

Abstract
The cyclodepsipeptide jasplakinolide (1) (a.k.a. jaspamide), isolated previously from the marine sponge Jaspis splendens, is a unique cytotoxin and molecular probe that operates through stabilization of filamentous actin (F-actin). We have recently disclosed that two analogues of 1, jasplakinolides B (3) and E, were referred to the National Cancer Institute's (NCI) Biological Evaluation Committee and the objective of this study was to re-investigate a Fijian collection of J. splendens in an effort to find jasplakinolide congeners with similar biological properties. The current efforts have afforded six known jasplakinolide analogues (4 - 7, 9 - 10), two structures requiring revision (8 and 14) and four new congeners of 1 (11 - 13, 15) including open chain derivatives and structures with modified β-tyrosine residues. Compounds were evaluated for biological activity in the NCI's 60 cell line screen and in a microfilament disruption assay in both HCT-116 and HeLa cells. These two phenotypic screens provide evidence that each cytotoxic analogue, including jasplakinolide B (3), operates by modification of microfilaments. The new structure jasplakinolide V (13) has also been selected for study by the NCI's Biological Evaluation Committee. In addition, the results of a clonogenic dose response study on jasplakinolide are presented.
The unique cyclodepsipeptide jasplakinolide (1) (a.k.a. jaspamide) has been a seed for continuous chemical and biological investigations since its discovery in 1986.1,2 This mixed polyketide-peptide compound (often referred to as a PKS-NRPS hybrid)3 was first isolated from Jaspis (syn: Doryplores) splendens4 (order Astrophorida, family Ancorinidae) sponges collected in Fiji and Palau, and subsequent investigations5-10 have shown that 1 can be obtained from Jaspis sponges collected throughout the Indo-Pacific. We recently disclosed seven new analogues of 1,10 and included an account of the 15 additional natural derivatives divided into two frameworks along with a bio-geographical outline of the two sponge orders that are the source of these metabolites. Other marine sponge genera are also the source of related cyclodepsipeptides named the geodiamolides (Geodia, and Cymbastela)11-16 and the seragamides (Suberites).17 Similar to the jasplakinolide family, the members of these two classes occur in taxonomically distant groups of sponges. The discovery of yet another related PKS-NRPS cyclodepsipeptide ring system represented in chondramides A - D (2a - 2d) isolated from Chondromyces crocatus (a terrestrial myxobacterium)18,19 adds additional insights to a structure vs. bioactivity pattern still being developed. Strikingly, each of the five framework types in this assemblage including the 19-membered macrolides of the jasplakinolide / geodiamolide (i.e. H - I)15 families, and the 18-membered macrolides of the chondramide18,19 / seragamide17 / geodiamolide (i.e. A - G, J - R, TA, and neosiphoniamolide)11-14,16 families, share an identical N-methyl and N-hydrogen pattern. All of these compounds also possess virtually identical absolute configurations at the sites of aryl and alkyl ring substituents. Further, there is remarkable halogenation parallelism between jasplakinolide (1, C36H45BrN4O6) and chondramide D (2d, C35H43ClN4O6).
The push to understand the biological properties of jasplakinolide (1) has been continuous and can be traced back to the initial remarkable spectrum of activities including anthelmenthic,1 insecticidal,2 fungicidal,2 and fish toxicity.20 Also noteworthy was an early observation of nM GI50s for 1 against human prostate carcinoma cell lines (PC-3 = 65, LnCap = 41, and TSU-Pr1 = 170) and that these effects were due to disruption of the actin cytoskeleton through the specific stabilizing interaction with polymerized F-actin (filamentous-actin) and not G-actin (globular-actin).21 Although attempts to prepare 1 tagged with fluorescent probes to facilitate fundamental micro-filament (MF)-based studies never matured, subsequent results showed that 1 exhibits an in vivo stabilizing effect against F-actin.22 The current understanding of the cytotoxicity profile of this scaffold is based on studies of natural jasplakinolide and its analogues conducted against colon (HT-29 or HCT-116) and breast (MCF-7) cancer cells,6-8,10 and complementary results were obtained against renal, prostate and CNS tumor cell lines in the National Cancer Institute's (NCI) 60 cell line screen (see later discussion in connection with the data of Tables 4 - 5).10 A list of structural features that must be present in the jasplakinolide skeleton to impart activity at a nM level can be enumerated as follows: (a) the PKS region of the macrolide must be maintained in a semi-rigid form, and the double bond can be modified to exist as an enone; and (b) there can be no oxidation or derivatization of the aromatic amino acid residues, but the aliphatic amino acids can be altered. In addition, it has also become evident that 1 and 2c have a shared cytotoxicity mechanism via F-actin binding.23
Table 4.
Bioassay Data for Compounds in this Study.
| cmpd. | NSC # | GI50 (μM) | Cell-based microfilament assayb | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HCT-116a | MDA-MB-231a | 0.01 μM | 0.05 μM | 0.1 μM | 0.5 μM | 1.0 μM | 5.0 μM | ||
| 1 | 613009 | 0.04 | 0.01 | - | - | - | +++ | +++ | +++ |
| 3 | 731258 | 0.14c | 0.04 | - | + | + | ++ | ++ | ++ |
| 4 | 751841 | 0.02 | 0.02 | NT | |||||
| 5 | 751842 | 0.11 | 0.26 | NT | |||||
| 6 | 751843 | 0.13 | 0.21 | NT | |||||
| 7 | 751844 | 0.05 | 0.07 | NT | |||||
| 8 | 751837 | 0.06 | 0.09 | - | - | - | +++ | +++ | +++ |
| 9 | 666654 | 13.7c | 2.50 | - | - | - | - | - | - |
| 10 | –d | 0.27c | NT | - | - | - | + | ++ | ++ |
| 11 | 751840 | 0.28c | 0.74 | - | - | - | + | ++ | ++ |
| 12 | 751838 | 11.1 | 22.3 | - | - | - | - | - | - |
| 13 | 751839 | 0.07 | 0.09 | - | + | ++ | +++ | +++ | +++ |
| 14 | 662466 | 3.84 | 2.32 | - | - | - | - | - | - |
| 15 | 752380 | NAe | NA | - | - | - | - | - | - |
GI50 data provided by the National Cancer Institute-Developmental Therapeutics Program (NCI-DTP). For the comprehensive data set against 60 cell lines use the NSCs above at http://dtp.nci.nih.gov/.
The microfilament-disrupting effects were evaluated based on image analysis of the actin cytoskeleton in fixed HCT-116 cells: (+) Loss of microfilament; (++) Additional loss of microfilament; (+++) Total loss of microfilament network; (-) inactive.
IC50 determined by the Valeriote group at Henry Ford Hospital.
10 was not selected for testing by the NCI-DTP.
15 displayed < 25% growth inhibition at 10 μM and was not selected for five-dose testing. NT=not tested, NA=not active.
Table 5.
Selected NCI 60 Cell Line Screening Results for Compounds 1, 4-9, 11 and 13.
| cell line | IGROV-1 | U25-1 | NCI-H522 | DU-145 | A498 | LOX-IMVI |
|---|---|---|---|---|---|---|
| cell type | Ovarian | CNS | Lung | Prostate | Renal | Melanoma |
| cmpd. | GI50 (μM) | |||||
| 1 | 0.02 | 0.07 | 0.03 | 0.03 | 0.03 | 0.01 |
| 4 | 0.008 | 0.01 | 0.03 | 0.02 | 0.002 | 0.003 |
| 5 | 0.02 | 0.05 | 0.61 | 0.29 | 0.21 | 0.03 |
| 6 | 0.03 | 0.06 | 0.20 | 0.25 | 0.17 | 0.02 |
| 7 | 0.01 | 0.03 | 0.16 | 0.10 | 0.08 | 0.02 |
| 8 | 0.03 | 0.03 | 0.003 | 0.31 | 0.04 | 0.04 |
| 9 | 1.76 | 0.60 | 0.12 | 18.8 | NTa | 0.28 |
| 11 | 0.17 | 0.05 | 0.04 | 3.72 | 0.44 | 0.16 |
| 13 | 0.03 | 0.04 | 0.06 | 0.08 | 0.01 | 0.007 |
NT=not tested
Recently published findings provide further insights on the jasplakinolide – chondramide pharmacophore with regards to actin-mediated cytotoxicity. These have been obtained through the examination of compounds obtained through total synthesis, and 10 such pathways exist to create analogues of 1,24-31 2a,32 or 2c.23 Some additional confirmatory results have been obtained through the screening of geodiamolides.33,34 Based on the current literature, we estimate that 45 synthetic congeners have been tested based on the jasplakinolide35-41 and chondramide40,42 scaffolds. Adding to the SAR pattern outlined above are the following: (1) no compound has been obtained with greater potency than 1; (2) C-9 S-configuration is required (in the 1 or 2 framework) for optimal activity;40 (3) modifying the E/Z geometry about the PKS double bond (C-11 and C-12, in the 1 or 2 framework) has a small effect;23,40 (4) inverting the methyl group configuration at C-13/C-15 (in 1) or at C-13/C-14 (in 2) results in only a 10-fold loss in activity;40,42 (5) removal of the N-methyl moiety decreases potency;8,40 and (6) the β-tyrosine unit is essential for binding to the actin target.40
Although a wealth of research has been invested in exploring the experimental therapeutics potential of actin-targeting compounds, the NCI has not identified any leads with a realistic in vivo therapeutic index.43 However, a new theme in the biological profiles for the jasplakinolide class appears to be on the horizon. The selection by the NCI's Biological Evaluation Committee of two analogues, jasplakinolide B (3) and jasplakinolide E, for follow-up in vivo assessment is the first encouraging outcome. Secondly, jasplakinolide (1) has been identified as an antimalarial lead, effecting the growth, invasion and actin cytoskeleton of Plasmodium falciparum.44 The goal of the current study was to further examine J. splendens extracts and compounds in our repository to uncover additional congeners with significant bioactivities; specifically, those with comparable responses in the NCI 60 cell line screen and the microfilament disruption assay vs. that of jasplakinolides B (3) and E. Our new results achieved this intent and involved a study of 12 additional analogues.

Results and Discussion
The decision to continue isolation efforts on the Fijian J. splendens coll no. 00101 proved to be worthwhile and led to several new outcomes. Previously, we analyzed two of its 11 semi-pure extract fractions,10 which afforded only two compounds, the unusual quinazoline analogue jasplakinolide T and the known compound jasplakinolide F.10,45 Further LC-MS scrutiny of the unstudied semi-pure fractions indicated the presence of a series of minor bromine-containing constituents with molecular ion peaks in the range of 700-800 amu. Establishing the identity of these as new or known jasplakinolide congeners proceeded by re-applying our dereplication scheme10 analyzing: (a) LC-MS profiles; (b) 1H NMR signature δs; (c) the pattern of heavy atom formulas for all reported jasplakinolide analogues (C35-37N4O6/7/9, C35-37BrN4O6-7, C36Br2N4O6); and (d) the 1H NMR δs of the C-11 PKS carbon substituents to assign the macrolides as belonging to Group 1 (see 1) or Group 2 (see 3).
All 12 of the compounds isolated herein were either members of Group 1 as macrocycles or their acyclic analogues. A set of four known compounds (4 - 7) was isolated from three different CH2Cl2-soluble preparative RP-HPLC fractions further separated by semi-preparative chromatography (individually coded in the experimental as H7, H8, and H10). The first three were bromine-containing and consisted of a C35 isomeric pair (C35H43BrN4O6: [M+H]+ m/z 695.2/697.2) plus a C37 homolog (C37H47BrN4O6: [M+H]+ m/z 723.2/725.2). These were identified as jasplakinolide D (4),6 jasplakinolide J (5),7 and jasplakinolide M (6)8 by using the 1H NMR patterns for the five methyl groups of jasplakinolide (1) along with the 2D NMR correlations to these methyls as a benchmark for comparison to the data sets for this trio. The fourth compound was assigned as the known debromo analogue of 1, named jasplakinolide Q (7),9 based on comparison to the ESI [M+H]+ peak at m/z 631.3 and the aromatic NMR fingerprint shown in Tables 1 and 2.
Table 1.
13C NMR Data for Compounds 7 - 12 in CD3OD at 150 MHz.
| cmpd. | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|
| pos # | δ C, typea | δC, typea | δ C, typea | δC, typea | δ C, typea | δC, typea |
| 1 | 172.1, C | 172.3, C | 174.8, C | 173.2, C | 172.6, C | |
| 2 | 42.1, CH2 | 42.1, CH2 | 41.9, CH2 | 41.6, CH2 | 41.9, CH2 | |
| 3 | 50.5, CH | 50.6, CH | 51.3, CH | 51.3, CH | 51.3, CH | |
| 4 | 171.3, C | 170.9, C | 171.1, C | 171.3, C | 171.2, C | 175.1, C |
| 5 | 57.9, CH | 56.9, CH | 58.0, CH | 58.1, CH | 58.0, CH | 57.7, CH |
| 6 | 174.9, C | 175.0, C | 176.1, C | 176.3, C | 176.2, C | 175.7, C |
| 7 | 47.1, CH | 47.0, CH | 46.9, CH | 47.0, CH | 47.0, CH | 46.9, CH |
| 8 | 177.9, C | 177.9, C | 179.1, C | 179.3, C | 179.2, C | 179.3, C |
| 9 | 40.8, CH | 40.6, CH | 39.0, CH | 39.0, CH | 39.1, CH | 39.4, CH |
| 10 | 42.6, CH2 | 42.8, CH2 | 44.9, CH2 | 45.1, CH2 | 45.1, CH2 | 44.8, CH2 |
| 11 | 134.1, C | 134.1, C | 131.9, C | 132.0, C | 131.9, C | 132.0, C |
| 12 | 130.6, CH | 130.7, CH | 134.8, CH | 135.0, CH | 134.9, CH | 134.8, CH |
| 13 | 30.5, CH | 30.6, CH | 30.5, CH | 30.6, CH | 30.6, CH | 30.5, CH |
| 14 | 44.1, CH2 | 44.2, CH2 | 48.1, CH2 | 48.2, CH2 | 48.2, CH2 | 48.2, CH2 |
| 15 | 71.7, CH | 71.7, CH | 66.7, CH | 66.9, CH | 66.9, CH | 66.8, CH |
| 16 | 132.7, C | 132.9, C | 133.7, C | 133.4, C | 133.4, C | |
| 17 | 128.2, CH | 128.3, CH | 128.9, CH | 129.0, CH | 129.0, CH | |
| 18 | 116.2, CH | 116.4, CH | 116.2, CH | 116.4, CH | 116.3, CH | |
| 19 | 157.7, C | 157.9, C | 157.8, C | 158.0, C | 158.0, C | |
| 20 | 25.2, CH2 | 25.2, CH2 | 24.5, CH2 | 24.5, CH2 | 26.6, CH2 | 24.8, CH2 |
| 21 | 110.6, C | 110.8, C | 110.8, C | 110.9, C | 110.8, C | 110.8, C |
| 22 | 128.5, C | 127.6, C | 128.8, C | 128.9, C | 128.8, C | 128.9, C |
| 23 | 119.3, CH | 120.9, CH | 119.0, CH | 119.2, CH | 119.1, CH | 119.2, CH |
| 24 | 119.8, CH | 123.8, CH | 120.4, CH | 120.6, CH | 120.5, CH | 120.6, CH |
| 25 | 122.4, CH | 116.5, C | 122.7, CH | 122.9, CH | 122.8, CH | 122.9, CH |
| 26 | 112.3, CH | 114.6, CH | 111.5, CH | 111.7, CH | 111.6, CH | 111.7, CH |
| 27 | 138.1, C | 138.7, C | 137.9, C | 138.0, C | 138.0, C | 138.1, C |
| 28 | 124.2, CH | 111.6, C | 110.3, C | 110.4, C | 110.4, C | 110.5, C |
| 29 | 18.5, CH3 | 18.4, CH3 | 16.0, CH3 | 15.7, CH3 | 15.8, CH3 | 16.3, CH3 |
| 30 | 20.0, CH3 | 20.1, CH3 | 17.0, CH3 | 17.1, CH3 | 17.1, CH3 | 17.5, CH3 |
| 31 | 18.2, CH3 | 18.3, CH3 | 15.7, CH3 | 16.1, CH3 | 16.1, CH3 | 16.2, CH3 |
| 32 | 22.0, CH3 | 22.1, CH3 | 21.4, CH3 | 21.5, CH3 | 21.5, CH3 | 21.5, CH3 |
| 33 | 19.6, CH3 | 19.8, CH3 | 23.3, CH3 | 23.5, CH3 | 23.5, CH3 | 23.5, CH3 |
| 34 | 31.1, CH3 | 31.8, CH3 | 32.8, CH3 | 33.0, CH3 | 33.0, CH3 | 32.7, CH3 |
| 35 | 52.4, CH3 | 61.8, CH2 | ||||
| 36 | 14.7, CH3 | |||||
Carbon type determined from gHMQC data.
Table 2.
1H NMR Data for Compounds 7 - 12 in CD3OD at 600 MHz
| cmpd. | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|
| pos # | δ H (J[Hz]) | δH (J[Hz]) | δH (J[Hz]) | δH (J[Hz]) | δH (J[Hz]) | δ H (J[Hz]) |
| 2 | 2.74, dd (15.2, 4.0) | 2.72, dd (15.4, 4.0) | 3.01, dd (15.6, 9.0) | 3.05, dd (15.8, 9.4) | 3.03, dd (15.8, 9.0) | |
| 2.63, dd (15.2, 9.0) | 2.69, dd (15.4, 8.6) | 2.78, dd (15.6, 6.0) | 2.83, dd (15.8, 5.8) | 2.83, dd (15.8, 5.6) | ||
| 3 | 5.26, dd (9.0, 4.0) | 5.25, dd (8.6, 4.0) | 5.37, dd (9.0, 6.0) | 5.36, dd (9.4, 5.8) | 5.36, dd (9.0, 5.6) | |
| 5 | 5.55, dd (9.0, 7.2) | 5.63, dd (9.5, 7.0) | 5.65, dd (11.4, 4.8) | 5.64, dd (11.4, 4.8) | 5.65, dd (11.4, 4.8) | 5.67, dd (11.6, 4.8) |
| 7 | 4.76, q (6.6) | 4.72, q (6.6) | 4.37, q (7.0) | 4.35, q (7.0) | 4.37, q (6.8) | 4.45, q (7.0) |
| 9 | 2.70, m | 2.69, m | 2.46, m | 2.46, m | 2.47, m | 2.46, m |
| 10 | 2.22, dd (15.4, 11.4) | 2.21, dd (15.4, 11.6) | 2.26, dd (13.4, 5.6) | 2.25, dd (13.4, 5.4) | 2.26, dd (13.4, 5.2) | 2.24, dd (13.5, 6.0) |
| 1.97, d (15.4) | 1.95, br d (15.4) | 1.89, dd (13.4, 9.6) | 1.86, dd (13.4, 9.4) | 1.88, dd (13.4, 9.6) | 1.90, dd (13.5, 8.0) | |
| 12 | 4.90a | 4.88a | 4.91, d (9.6) | 4.89a | 4.91a | 4.93a |
| 13 | 2.34, dddq (9.6, 9.6, 8.4, 5.4) | 2.32, dddq (9.0, 9.0, 8.4, 5.4) | 2.46, m | 2.46, m | 2.47, m | 2.46, m |
| 14 | 1.54, ddd (13.2, 9.8, 5.8) | 1.52, ddd (13.2, 9.6, 6.0) | 1.44, ddd (13.4, 8.4, 6.6) | 1.44, ddd (13.2, 8.4, 6.4) | 1.45, ddd (13.2, 8,4, 6.6) | 1.42, ddd (13.4, 8.2, 6.6) |
| 1.23, ddd (13.2, 8.2, 5.4) | 1.22, ddd (13.2, 8.4, 5.4) | 1.29, dd (13.4, 6.8) | 1.28, ddd (13.2, 6.6, 6.6) | 1.29, ddd (13.2, 6.6, 6.6) | 1.28, ddd (13.4, 6.6, 6.6) | |
| 15 | 4.71, qdd (6.2, 6.0, 6.0) | 4.72, m | 3.69, ddq (6.8, 6.6, 6.2) | 3.68, ddq (6.6, 6.4, 6.2) | 3.64, ddq (6.6, 6.6, 6.2) | 3.68, ddq (6.6, 6.6, 6.0) |
| 17 | 6.91, d (8.5) | 6.95, d (8.5) | 7.18, d (8.5) | 7.16, d (8.5) | 7.16, d (8.5) | |
| 18 | 6.66, d (8.5) | 6.68, d (8.5) | 6.71, d (8.5) | 6.71, d (8.5) | 6.72, d (8.5) | |
| 20 | 3.28a | 3.25, dd (15.0, 7.0) | 3.43, dd (15.2, 4.8) | 3.43, dd (15.2, 4.8) | 3.43, dd (15.0, 4.8) | 3.39, dd (15.2, 4.8) |
| 3.15, dd (15.0, 7.2) | 3.16, dd (15.0, 9.5) | 3.13, dd (15.2, 11.4) | 3.12, dd (15.2, 11.4) | 3.13, dd (15.0, 11.4) | 3.18, dd (11.6, 15.2) | |
| 23 | 7.59, dt (8.0, 1.0) | 7.48, d (8.5) | 7.50, dt (8.0, 1.0) | 7.49, dt (8.0, 0.6) | 7.50, d (8.0) | 7.53, dt (8.0, 0.8) |
| 24 | 7.01, td (8.0, 1.0) | 7.12, dd (8.5, 1.5) | 6.99, td (8.0, 1.0) | 6.99, td (8.0, 1.0) | 7.01, td (8.0, 0.8) | 7.01, td (8.0, 0.8) |
| 25 | 7.09, td (8.0, 1.0) | 7.06, td (8.0, 1.0) | 7.05, td (8.0, 1.0) | 7.07, td (8.0, 0.8) | 7.07, td (8.0, 0.8) | |
| 26 | 7.32, dt (8.0, 1.0) | 7.42, d (1.5) | 7.23, dt (8.0, 1.0) | 7.23, dt (8.0, 0.6) | 7.23, d (8.0) | 7.24, dt (8.0, 0.8) |
| 28 | 7.00, s | |||||
| 29 | 0.92, d (6.6) | 0.79, d (6.6) | 0.65, d (7.0) | 0.66, d (7.0) | 0.66, d (6.8) | 0.58, d (7.0) |
| 30 | 1.11, d (6.8) | 1.09, d (6.8) | 0.98, d (7.0) | 0.97, d (6.8) | 0.98, d (6.8) | 0.97, d (7.0) |
| 31 | 1.61, s | 1.60, s | 1.57, d (1.0) | 1.57, d (1.2) | 1.58, s | 1.57, d (1.2) |
| 32 | 0.87, d (6.6) | 0.86, d (6.6) | 0.91, d (6.6) | 0.90, d (6.6) | 0.91, d (6.6) | 0.89, d (6.6) |
| 33 | 1.06, d (6.2) | 1.08, d (6.6) | 1.11, d (6.2) | 1.11, d (6.2) | 1.12, d (6.2) | 1.11, d (6.0) |
| 34 | 3.09, s | 3.11, s | 3.08, s | 3.06, s | 3.07, s | 3.06, s |
| 35 | 3.65, s | 4.11, q (7.2) | ||||
| 36 | 1.22, t (7.0) | |||||
Shift assigned based on gHMQC and gHMBC data.

An additional compound was isolated from the previously described semi-pure fractions (the one noted above as H10). The properties of this dibromo analogue (C36H44Br2N4O6: [M+H]+ m/z 787.2/789.2/791.2, 1:2:1) of 1 were in agreement with its assignment as jasplakinolide R9 (see regioisomer B in Figure 1), though the literature for this compound included only an 1H NMR data set in CDCl3. While the 1H shifts in CDCl3 of our material were superimposable with those previously published, on completion of our 2D NMR analysis it was clear that this structure was assigned in error. The process we used to clarify the ambiguity in the bromo-indole region chemistry is shown in Figure 1 and it required collecting 1H data in two additional solvents. One powerful result included the 3JH-C gHMBC46 from H-23 (δH 7.48, d, 3JH=8.5, CD3OD) to C-21 (δC 110.8), which required the arrangement of regioisomer A. Apparently spectra obtained in CDCl3 where there is close overlap of H-23 (δH 7.43 (d, JH=8.5) (this work) vs. δH 7.42 (d, JH=8.2)9) and H-26 (δH 7.42 (d, JH=1.2) (this work) vs. δH 7.41 (bs)9) led to an unclear interpretation of the NOE correlations shown for the putative regioisomer B in Figure 1. However in DMSO-d6 these signals were well resolved. Irradiation of the NH-Tyr (δH 8.23 (d, JH = 8.7)), revealed a clear correlation to H-23 (δH 7.59 (d, JH= 8.4): Figure S1, Supporting Information) and provided the final basis for our assignment of the revised structure, named (+)-jasplakinolide R1 (8). The absolute configuration shown for this compound and those following below are based on the obvious biosynthetic analogy to 1.
Figure 1.

Clarifying the bromination regiochemistry for 8 (this work) vs. literature.9
The next phase of this work focused on two macrocyclic and five acyclic analogues isolated from three different CH2Cl2-derived chromatographic fractions and one butanol sub-fraction. One of the most polar compounds, the free acid form of jasplakinolide named (+)-jasplakinolide Z1 (9, C36H47BrN4O7: [M+H]+ m/z 727.3/729.3), was obtained from the butanol solvent partition fraction. This compound, without accompanying physical properties, was first described in 1986,2 as the saponification product of 1, but it was reported again in 2006 accompanied by detailed 1H NMR data.17 Although the careful storage conditions of semi-pure extracts (4°C) containing 9 suggest that it could have been present in the sponge tissue, it is also likely that 9 is a hydrolysis product of 1. Two esters, undoubtedly created from 9 during storage or work-up were also obtained (from CH2Cl2 solvent partition fractions). These consisted of (+)-jasplakinolide Z2 (10, C37H49BrN4O7), with the −OCH3 group identified by a methyl (H3) singlet at δH 3.65, and (+)-jasplakinolide Z3 (11, C38H51BrN4O7), with the −OCH2CH3 group identified by two 1H signals, δH 4.11 (H2, q, JH = 7.2) / 1.22 (H3, t, JH = 7.0). Another truncated acyclic analogue, (-)-jasplakinolide Z4 (12, C27H39BrN4O4: [M+H]+ m/z 563.2/565.2) lacking the β-tyrosine moiety, but with the 2-bromoabrine retained, was straightforwardly characterized. None of the NMR signals for the R2 group of 9 were present in the data for 12 as shown in Tables 1 and 2.
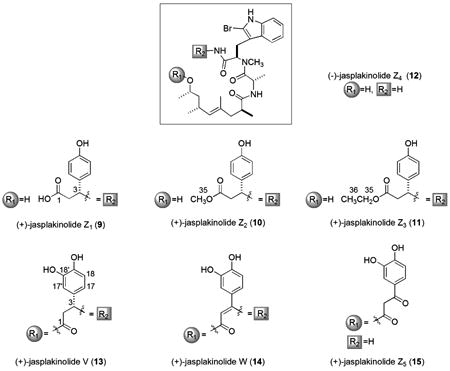
Our characterization of two unique and closely related macrolides (+)-jasplakinolide V (13, C36H45BrN4O7) and (+)-jasplakinolide W (14, C36H43BrN4O7) was streamlined by scrutiny of their MS-derived molecular formulas accompanied by NMR-derived count (Table 3) of the benzenoid ring oxygens. The low-field 1H/13C NMR δs of 13 and 14 clearly showed that the β-tyrosine of 1 was further functionalized with an extra oxygen atom. The molecular formula of the latter was identical to that of jasplakinolide B possessing a hydroquinone,21 described by our laboratory in 1995 (based on unreported 1D NMR data, and not identical to jasplakinolide B / jaspamide B (3), reported in 19995). Assigning the oxy-aryl ring in 13 and 14 as a catechol rather than the hydroquinone was based on the gHMBC correlations from H-17 to C-3 and H-17′ to C-3 shown in Figure 2. Additional three-bond gHMBC correlations from the hydroxy protons observed in DMSO-d6 (18′-OH, δH =8.84, br s; 19-OH δH =8.81, br s) further confirmed the placement of these substituents (18′-OH to C-17′, C-19 and 19-OH to C-18, C-18′). Although an authentic sample of the proposed hydroquinone compound21 was not available from our repository to examine by 2D NMR, we believe that the structure was mis-assigned and must be revised to 14. While the aromatic 13C chemical shifts (C-16 through C-19) matched closely for compounds 13 and 14, there was great disparity for positions C-2 and C-3, as shown in Table 3 (also see Figure S2, Supporting Information). Compound 13 contained methylene and methine carbons (C-2 δC=41.9; C-3 δC=50.7), whereas the shifts of 14 were indicative of a tri-substituted alkene (C-2 δC=101.8; C-3 δC=155.1), allowing the structural assignment shown here. Interestingly, multiple examples of halogenation exist at the same site (C-18′) in other marine sponge metabolites such as the geodiamolides16 and seragamides.17
Table 3.
1H and 13C NMR Data for Compounds 13 - 15 in CD3OD at 600 and 150 MHz, respectively.
| cmpd. | 13 | 14 | 15a | |||
|---|---|---|---|---|---|---|
| pos# | δC, typeb | δH (J[Hz]) | δC, typeb | δH (J[Hz]) | δ C, typeb | δH (J[Hz]) |
| 1 | 172.5, C | 169.5, C | 167.7, C | |||
| 2 | 41.9, CH2 | 2.73, dd (15.2, 4.2) | 101.8, CH | 5.22, s | 46.8, CH2 | 3.90, d (14.8) |
| 2.67, dd (15.2, 8.0) | 3.78, d (14.8) | |||||
| 3 | 50.7, CH | 5.21, dd (8.0, 4.2) | 155.1, C | 191.6, C | ||
| 4 | 171.2, C | 170.6, C | 172.1, C | |||
| 5 | 57.0, CH | 5.71, dd (9.8, 6.4) | 57.9, CH | 6.07, dd (11.4, 4.8) | 56.4, CH | 5.74, dd (11.2, 4.8) |
| 6 | 175.2, C | 175.6, C | 174.2, C | |||
| 7 | 47.2, CH | 4.68, m | 47.3, CH | 4.77, m | 45.7, CH | 4.40, q (6.6) |
| 8 | 177.9, C | 177.8, C | 177.3, C | |||
| 9 | 40.4, CH | 2.65, m | 38.7, CH | 2.71, dqd (11.8, 7.2, 1.2) | 39.7, CH | 2.33, m |
| 10 | 42.6, CH2 | 2.21, dd (15.2, 11.4) | 41.3, CH2 | 2.35, dd (17.0, 11.8) | 43.5, CH2 | 2.19, dd (13.8, 9.2) |
| 1.93, d (15.2) | 1.85, d (17.0) | 1.82, dd (13.8, 4.8) | ||||
| 11 | 134.1, C | 134.7, C | 131.4, C | |||
| 12 | 130.4, CH | 4.83c | 127.7, CH | 4.80c | 132.5, CH | 4.74, d (8.8) |
| 13 | 30.6, CH | 2.30, m | 30.7, CH | 2.41, m | 28.9, CH | 2.03, m |
| 14 | 44.2, CH2 | 1.50, m | 44.8, CH2 | 1.55, ddd (12.2, 12.0, 4.0) | 43.2, CH2 | 1.39, ddd (13.8, 9.0, 6.6) |
| 1.18, m | 1.33, ddd (12.0, 10.8, 3.6) | 1.20, ddd (13.8, 7.8, 4.2) | ||||
| 15 | 71.8, CH | 4.68, m | 70.7, CH | 4.81, m | 71.3, CH | 4.85, dqd (9.0, 6.2, 4.2) |
| 16 | 133.6, C | 127.8, C | 129.3, C | |||
| 17 | 118.5, CH | 6.49, dd (8.2, 2.0) | 121.1, CH | 6.86, dd (8.2, 2.0) | 123.4, CH | 7.41, d (8.2, 1.2) |
| 17′ | 114.5, CH | 6.70, d (2.0) | 116.0, CH | 6.91, d (2.0) | 115.0, C | |
| 18 | 116.5, CH | 6.67, d (8.2) | 116.0, CH | 6.79, d (8.2) | 115.3, CH | 7.52, d (1.2) |
| 18′ | 146.5, C | 146.1, C | 144.5, C | |||
| 19 | 145.8, C | 149.0, C | 150.4, CH | 6.84, d (8.2) | ||
| 20 | 25.1, CH2 | 3.22, m | 24.0, CH2 | 3.26, dd (15.4, 4.8) | 23.5, CH2 | 3.48, dd (15.4, 4.8) |
| 3.18, dd (15.4, 11.6) | 3.17, dd (15.4, 11.2) | |||||
| 21 | 110.3, C | 110.3, C | 110.8, C | |||
| 22 | 128.7, C | 128.9, C | 127.6, C | |||
| 23 | 119.3, CH | 7.54, d (8.0) | 119.2, CH | 7.55, dt (8.0, 0.6) | 118.3, CH | 7.53, d (8.0) |
| 24 | 120.6, CH | 7.00, td (8.0, 1.0) | 120.6, CH | 7.03, td (8.0, 0.8) | 120.5, CH | 7.09, td (8.0, 0.8) |
| 25 | 123.0, CH | 7.07, td (8.0, 1.0) | 123.0, CH | 7.09, td (8.0, 0.8) | 122.7, CH | 7.14, td (8.0, 0.8) |
| 26 | 111.8, CH | 7.25, d (8.0) | 111.7, CH | 7.25, dt (8.0, 0.6) | 110.7, CH | 7.25c |
| 27 | 138.1, C | 138.1, C | 136.2, C | |||
| 28 | 110.6, C | 110.5, C | 109.1, C | |||
| 29 | 18.1, CH3 | 0.66, d (6.6) | 17.9, CH3 | 0.49, d (6.6) | 16.2, CH3 | 0.72, d (6.6) |
| 30 | 20.2, CH3 | 1.08, d (6.8) | 21.3, CH3 | 1.06, d (7.2) | 18.4, CH3 | 1.04, d (6.8) |
| 31 | 18.5, CH3 | 1.59, d (0.8) | 18.9, CH3 | 1.62, s | 16.2, CH3 | 1.29, s |
| 32 | 22.2, CH3 | 0.85, d (6.6) | 22.7, CH3 | 0.87, d (6.6) | 20.5, CH3 | 0.72, d (6.6) |
| 33 | 19.7, CH3 | 1.07, d (6.2) | 19.3, CH3 | 1.14, d (6.2) | 20.6, CH3 | 1.15, d (6.2) |
| 34 | 31.8, CH3 | 3.08, s | 32.1, CH3 | 2.98, s | 32.1, CH3 | 3.10, s |
Spectra recorded in CDCl3
Carbon type determined from gHMQC data
Shift assigned based on gHMQC and gHMBC data
Figure 2.

Choosing between catechol and hydroquinone substructures of 13 and 14 using 1H shifts (14. CD3OD, 600 MHz) and selected gHMBC correlations (CD3OD, DMSO-d6).
The last open-chain congener (+)-jasplakinolide Z5 (15, C36H45BrN4O8) was immediately seen as being different than the other acylics, 9 - 12, described above. The two extra oxygens vs. that of 1 were rationalized by proposing the R1 substructure of 3-(3,4-dihydroxyphenyl)-3-oxopropanoate. The parallel 13C NMR shifts (Table 3 and Figure S2, Supporting Information) at C-16 to C-19 among 13 - 15 supported the catechol (see Figure 2) ring assignment. A series of 2D gHMBC NMR data sets were used to interconnect additional key proximal residues of functional group R1 as follows: (a) correlations from H-17, H-17′ and H2-2 to the low-field carbon (δC=191.6, C-3), and (b) from H2-2 to the ester carbonyl (δC=167.7, C-1). The 2D gHMBC correlation from H-15 to C-1 was also observed and remaining NMR shifts of 15 overlapped with those of 1. We next considered the plausible source of this compound and deemed it unlikely that 15 was a true natural product. Insights from the biosynthetic gene cluster defined for chondramide C (2c),47 suggest the PKS portion of the molecule is assembled first, followed by the final incorporation of the (R)-β-Tyr unit, biosynthesized from L-Tyr.48 Tailoring enzymes subsequently carry out ring closure to create the macrocycle; thus it would be unexpected for a second group of tailoring enzymes to re-open the 19-membered ring at the C3-N bond.
Experiments were launched to pinpoint the pathway responsible for the production of 15. A plausible important clue was provided by the observation that this open chain congener was co-isolated with 14 from the same HPLC sub-fraction. Subsequently, we successfully used 1H NMR data to track the conversion of 14 to 15. Compound 14 was treated with 1% TFA-d in CDCl3 and after six days diagnostic proton shifts for 15 were visible (H2-2 δH=3.78, 3.90; H-5 δH=5.74) as seen in Figure 3 and these two were present in a ratio of 14:15 = 87:13. We conclude that the enamine functionality of 14 is labile and can be slowly hydrolyzed to yield 15.
Figure 3.

Conversion of 14 to 15 with 1% TFA-d.
The combination of prolonged storage for several years at 4 ºC with our standard work-up procedures was the serendipitous recipe for expanding the set of compounds we were able to isolate. An overview of these events is shown in Figure S3, Supporting Information. Jasplakinolide (1) served as the precursor to the acyclic acid 9, which underwent further esterification to produce 10 and 11. As demonstrated above 14 is the precursor for 15 which must be the starting point for further solvolysis providing the dipeptide-PKS analogue 12.
The bilateral approach we employed to probe the bioactivity properties of our first library of jasplakinolide analogues10 was again used to scan for cytotoxic and MF effects of the six known and six new compounds described above. This involved side-by-side tabulation of cytotoxicity data obtained from the National Cancer Institute-Developmental Therapeutic Program (NCI-DTP) 60 cell line screen49 vs. MF-disruption properties provided by the UCSC Chemical Screening Center10 for compounds 1, 4 - 15. A comprehensive array of GI50 parameters was obtained and compounds were also tested at six concentrations in the microfilament assay, in an effort to gain a broad view of their biological responses in this arm of the screening.
A subset of these data are shown in Tables 4 and 5 (the expanded data set from the NCI-DTP screen, including GI50, TGI, and LC50 values for all cell lines can be viewed in the Supporting Information). Our results provide a format to re-evaluate the SAR patterns summarized above and provide some important new insights. In regards to cytotoxicity the following trends merit further discussion. (1) Consistent with the observation that the aliphatic peptide residue can be modified and still retain potency is that the substitution of a 2-Aba (aminobutyric acid) unit in 4 vs. for Ala in 1 is well tolerated6 and enhances potency 10-fold vs. 1 in some human cell lines (Table 5: ovarian adenocarcinoma (IGROV-1), renal carcinoma (A498), melanoma (LOX-IMVI)). (2) While oxidation of the Trp is ruinous to nM activity8,10 this does not apply to the β-Tyr residue as activity is retained in 13 in spite of further aryl hydroxylation. (3) Removal or addition of halogens to the Trp is also tolerated as shown by the data of 7 and 8 in comparison to that of 1.9,40 (4) A conformation change in the macrolide ring induced by the Δ2,3 double bond (13 vs. 14) obliterates nM activity. (5) Our new data suggest a refined view of the SAR at the Trp-N-methyl group (see 6) and C-9 methyl group (see 5), displaying equally potent nM activity in ovarian, CNS, and melanoma cell lines (Table 5: IGROV-1, glioma (U25-1), LOX-IMVI), but diminished activity in other cell lines (Tables 4, 5) vs. 1.7,8,40 (7) Inexplicably, the acyclic analogue jasplakinolide Z3 11 exhibits similar potency to jasplakinolide (1) in colon, CNS, and lung lines (HCT-116, U25-1, NCI-H522) and another acyclic analogue, 9, also showed selectivity for colon, CNS, lung and melanoma cells.
The microfilament disruption effects of analogues 8-15 were first tested in HeLa cells, at concentrations ranging from 10 nM to 5 μM, with the goal of discovering another compound with muted MF properties similar to those of jasplakinolide B (3).10 This first data set in HeLa cells indicated that the acyclic analogue 11 possibly operated through a mechanism not explained through MF association, as a 100-fold divergence between cytotoxicity (HCT-116) and MF activity (HeLa) was observed. However, a second screen in HCT-116 cells with the same concentration gradient provided important additional insights and selected results of both MF screens can be viewed in Figure 4. Compounds 3 and 11 interact with microfilaments at their GI50 values in HCT-116 cells, but the phenotype differs from that of 1, as a complete loss of the microfilament network is not detected as high as 5 μM. Among the four compounds displaying nM cytotoxicity (8, 10, 11, 13 (HCT-116)) only analogues 8 and 13 caused complete loss of the microfilament network, similar to jasplakinolide (1). These MF responses for macrolides 8 and 13 may be attributed to retaining a conformation similar to 1, as the point substitutions in these molecules should not affect the geometry of the Ala-N-Me-2BrTrp-β-Tyr segment. This hypothesis corresponds well with the binding model describing the actin association with chondramide C to F-actin.23,40 Key points in this active site interaction model involve the Trp ring system of chondramide C with aromatic amino acids of F-actin and contact of the Tyr-OH with an actin threonine residue. Thus, it is noteworthy that acyclic compounds 10 and 11 interact with the MF network, while the slightly modified macrocycle 14 loses all binding activity.
Figure 4.

Anti-microfilament effects of compounds 1, 3, 8, 11 and 13 in HCT-116 and HeLa cells. F-actin is colored red and nuclei and chromosomes are colored green. Jasplakinolide (1) is used as a positive control10,22 and negative controls are treated with DMSO. Screens were performed in duplicate and representative images are shown. Multiple images of control wells are included to demonstrate experiment variability.
We have just begun to consider secondary screening work on the jasplakinolide scaffold in a complementary approach designed to develop solid-tumor selective anticancer agents from natural products.50 The first step in assembling a relevant therapeutics data set is to interrogate a compound's solid tumor selectivity beyond that observed in the disk diffusion assay. This assessment involves a clonogenic dose-response study to determine the cytotoxic effect at varying concentrations of the compound for exposure times of two hours, 24 hours and seven days. Finally, a maximum tolerated dose (MTD) is measured and the pharmacokinetic behavior of the compound is measured in both plasma and tumor at the MTD. The combination of these results determines whether a compound shows therapeutic efficacy and if a mechanism of action study should be initiated. Molecules from our laboratory that have shown a favorable profile in this evaluation are fascaplysin A50 and fijianolide B.51
Our results from the clonogenic assay for jasplakinolide (1) are shown in Figure 5. This study determines the required time-concentration profile to obtain a 90% kill of tumor cells. Little toxicity to HCT-116 cells was shown at two hour and 24 hour dosing schedules of 1, however the desired therapeutic effect was achieved at 0.004 μM with continuous exposure for seven days. This outcome indicates that jasplakinolide would require a chronic dosing schedule for maximum therapeutic effectiveness in vivo. The MTD of 1 has been determined as 0.625 mg/kg, and the subsequent pharmacokinetic studies are in progress. In the future similar follow-up work will be launched on jasplakinolides B (3), E, and V (13).
Figure 5.

Results of a clonogenic dose-response study with jasplakinolide (1) in HCT-116 cells. Cells were exposed to different concentrations of 1 for either 2 hrs, 24 hrs, or 7 days (continuous exposure).
Conclusions
In summary, we have extended the structure-activity understanding for the jasplakinolide family of compounds through a side-by-side study of seven macrolides and five closely related acyclic analogues. It is important to reiterate that in addition to jasplakinolide, the solid tumor selective jasplakinolide B (3), jasplakinolide E and jasplakinolide V (13), pinpointed by the NCI Biological Evaluation Committee,10 constitute targets for further therapeutic evaluation. In this work, previously un-scrutinized biological functionality was represented in the structures of the known macrolides 4 – 7, the unique macrolide 13, and compounds 8 and 14 whose structures have been revised. Further, access to the five acyclics 9 - 12 and 15, named as the jasplakinolide Z family, also provided useful data. The nM cytotoxicity and potent microfilament disruption activity of macrolides 4, 7, 8 and 13 vs. the diminished cytotoxicity (≈ 1 uM) for 5 and 6 extend the known structure-activity requirements and actin-binding models based on jasplakinolide (1) and chondramide C (2c). In addition the inactivity of 14 underscores the importance of the macrolide geometry for this binding model. It is significant that 3 and the acyclic analogues 10 and 11 demonstrated broad spectrum sub-micromolar cytotoxicity and associate with microfilaments without complete loss of the actin network. By contrast, the diminished activity of other acyclics such as 12, and inactivity of 15 underlines the importance in the connectivity sequence for the PKS and NPRS subunits. It is also important to emphasize the outcomes of the MF screening in HCT-116 cells. This information provided a realistic view of each compound's cellular actions when viewed in conjunction with the GI50 results in the matched cell line. The new evidence that jasplakinolide B (3) operates by interaction with MF counters our previous hypothesis10 based on HeLa cell screening at 80 nM. It is possible that contradictory results could arise from similar testing of the latrunculin analogues 18-epi-latrunculol A52 and oxalatrunculin B,53 which were reported to have non-MF cytotoxic mechanisms. Overall, the results discussed above highlight that the frameworks of jasplakinolide B (3), jasplakinolide E and jasplakinolide V (13) represent an ideal starting point for scaffold mining and we believe that additional screening of natural and unnatural jasplakinolide compounds will provide a route for significant future innovation.
Experimental Section
General Experimental Procedures
The optical rotations were determined on a Jasco P-2000 digital polarimeter and UV data were obtained on an Agilent 8453 UV-Vis photodiode array spectrophotometer. All NMR spectra were recorded on a Varian Unity Inova 600 spectrometer in CD3OD, CDCl3, or DMSO-d6 with a 5 mm triple resonance (HCN) probe. Chemical shifts are reported in ppm relative to CD3OD (δH 3.31 and δC 49.0), CDCl3 (δH 7.27 and δC 77.4), or DMSO-d6 (δH 2.50 and δC 39.5). A Mariner ESITOF mass spectrometer was used for low- and high-resolution mass measurements. Preparative reversed-phase (RP) separation was carried out utilizing a Waters 600E Prep LC 25 mm radial compression column using 25 × 100 mm C18 Nova-Pak HR16 (6 μm) cartridges. Both ELSD and UV (254 nm) were used for peak detection. Semipreparative RP HPLC used a Phenomenex Synergi C18 4 μm column, 10 × 250 mm, and UV peak detection (230 nm). Compound purity (>95%) was confirmed using both 1H and 13C NMR and LC-MS (UV and ELSD detection) experiments.
Animal Material
Samples of Jaspis splendens (coll. no. 00101, 1.9 kg wet weight) were collected in Fiji (S 18°22.28′, E 177°58.87′) February 2000. Taxonomic identification of Jaspis splendens was performed by Dr. Rob van Soest of the Zoological Museum of Amsterdam. Underwater pictures are in Figure S11, Supporting Information, and voucher specimens are available from the corresponding author (P.C.).
Extraction and Isolation
Sponge materials were preserved in the field in 1:1 CH3OH : H2O, decanted, and shipped back to UC Santa Cruz where they were then submersed in 100% CH3OH and stored at 4 °C. For extraction, the CH3OH was decanted and the sponge was soaked in 1L CH3OH four more times to obtain the crude CH3OH extract. The extract was partitioned between 1:1 hexanes and CH3OH-H2O (9:1). After separation from the hexane layer, H2O was added to the CH3OH-H2O layer adjusting the solution to 1:1. The aqueous layer was then extracted with CH2Cl2 to give two crude extracts called FM (CH3OH-H2O soluble, 1.6 grams) and FD (CH2Cl2-soluble, 2.1 g). The FM extract was then partitioned between H2O and butanol to provide the WB extract (butanol-soluble, 150.1 mg).
Next, 00101 FD was subjected to preparative RP HPLC (10 – 65% CH3CN : H2O, 50 min) yielding 11 fractions (H1 - H11). Fractions H4 (105.5 mg), H7 (145.3 mg), H8 (159.2 mg), H10 (198.4 mg) and H11 (192.5 mg) were purified further by RP HPLC utilizing isocratic conditions. Fraction H4 provided 12 (2.1 mg, H4 H4 H2) through repetitive semi-preparative HPLC (70% CH3OH : H2O). Fraction H7 was treated in a similar manner (50% CH3CN : H2O, 2 iterations) to give 10 (13.6 mg, H7 H8 H2) and 6 (1.2 mg, H7 H10 H3 H4). Fraction H8 (159.2 mg) was also separated via multiple rounds of isocratic (77% CH3OH : H2O) RP HPLC to yield 11 (2.8 mg, H8 H4 H2), 13 (16.6 mg, H8 H4 H3), 7 (0.4 mg, H8 H5 H6 H3 H4 H2), and 5 (1.1 mg, H8 H5 H6 H3 H3 H4 H3). Fraction H10 was first subjected to preparative isocratic (57% CH3CN : H2O) RP HPLC resulting in 10 fractions, of which fractions H10 H4 (7.8 mg) and H10 H6 (3.9 mg) were further purified by semi-preparative RP HPLC (80% CH3OH : H2O) to give 4 (0.6 mg, H10 H4 H6) and 8 (1.5 mg, H10 H6 H4). Fraction H11 was purified by one round of preparative isocratic (60% CH3CN : H2O) RP HPLC and two rounds of semi-preparative isocratic (85% CH3OH : H2O, 60% CH3CN : H2O) RP HPLC resulting in 15 (3.5 mg, H11 H4 H1) and 14 (13.0 mg, H11 H4 H3).
Similarly, the WB extract was separated by RP HPLC (10-65% CH3CN : H2O, 60 min) giving 11 fractions (H1 – H11). Fraction H9 (37.8 mg) was then subjected to isocratic (44% CH3CN : H2O) semi-preparative HPLC to yield 9 (4.0 mg, H9 H2).
(+)-Jasplakinolide R1 (8)
White solid (1.5 mg). [α]24D +48 (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 203 (1.43), 227 (1.24), 281 (0.27) nm; 1H and 13C NMR (see Tables 1 and 2); HRESITOFMS [M+Na]+ m/z 809.1482 (calcd for C36H44Br2N4O6Na, 809.1520).
(+)-Jasplakinolide Z1 (9)
White solid (4.0 mg). [α]23D +37 (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 202 (0.76), 222 (0.59), 280 (0.12) nm; 1H and 13C NMR (see Tables 1 and 2); HRESITOFMS [M+Na]+ m/z 749.2590 (calcd for C36H47BrN4O7Na, 749.2520).
(+)-Jasplakinolide Z2 (10)
White solid (13.6 mg). [α]23D +46 (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 218 (1.30), 283 (0.38), 290 (0.33) nm; 1H and 13C NMR (see Tables 1 and 2); HRESITOFMS [M+H]+ m/z 741.2866 (calcd for C37H50BrN4O7, 741.2857).
(+)-Jasplakinolide Z3 (11)
White solid (2.8 mg). [α]23D +64 (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 220 (0.41), 283 (0.15), 290 (0.16); 1H and 13C NMR (see Tables 1 and 2); HRESITOFMS [M+Na]+ m/z 777.2885 (calcd for C38H51BrN4O7Na, 777.2833).
(-)-Jasplakinolide Z4 (12)
White solid (2.1 mg). [α]22D -32 (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 204 (1.48), 220 (1.37), 282 (0.32); 1H and 13C NMR (see Tables 1 and 2); HRESITOFMS [M+H]+ m/z 563.2230 (calcd for C27H40BrN4O4, 563.2227).
(+)-Jasplakinolide V (13)
White solid (16.6 mg). [α]23D +120 (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 203 (0.95), 220 (0.60), 282 (0.16); 1H and 13C NMR (see Table 3); HRESITOFMS [M+Na]+ m/z 747.2319 (calcd for C36H45BrN4O7Na, 747.2364).
(+)-Jasplakinolide W (14)
White solid (13.0 mg). [α]23D +60 (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 204 (0.85), 218 (0.75), 281 (0.29); 1H and 13C NMR (see Table 3); HRESITOFMS [M+H]+ m/z 723.2397 (calcd for C36H44BrN4O7, 723.2388).
(+)-Jasplakinolide Z5 (15)
White solid (3.5 mg). [a]23D +15° (c, 0.05, MeOH); UV (MeOH) λmax (log ε): 203 (0.55), 222 (0.45), 280 (0.18); 1H and 13C NMR (see Table 3); HRESITOFMS [M+Na]+ m/z 763.2322 (calcd for C36H45BrN4O8Na, 763.2313).
Conversion of 14 to 15
Compound 14 (2 mg) was dissolved in 1.4 mL of CDCl3 and the solution was divided between two 5 mm NMR tubes. Using a gas-tight syringe, 7 μL of TFA-d was added to one tube. 1H NMR spectra were acquired for both samples on the first day and again after six days. Spectra are displayed in Figure 3.
Microfilament Disruption Assay
HCT-116 or HeLa cells were plated in 384-well tissue culture treated plates at a density of 1500 cells per well. After incubating at 37 °C with 5% CO2 overnight compounds were pinned into plates using an automated liquid handler. After 24 h cells were fixed in 4% formaldehyde for 20 min then washed with PBS using an automated plate washer. The cells were then treated with 0.5% TritonX-100 in PBS for 10 min, washed, and then blocked with a 2% BSA PBS solution for 20 min. Actin was stained with rhodamine-phalloidin for 20 min and then washed. Lastly, the DNA was stained with Hoechst 33342 (AnaSpec Inc.) followed by a wash with the automated plate washer. The plates are then stored in a 0.1 % azide PBS solution and images were taken using an automated fluorescence microscope at 20× magnification after 24 h.
Jasplakinolide (1) Clonogenic Dose-Response Analysis
Concentration and time-survival studies of 1 were carried out against HCT-116 cells. HCT-116 cells were seeded at 200 to 20 000 cells in 60 mm dishes. Drug was added to the medium (RPMI + 10% FBS) at concentrations of 1 mg/mL and 10-fold dilutions thereof. At either 2 h or 24 h, the drug-containing media was removed and fresh media without drug was added. For continuous exposure to the drug, it remained in contact with the cells for the entire incubation period. The dishes were incubated for 7 days, media was removed, and the colonies were stained with methylene blue. Colonies containing 50 cells or more were counted. The results were normalized to an untreated control. Plating efficiency for the untreated cells was about 90%. Repeat experiments were carried out to define the cell survival range between 100% and 0.1% survival. The results of this analysis are shown in Figure 5.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (RO1 CA 047135 and U19 CA52955), by NMR equipment grants NSF CHE 0342912 and NIH S10 RR19918, by the U.S. Civilian Research and Development Foundation (GTR-G7-044) and the California Institute for Quantitative Biomedical Research. We thank Dr. R. van Soest for taxonomic identification.
Footnotes
Dedicated to Dr. Koji Nakanishi of Columbia University for his pioneering work on bioactive natural products.
Supporting Information Available: 1H, 13C, gHMQC and gHMBC NMR spectra for compounds 6-15, a sponge picture of 00101, NCI 60 cell line results, and microfilament effects of 10. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Crews P, Manes LV, Boehler M. Tetrahedron Lett. 1986;27:2797–2800. [Google Scholar]
- 2.Zabriskie TM, Klocke JA, Ireland CM, Marcus AH, Molinski TF, Faulkner DJ, Xu C, Clardy JC. J Am Chem Soc. 1986;108:3123–3124. [Google Scholar]
- 3.Du L, Shen B. Curr Opin Drug Discovery Dev. 2001;4:215–228. [PubMed] [Google Scholar]
- 4.These sponges were once designated as Jaspis johnstoni by our lab, but their identification was revised to Jaspis splendens: Sanders M, Diaz MC, Crews P. Mem Queens Museum. 1999;44:525–532.
- 5.Zampella A, Giannini C, Debitus C, Roussakis C, D'Auria MV. J Nat Prod. 1999;62:332–334. doi: 10.1021/np9803225. [DOI] [PubMed] [Google Scholar]
- 6.Gala F, Zampella A, De Marino S, Zollo F, Smith CD, Copper JE, D'Auria MV. Tetrahedron. 2007;63:5212–5219. [Google Scholar]
- 7.Gala F, D'Auria MV, De Marino S, Sepe V, Zollo F, Smith CD, Copper JE, Zampella A. Tetrahedron. 2008;64:7127–7130. [Google Scholar]
- 8.Gala F, D'Auria MV, De Marino S, Sepe V, Zollo F, Smith CD, Keller SN, Zampella A. Tetrahedron. 2009;65:51–56. [Google Scholar]
- 9.Ebada S, Wray V, de Voogd N, Deng Z, Lin W, Proksch P. Mar Drugs. 2009;7:435–444. doi: 10.3390/md7030435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson SJ, Morinaka BI, Amagata T, Tenney K, Bray WM, Gassner NC, Lokey RS, Crews P. J Med Chem. 2010;53:1651–1661. doi: 10.1021/jm9013554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan WR, Tinto WF, Manchand PS, Todaro LJ. J Org Chem. 1987;52:3091–3093. [Google Scholar]
- 12.de Silva ED, Andersen RJ, Allen TM. Tet Lett. 1990;31:489–492. [Google Scholar]
- 13.Talpir R, Benayahu Y, Kashman Y, Pannell L, Schleyer M. Tetrahedron Lett. 1994;35:4453–4456. [Google Scholar]
- 14.Coleman JE, de Silva ED, Kong F, Andersen RJ, Allen TM. Tetrahedron. 1995;51:10653–10662. [Google Scholar]
- 15.Tinto WF, Lough AJ, McLean S, Reynolds WF, Yu M, Chan WR. Tetrahedron. 1998;54:4451–4458. [Google Scholar]
- 16.Coleman JE, Van Soest R, Andersen RJ. J Nat Prod. 1999;62:1137–1141. doi: 10.1021/np990155o. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka C, Tanaka J, Bolland RF, Marriott G, Higa T. Tetrahedron. 2006;62:3536–3542. [Google Scholar]
- 18.Kunze B, Jansen R, Sasse F, Hoefle G, Reichenbach H. J Antibiot. 1995;48:1262–1266. doi: 10.7164/antibiotics.48.1262. [DOI] [PubMed] [Google Scholar]
- 19.Jansen R, Kunze B, Rieichenbach H, Hoefle G. Liebigs Ann. 1996:285–290. [Google Scholar]
- 20.Braekman JC, Daloze D, Moussiax B, Riccio R. J Nat Prod. 1987;50:994–995. [Google Scholar]
- 21.Senderowicz AMJ, Kaur G, Sainz E, Laing C, Inman WD, Rodrigues J, Crews P, Malspeis L, Grever MR, Sausville EA, Duncan KLK. J Natl Cancer Inst. 1995;87:46–51. doi: 10.1093/jnci/87.1.46. [DOI] [PubMed] [Google Scholar]
- 22.Bubb MR, Senderowicz AMJ, Sausville EA, Duncan KLK, Korn ED. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- 23.Waldmann H, Hu TS, Renner S, Menninger S, Tannert R, Oda T, Arndt HD. Angew Chem Int Ed. 2008;47:6473–6477. doi: 10.1002/anie.200801010. [DOI] [PubMed] [Google Scholar]
- 24.Grieco PA, Hon YS, Perezmedrano A. J Am Chem Soc. 1988;110:1630–1631. [Google Scholar]
- 25.Schmidt U, Siegel W, Mundinger K. Tetrahedron Lett. 1988;29:1269–1270. [Google Scholar]
- 26.Chu KS, Negrete GR, Konopelski JP. J Org Chem. 1991;56:5196–5202. [Google Scholar]
- 27.Imaeda T, Hamada Y, Shioiri T. Tetrahedron Lett. 1994;35:591–594. [Google Scholar]
- 28.Hirai Y, Yokota K, Momose T. Heterocycles. 1994;39:603–612. [Google Scholar]
- 29.Ghosh AK, Moon DK. Org Lett. 2007;9:2425–2427. doi: 10.1021/ol070855h. [DOI] [PubMed] [Google Scholar]
- 30.Ashworth P, Broadbelt B, Jankowski P, Kocienski P, Pimm A, Bell R. Synthesis. 1995:199–206. [Google Scholar]
- 31.Tannert R, Hu TS, Arndt HD, Waldmann H. Chem Commun. 2009:1493–1495. doi: 10.1039/b900342h. [DOI] [PubMed] [Google Scholar]
- 32.Schmauder A, Sibley LD, Maier ME. Chem Eur J. 2010;16:4328–4336. doi: 10.1002/chem.200903500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rangel M, Prado MP, Konno K, Naoki H, Freitas JC, Machado-Santelli GM. Peptides. 2006;27:2047–2057. doi: 10.1016/j.peptides.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 34.Freitas VM, Rangel M, Bisson LF, Jaeger RG, Machado-Santelli GM. J Cell Physiol. 2008;216:583–594. doi: 10.1002/jcp.21432. [DOI] [PubMed] [Google Scholar]
- 35.Terracciano S, Bruno I, Bifulco G, Copper JE, Smith CD, Gomez-Paloma L, Riccio R. J Nat Prod. 2004;67:1325–1331. doi: 10.1021/np049955b. [DOI] [PubMed] [Google Scholar]
- 36.Terracciano S, Bruno I, Bifulco G, Avallone E, Smith CD, Gomez-Paloma L, Riccio R. Bioorg Med Chem. 2005;13:5225–5239. doi: 10.1016/j.bmc.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 37.Marimganti S, Yasmeen S, Fischer D, Maier ME. Chem Eur J. 2005;11:6687–6700. doi: 10.1002/chem.200500319. [DOI] [PubMed] [Google Scholar]
- 38.Celanire S, Descamps-Francois C, Lesur B, Guillaumet G, Joseph B. Lett Org Chem. 2005;2:528–531. [Google Scholar]
- 39.Terracciano S, Bruno I, D'Amico E, Bifulco G, Zampella A, Sepe V, Smith CD, Riccio R. Bioorg Med Chem. 2008;16:6580–6588. doi: 10.1016/j.bmc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 40.Tannert R, Milroy LG, Ellinger B, Hu TS, Arndt HD, Waldmann H. J Am Chem Soc. 2010;132:3063–3077. doi: 10.1021/ja9095126. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh AK, Dawson ZL, Moon DK, Bai R, Hamel E. Bioorg Med Chem Lett. 20:5104–5107. doi: 10.1016/j.bmcl.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eggert U, Diestel R, Sasse F, Jansen R, Kunze B, Kalesse M. Angew Chem Int Ed. 2008;47:6478–6482. doi: 10.1002/anie.200801156. [DOI] [PubMed] [Google Scholar]
- 43.Newman DJ, Cragg GM, Holbeck S, Sausville EA. Curr Cancer Drug Targets. 2002;2:279–308. doi: 10.2174/1568009023333791. [DOI] [PubMed] [Google Scholar]
- 44.Fotie J, Morgan RE. Mini Rev Med Chem. 2008;8:1088–1094. doi: 10.2174/138955708785909916. [DOI] [PubMed] [Google Scholar]
- 45.The structure for jasplakinolide F (C-30 demethyl, C-31 methyl) was incorrectly displayed in our previous paper as jasplakinolide J (C-30 methyl, C-31 demethyl).10 Jasplakinolide F and J were both isolated from HPLC fraction H8.
- 46.Rinaldi PL, Keifer P. (A).J Magn Reson. 1994;108:259–62. [Google Scholar]
- 47.Rachid S, Krug D, Kunze B, Kochems I, Scharfe M, Zabriskie TM, Blocker H, Muller R. Chem Biol. 2006;14:667–681. doi: 10.1016/j.chembiol.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Rachid S, Krug D, Weissman KJ, Muller R. J Biol Chem. 2007;282:21810–21817. doi: 10.1074/jbc.M703439200. [DOI] [PubMed] [Google Scholar]
- 49.Shoemaker RH. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 50.Subramanian B, Nakeff A, Tenney K, Crews P, Gunatilaka L, Valeriote F. J Exp Therapeut Oncol. 2006;5:195–204. [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson TA, Tenney K, Cichewicz RH, Morinaka BI, White KN, Amagata T, Subramanian B, Media J, Mooberry SL, Valeriote FA, Crews P. J Med Chem. 2007;50:3795–3803. doi: 10.1021/jm070410z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amagata T, Johnson TA, Cichewicz RH, Tenney K, Mooberry SL, Media J, Edelstein M, Valeriote FA, Crews P. J Med Chem. 2008;51:7234–7242. doi: 10.1021/jm8008585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed SA, Odde S, Daga PR, Bowling JJ, Mesbah MK, Youssef DT, Khalifa SI, Doerksen RJ, Hamann MT. Org Lett. 2007;9:4773–4776. doi: 10.1021/ol7020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.