effective when used in combination to target the MUC1-C-STAT3 auto-inductive loop.
Materials and Methods
Cell culture
Human ZR-75-1, ZR-75-1/vector and ZR-75-1/MUC1siRNA (23,24) breast cancer cells were grown in RPMI 1640 medium (ATCC) containing 10% heat-inactivated fetal bovine serum (HI-FBS), 100 μg/ml streptomycin, 100 units/ml penicillin and 2 mM L-glutamine. MCF-7, MCF-7/vector and MCF-7/MUC1siRNA breast cancer cells (23), 293 cells and WI-38 fibroblasts were maintained in Dulbecco's modified Eagle's medium with 10% HI-FBS, antibiotics and L-glutamine. Human MCF-10A breast epithelial cells were grown in mammary epithelial cell growth medium (MEGM; Lonza, Walkersville, MD). Transfection of cells with (i) siRNA pools (Dharmacon, Lafayette, CO) or (ii) pIRES-MUC1 (27) and Flag-STAT3 or Flag-STAT3(Y705F) (Addgene, Cambridge, MA) was performed in the presence of Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells were stimulated with 20 ng/ml IL-6, IL-10, and IL-22 (R&D Systems, Minneapolis, MN). Cells were treated with the MUC1-C inhibitor GO-201 or the control peptide CP-1 as described (34).
Immunoprecipitation and immunoblotting
Lysates from subconfluent cells were prepared as described (27) by incubating cell pellets in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 10 mM NaF, 1 mM NaV, 1 mM DTT, 1 mM PMSF, 1% NP-40, 1 μg/ml aprotinin and 1 μg/ml leupeptin) for 15 min on ice. Soluble proteins isolated by centrifugation at 14,000 rpm for 15 min were precipitated with anti-STAT3, anti-JAK1, anti-JAK2 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-p-STAT3 (Cell Signaling Technology, Denver, MA), anti-MUC1-C (Ab5; LabVision, Fremont, CA), anti-Flag (Sigma, St. Louis, MO) or control IgGs. The precipitates and cell lysates were immunoblotted with anti-MUC1-C, anti-gp130 (Santa Cruz Biotechnology), anti-STAT3, anti-p-STAT3, anti-JAK1, anti-p-JAK1 (Abcam, Cambridge, MA), anti-JAK1-KD (Millipore, Billerica, MA), anti-JAK2 (Santa Cruz Biotechnology), anti-p-JAK2 (Cell Signaling Technology), anti-β-actin (Sigma), anti-lamin B (Calbiochem, San Diego, CA) and anti-IκBα (Santa Cruz Biotechnology). Immune complexes were detected with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (GE Healthcare Biosciences, Piscataway, NJ). Each experiment was performed 2-3 times to confirm results.
In vitro binding assays
GST, GST-MUC1-CD, GST-MUC1-CD(1-45), and GST-MUC1-CD(46-72) were prepared as described (23) and incubated with cell lysates, purified recombinant STAT3, the JAK1 kinase domain (JAK1-KD; amino acids 870-1,152) (13) or JAK2 kinase domain (JAK2-KD; Cell Signaling Technology, Denver, MA). GST-STAT3, GST-STAT3 deletion mutants, GST-JAK1-KD, and GST-JAK2-KD were incubated with purified MUC1-CD that had been cleaved with thrombin to remove the GST moiety. The binding reactions were performed in 50 mM Tris-HCl, pH 7.5, 6 mM NaCl and 1% NP-40 for 6 h. Adsorbates to glutathione-conjugated beads were analyzed by immunoblotting. Input of purified proteins was assessed by Coomassie blue staining as described (24).
Assays of JAK1 activity
GST-JAK1-KD was incubated with recombinant STAT3 in kinase buffer containing 3 mM MnCl2 and 15 μM ATP as described (29). After incubation for 15 min at room temperature, the samples were washed in ATP-free kinase buffer and the reaction products were immunoblotted with anti-p-STAT3 and anti-STAT3.
Immunofluorescence confocal microscopy
Cells were grown on coverslips, fixed with 4% paraformaldehyde for 30 min and permeabilized in 1% BSA-supplemented PBS containing 0.5% Triton-X100 for 20 min. The fixed cells were incubated with anti-MUC1-C overnight at 4°C followed by Alexa fluor 488 secondary anti-hamster antibody (Invitrogen). The cells were then incubated with anti-STAT3 followed by Alexa fluor 568 secondary anti-rabbit antibody (Invitrogen). Nuclei were stained with 1 mg/ml DAPI (Invitrogen). In certain experiments, the cells were incubated with 20 ng/ml IL-6 for 24 h before fixation.
ChIP assays
Soluble chromatin was isolated as described (26) and precipitated with anti-STAT3, anti-MUC1-C, or a control non-immune IgG. For re-ChIP assays, complexes from the initial ChIP were eluted with 10 mM DTT, diluted 50-fold in ChIP dilution buffer and reimmunoprecipitated with a second antibody. For PCR, 2 μl from a 50 μl DNA sample was used with 25-35 cycles of amplification. Each ChIP-PCR experiment was performed 2-3 times to confirm results. For real time qPCR, 2 μl of the ChIP DNA samples was analyzed using the SYBR green Champion ChIP qPCR assay kit (SABiosciences, Frederick, MD) and the ABI Prism 7000 Sequence Detector (Applied Biosystems, Carlsbad, CA). Standardized sets of primers for the MUC1 promoter STAT binding region and a 5 kb upstream control region were purchased from SABiosciences. Fold enrichment (that is to say, difference in protein binding at the STAT region as compared to the control region) was calculated as described (48). The results are expressed as the mean±SD of triplicate values obtained for each DNA sample. The qPCR analysis was performed using the DNA samples shown for the ChIP-PCR studies.
Luciferase assays
Cells were transfected with pMUC1-Luc (49) and SV-40-Renilla-Luc (Promega, Madison, WI) in the presence of Lipofectamine. After 48 h, the cells were lysed in passive lysis buffer. Lysates were analyzed for firefly and Renilla luciferase activities using the dual luciferase assay kit (Promega).
Statistical analysis
Analyses were performed two to three times. The Student's t-test was used for determination of statistical significance.
Supplementary Material
Fig. S1. Immunoblot analysis of MUC1-C in lysates from WI-38 fibroblasts and MCF-7 breast cancer cells.
Fig. S2. Association of STAT3 and MUC1-C with the STAT binding region of the MUC1 promoter in MCF-7 cells.
Fig. S3. IL-6 induces STAT3 association with the MUC1 promoter in MCF-10A cells.
Fig. S4. IL-6 increases MUC1 mRNA in MCF-10A cells.
Fig. S5. Effects of GO-201 on IL-6-induced association of STAT3 and MUC1-C with the MUC1, STAT3 and cyclin D1 promoters in MCF-10A cells.
Fig. S6. Effects of GO-201 on EGFR phosphorylation in ZR-75-1 cells.
Acknowledgments
Funding: This work was supported by Grants CA97098, CA42802 and CA100707 awarded by the National Cancer Institute.
Footnotes
Competing interests: S. Kharbanda is an employee of Genus Oncology, a company that focuses on the development of MUC1 therapeutics. D. Kufe holds equity in Genus Oncology and is a consultant to the company. The other authors disclosed no potential conflicts of interest.
Contributions: R.A., H.R., M.D.J., M.A. and T.K. performed research and analyzed data. B.V. contributed clinical samples. S.K. provided vital new reagents. D.K. designed the research and wrote the paper.
References and Notes
- 1.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S, Sung B. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wegenka UM, Lütticken C, Buschmann J, Yuan J, Lottspeich F, Müller-Esterl W, Schindler C, Roeb E, Heinrich PC, Horn F. The interleukin-6-activated acute-phase response factor is antigenically and functionally related to members of the signal transducer and activator of transcription (STAT) family. Mol Cell Biol. 1994;14:3186–3196. doi: 10.1128/mcb.14.5.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–5062. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- 5.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 6.Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 7.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 8.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–947. [PubMed] [Google Scholar]
- 10.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad R, Raina D, Meyer C, Kufe D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)-->signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008;68:2920–2926. doi: 10.1158/0008-5472.CAN-07-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13:5665–5669. doi: 10.1158/1078-0432.CCR-06-2491. [DOI] [PubMed] [Google Scholar]
- 15.Buerger C, Nagel-Wolfrum K, Kunz C, Wittig I, Butz K, Hoppe-Seyler F, Groner B. Sequence-specific peptide aptamers, interacting with the intracellular domain of the epidermal growth factor receptor, interfere with Stat3 activation and inhibit the growth of tumor cells. J Biol Chem. 2003;278:37610–37621. doi: 10.1074/jbc.M301629200. [DOI] [PubMed] [Google Scholar]
- 16.Kufe D. Mucins in cancer: function, prognosis and therapy. Nature Reviews Cancer. 2009;9:874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Liu D, Chen D, Kharbanda S, Kufe D. Human DF3/MUC1 carcinoma-associated protein functions as an oncogene. Oncogene. 2003;22:6107–6110. doi: 10.1038/sj.onc.1206732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L, Chen D, Liu D, Yin L, Kharbanda S, Kufe D. MUC1 oncoprotein blocks GSK3beta-mediated phosphorylation and degradation of beta-catenin. Cancer Res. 2005;65:10413–10422. doi: 10.1158/0008-5472.CAN-05-2474. [DOI] [PubMed] [Google Scholar]
- 19.Ligtenberg MJ, Kruijshaar L, Buijs F, van Meijer M, Litvinov SV, Hilkens J. Cell-associated episialin is a complex containing two proteins derived from a common precursor. J Biol Chem. 1992;267:6171–7. [PubMed] [Google Scholar]
- 20.Macao B, Johansson DG, Hansson GC, Hard T. Autoproteolysis coupled to protein folding in the SEA domain of the membrane-bound MUC1 mucin. Nat Struct Mol Biol. 2006;13:71–6. doi: 10.1038/nsmb1035. [DOI] [PubMed] [Google Scholar]
- 21.Ramasamy S, Duraisamy S, Barbashov S, Kawano T, Kharbanda S, Kufe D. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol Cell. 2007;27:992–1004. doi: 10.1016/j.molcel.2007.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leng Y, Cao C, Ren J, Huang L, Chen D, Ito M, Kufe D. Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. J Biol Chem. 2007;282:19321–19330. doi: 10.1074/jbc.M703222200. [DOI] [PubMed] [Google Scholar]
- 23.Ahmad R, Raina D, Trivedi V, Ren J, Rajabi H, Kharbanda S, Kufe D. MUC1 oncoprotein activates the I|B kinase β complex and constitutive NF-|B signaling. Nat Cell Biol. 2007;9:1419–1427. doi: 10.1038/ncb1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmad R, Raina D, Joshi MD, Kawano T, Kharbanda S, Kufe D. MUC1-C oncoprotein functions as a direct activator of the NF-kappaB p65 transcription factor. Cancer Res. 2009;69:7013–7021. doi: 10.1158/0008-5472.CAN-09-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei X, Xu H, Kufe D. Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancer Cell. 2005;7:167–178. doi: 10.1016/j.ccr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Wei X, Xu H, Kufe D. MUC1 oncoprotein stabilizes and activates estrogen receptor α. Mol Cell. 2006;21:295–305. doi: 10.1016/j.molcel.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 27.Ren J, Agata N, Chen D, Li Y, Yu WH, Huang L, Raina D, Chen W, Kharbanda S, Kufe D. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anti-cancer agents. Cancer Cell. 2004;5:163–175. doi: 10.1016/s1535-6108(04)00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren J, Bharti A, Raina D, Chen W, Ahmad R, Kufe D. MUC1 oncoprotein is targeted to mitochondria by heregulin-induced activation of c-Src and the molecular chaperone HSP90. Oncogene. 2006;25:20–31. doi: 10.1038/sj.onc.1209012. [DOI] [PubMed] [Google Scholar]
- 29.Neilson LM, Zhu J, Xie J, Malabarba MG, Sakamoto K, Wagner KU, Kirken RA, Rui H. Coactivation of janus tyrosine kinase (Jak)1 positively modulates prolactin-Jak2 signaling in breast cancer: recruitment of ERK and signal transducer and activator of transcription (Stat)3 and enhancement of Akt and Stat5a/b pathways. Mol Endocrinol. 2007;21:2218–32. doi: 10.1210/me.2007-0173. [DOI] [PubMed] [Google Scholar]
- 30.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. 2002;277:33676–33682. doi: 10.1074/jbc.M204204200. [DOI] [PubMed] [Google Scholar]
- 32.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 33.Gaemers IC, Vos HL, Volders HH, van der Valk SW, Hilkens J. A STAT-responsive element in the promoter of the episialin/MUC1 gene is involved in its overexpression in carcinoma cells. J Biol Chem. 2001;276:6191–6199. doi: 10.1074/jbc.M009449200. [DOI] [PubMed] [Google Scholar]
- 34.Raina D, Ahmad R, Joshi M, Yin L, Wu Z, Kawano T, Vasir B, Avigan D, Kharbanda S, Kufe D. Direct targeting of the MUC1 oncoprotein blocks survival and tumorigenicity of human breast carcinoma cells. Cancer Res. 2009;69:5133–5141. doi: 10.1158/0008-5472.CAN-09-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J Biol Chem. 1998;273:6132–6138. doi: 10.1074/jbc.273.11.6132. [DOI] [PubMed] [Google Scholar]
- 36.Leslie K, Lang C, Devgan G, Azare J, Berishaj M, Gerald W, Kim YB, Paz K, Darnell JE, Albanese C, Sakamaki T, Pestell R, Bromberg J. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006;66:2544–2552. doi: 10.1158/0008-5472.CAN-05-2203. [DOI] [PubMed] [Google Scholar]
- 37.Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, Bornmann W, Bromberg JF. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007;9:R32. doi: 10.1186/bcr1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collum RG, Brutsaert S, Lee G, Schindler C. A Stat3-interacting protein (StIP1) regulates cytokine signal transduction. Proc Natl Acad Sci U S A. 2000;97:10120–10125. doi: 10.1073/pnas.170192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shuai K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 2000;19:2638–2644. doi: 10.1038/sj.onc.1203522. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Wrzeszczynska MH, Horvath CM, Darnell JE., Jr Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol Cell Biol. 1999;19:7138–7146. doi: 10.1128/mcb.19.10.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Z, Kone BC. The STAT3 DNA-binding domain mediates interaction with NF-|B p65 and inducible nitric oxide synthase transrepression in mesangial cells. J Am Soc Nephrol. 2004;15:585–591. doi: 10.1097/01.asn.0000114556.19556.f9. [DOI] [PubMed] [Google Scholar]
- 42.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khodarev N, Ahmad R, Rajabi H, Pitroda S, Kufe T, McClary C, Joshi MD, MacDermed D, Weichselbaum R, Kufe D. Cooperativity of the MUC1 oncoprotein and STAT1 pathway in poor prognosis human breast cancer. Oncogene. 2010;29:920–929. doi: 10.1038/onc.2009.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joshi MD, Ahmad R, Raina D, Rajabi H, Bubley G, Kharbanda S, Kufe D. MUC1 oncoprotein is a druggable target in human prostate cancer cells. Mol Cancer Ther. 2009;8:3056–3065. doi: 10.1158/1535-7163.MCT-09-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herskowitz I. Functional inactivation of genes by dominant negative mutations. Nature. 1987;329:219–222. doi: 10.1038/329219a0. [DOI] [PubMed] [Google Scholar]
- 46.Veitia RA. Dominant negative factors in health and disease. J Pathol. 2009;218:409–418. doi: 10.1002/path.2583. [DOI] [PubMed] [Google Scholar]
- 47.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19:631–642. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 49.Yin L, Kufe D. Human MUC1 carcinoma antigen regulates intracellular oxidant levels and the apoptotic response to oxidative stress. J Biol Chem. 2003;278:35458–64. doi: 10.1074/jbc.M301987200. [DOI] [PubMed] [Google Scholar]
- 50.Kawano T, Ito M, Raina D, Wu Z, Rosenblatt J, Avigan D, Stone R, Kufe D. MUC1 oncoprotein regulates Bcr-Abl stability and pathogenesis in chronic myelogenous leukemia cells. Cancer Res. 2007;67:11576–11584. doi: 10.1158/0008-5472.CAN-07-2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunoblot analysis of MUC1-C in lysates from WI-38 fibroblasts and MCF-7 breast cancer cells.
Fig. S2. Association of STAT3 and MUC1-C with the STAT binding region of the MUC1 promoter in MCF-7 cells.
Fig. S3. IL-6 induces STAT3 association with the MUC1 promoter in MCF-10A cells.
Fig. S4. IL-6 increases MUC1 mRNA in MCF-10A cells.
Fig. S5. Effects of GO-201 on IL-6-induced association of STAT3 and MUC1-C with the MUC1, STAT3 and cyclin D1 promoters in MCF-10A cells.
Fig. S6. Effects of GO-201 on EGFR phosphorylation in ZR-75-1 cells.