

Abstract
Ligands from the naltrexamine series have consistently demonstrated agonist activity at kappa opioid receptors (KOR), with varying activity at the mu opioid receptor (MOR). Various 6β-cinnamoylamino derivatives were made with the aim of generating ligands with a KOR agonist/MOR partial agonist profile, as ligands with this activity may be of interest as treatment agents for cocaine abuse. The ligands all displayed the desired high affinity, non-selective binding in vitro and in the functional assays were high efficacy KOR agonists with some partial agonist activity at MOR. Two of the new ligands (12a, 12b) have been evaluated in vivo, with 12a acting as a KOR agonist, and therefore somewhat similar to the previously evaluated analogues 3–6, while 12b displayed predominant MOR agonist activity.
Introduction
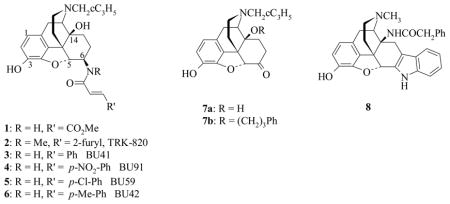
Various studies have demonstrated that kappa opioid receptor (KOR) agonists can reduce, or even prevent, some of the behavioural effects caused by cocaine.1–4 Selective KOR agonists are, however, unlikely to be seriously considered as treatment agents as they themselves cause dysphoria, both in drug naïve individuals and in those with a history of drug use.5 Preclinical evidence suggests that a more effective approach may be to target KOR-agonists that also display partial agonist activity at the mu opioid receptor (MOR).4,6 Compounds of this type, such as EKC, are more effective in reducing cocaine self-administration, and display fewer side effects, than their more selective counterparts such as enadoline and spiradoline.4 Derivatives of β-naltrexamine (β-NTA) have consistently demonstrated KOR agonist properties. The most well known of this series is β-FNA (1), a ligand widely utilized for its irreversible MOR antagonist properties, but which also displays short-lived KOR agonist effects.7–9 More recently, in a search for potent KOR agonists from the β-NTA series, TRK-820 (2) emerged as the lead compound identified from a comprehensive study of such derivatives.10 However TRK-820 was the result of optimisation for high KOR selectivity and potency and therefore is not an ideal candidate for further development against cocaine. Independently we found the cinnamoyl derivatives (3–6) to have KOR agonist activity in vivo,11,12 but we did not seek to optimise for KOR selectivity and found that with no methyl group on the 6β-nitrogen (where a methyl group is found in TRK-820) the ligands display equal affinity at MOR and KOR and are therefore potentially more interesting. The effect of the NMe group replicates the importance of having an N-alkyl group (usually NMe), rather than NH in the arylacetamide series of KOR agonists.13
The cinnamoyl derivatives (3–6) have KOR agonist activity in vivo,11,12 with 6 and 5 similar to β-FNA, having short-lived KOR agonist effects and then substantial MOR antagonist activity, while 3 and 4 were potent KOR agonists in both tail withdrawal (TW) and antiwrithing (AW) assays.11,12 The aim of the current work was therefore to increase the MOR efficacy of this series to achieve the desired, mixed KOR/MOR profile.
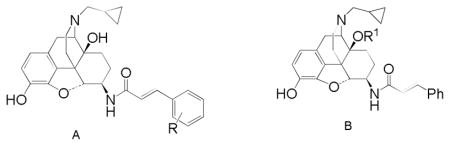
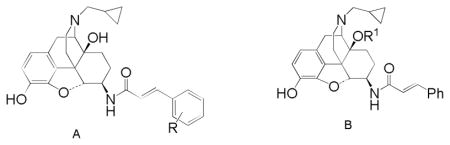
In related series of 14-cinnamoylaminomorphinones,14 substitution at the 2′-position of the cinnamoyl aryl ring produced ligands with significantly altered efficacy compared to their 4′-substituted homologues and so the 2′-substituted analogues of 3–6 were targeted to see if a similar effect was observed in the β-NTA series.
It has also become clear that introduction of substituents to the C14-hydroxyl group can lead to an increase in MOR affinity and efficacy. The effect is most robust when a three carbon chain, or substituted three carbon chain is used. The most dramatic example of this effect is provided by Schmidhammer and colleagues15 who converted the MOR antagonist naltrexone (7a) into a powerful MOR agonist by substitution of a phenylpropyl group in the C14-hydroxyl (7b). In addition, the 14-phenylacetyloxy derivatives were targeted as introduction of the phenylacetyl group in 14-amino analogues of naltrindole (8) and in 14-aminodihydromorphinones and codeinones also raised MOR efficacy.16,26
Chemistry
The synthesis of the target ligands relied on the efficient production of the key intermediate β-NTA 10 which can be achieved by reductive amination of naltrexone 7a with dibenzylamine followed by hydrogenolysis of the benzyl groups (Scheme 1).17 Although this synthetic approach is well established, in our hands removal of the benzyl group proved problematic and we opted for an alternative method. Portoghese and colleagues have reported a 9:1 mixture of β:α-diastereoisomers obtained from the reduction of naltrexone oxime (11a) with BH3-THF.18 In our hands, using BH3-Me2S, the result was 7:1 and required rigorous silica gel chromatography to obtain the pure β-NTA 10. It is proposed that in the course of the reaction the boron-complexed oxime mimics the bulky dibenzyliminium formed during the preparation of 9 and exists in a pseudochair conformation, hence exposing the α-face to preferential reduction.17,18 Therefore, we chose to add bulk to the oxime and synthesised ethers 11b, 11c by quantitative oximation of naltrexone with the relevant alkoxylamines. Upon reduction of the methyl oxime ether 11b with ZrCl4/NaBH4 (scheme 1) the stereoselectivity was improved to 13:1 in favour of the desired β-diastereoisomer 10. Reducing the benzyl ether 11c gave almost equal stereoselectivity (12:1). This new method, via 11c, was adopted for the present study and gram quantities of 10 could be reproducibly obtained in 2 steps with one chromographic purification. The β-configuration of the 6-amino group was confirmed by comparison with previously reported data on this compound.17,18 In particular, a coupling constant of 6.8 Hz was determined for H-5.
Scheme 1.

Reagents and conditions: (a) HNBn2, PhCOOH, TsOH then NaCNBH3, EtOH, Δ, 20 h, (69%); (b) 10% Pd/C, 40 PSI, H2, MeOH/HCl, 3 days; c) 10% Pd/C, 1:1 Cyclohexene/MeOH, Δ, 24 h; (d) o-Benzylhydroxylamine, NaOAc, EtOH/H2O, Δ, 20h; (e) ZrCl4/NaBH4, THF, Δ, 24h 42% (for R = Bn).
Synthesis of the target C-6-β amides 3, 12a–d was achieved by acylation of β-NTA (10) with the relevant cinnamoyl chlorides (Scheme 2). The 14-O-acyl targets (15) could be accessed from 3 by protection of the 3-phenol and then acylation of the C14-hydroxyl (Scheme 2).
Scheme 2.

Reagents and conditions: (a) RC6H4CH=CHCOCl, NEt3, CH2Cl2, 25°C, 0.5h (b) TBDMSCl, imidazole, DCM, (73%); (c) anhydride (R′OR′), toluene, reflux, 24h (d) TBAF, THF, RT.
Due to the low reactivity of the tertiary alcohol, alkylation of the C14-hydroxyl required forcing conditions and protection of the 3-phenol as the methyl ether (Scheme 3). Introduction of the 6β-amino group (17) was again accomplished using the method of asymmetric reduction of the ketoxime (16).
Scheme 3.

Reagents and conditions: a) MeI, K2CO3, MeCOMe/H2O, (73 %); b) o-Benzylhydroxylamine, NaOAc, EtOH/H2O; (c) ZrCl4/NaBH4, THF, Δ, 24h, (69%); (d) Cinnamoyl chloride, Et3N, DCM, RT, (83%); (e) R1Br, NaH, DMF; (f) BBr3, DCM, 0 °C.
Results
Binding affinities of the new compounds for MOR and KOR were determined by competition with [3H]-diprenorphine binding in C6-rat glioma cells expressing recombinant rat MOR and CHO cells expressing recombinant human KOR.19 DOR activity of the new ligands was not determined as in no case has DOR activity proved relevant to the pharmacological profile of ligands with significant MOR and/or KOR efficacy. All of the ligands bound with high to very high affinity to both KOR and MOR, with little selectivity for either receptor (Table 1). Neither the nature, nor position of substituents on the phenyl ring of the cinnamoyl group substantially affected affinities and similarly, introduction of substituents at C14 also had relatively little effect.
Table 1.
Binding affinities determined by displacement of [3H]diprenorphine binding to C6-μ and CHO-κ membranes
 | |||
|---|---|---|---|
| Comp’d | Type & Substituent | Ki/nM | |
| Mu | Kappa | ||
| 3 | A: R = H | 0.17 ± 0.091 | 0.043 ± 0.0022 |
| 12a | A: R = o-NO2 | 0.35 ± 0.13 | 0.30 ± 0.094 |
| 12b | A: R = m-NO2 | 0.11 ± 0.036 | 0.36 ± 0.32 |
| 12c | A: R = o-Cl | 0.079 ± 0.027 | 0.27 ± 0.062 |
| 12d | A: R = o-Me | 0.085 ± 0.033 | 0.34 ± 0.16 |
| 20a | B: R1 = CH2C6H5 | 0.16 ± 0.010 | 0.86 ± 0.22 |
| 20b | B: R1 = CH2CH=CH2 | 0.25 ± 0.074 | 0.12 ± 0.014 |
| 15a | B: R1 = COCH3 | 0.21 ± 0.059 | 0.59 ± 0.069 |
| 15b | B: R1 = COC6H5 | 0.42 ± 0.19 | 1.1 ± 0.097 |
| 15c | B: R1 = COCH2C6H5 | 0.28 ± 0.055 | 0.26 ± 0.0098 |
Values are an average ± SEM from three separate experiments
The in vitro assay used to determine opioid receptor functional activity was the [35S]GTPγS binding stimulation assay which, like the binding assays, was performed on recombinant opioid receptors transfected into C6-rat glioma cells (for MOR) and CHO cells (for KOR). Assays were performed as previously described by Traynor and Nahorski.20 Agonist efficacy at these opioid receptors was determined in comparison to the standard selective agonists DAMGO (MOR), and U69593 (KOR) (Table 2). The ligands were, as expected, agonists at the KOR, efficacies mostly ranging from 76% to 120% relative to U69593, the exception being 15b which had only low efficacy and potency KOR antagonist activity; otherwise potencies were in the low to sub-nanomolar range. The ring substituented analogues (12a – 12d) had very similar efficacy at KOR, though at 81% stimulation 12a, the o-nitro analogue, was somewhat lower than the rest. There was slightly greater variation in the KOR efficacy of the C14-substituted analogues, with the allyl substituted (20b) displaying the highest efficacy of the compounds in this study.
Table 2.
Stimulation of [35S]GTPγS binding to C6-μ and CHO-κ membranes
 | |||||
|---|---|---|---|---|---|
| Comp’d | Type & Substituent | Mu | Kappa | ||
| EC50/nM | %DAMGO | EC50/nM | %U69593 | ||
| 3 | A: R = H | 1.8 ± 0.79 | 21 ± 1.5 | 0.12 ± 0.037 | 100 ± 4.5 |
| 12a | A: R = o-NO2 | 0.69 ± 0.31 | 6.7 ± 0.73 | 1.9 ± 0.055 | 81 ± 2.4 |
| 12b | A: R = m-NO2 | 0.18 ± 0.062 | 20 ± 3.4 | 0.12 ± 0.039 | 100 ± 3.3 |
| 12c | A: R = o-Cl | 12 ± 8.7 | 17 ± 1.6 | 0.40 ± 0.16 | 97 ± 3.1 |
| 12d | A: R = o-Me | 2.1 ± 2.0 | 21 ± 1.4 | 0.95 ± 0.69 | 105 ± 6.0 |
| 20a | B: R1 = CH2C6H5 | 2.0 ± 0.38 | 19 ± 2.1 | 0.78 ± 0.16 | 97 ± 6.2 |
| 20b | B: R1 = CH2CH=CH2 | 0.60 ± 0.10 | 39 ± 1.8 | 0.079 ± 0.012 | 120 ± 3.0 |
| 15a | B: R1 = COCH3 | 1.0 ± 0.15 | 19 ± 0.86 | 2.4 ± 0.99 | 97 ± 7.1 |
| 15b | B: R1 = COC6H5 | - | N.Sa | 9.8 ± 5.3 | 26 ± 6.0 |
| 15c | B: R1 = COCH2C6H5 | 1.3 ± 0.21 | 41 ± 3.9 | 0.49 ± 0.053 | 76 ± 2.1 |
Values are an average ± SEM from three separate experiments
N.S. = No stimulation; 15b was evaluated as an antagonist at MOR, Ke (vs DAMGO) 0.41 ± 0.12 nM
All of the new ligands were low efficacy partial agonists at MOR, with the 14-substituted series providing the highest efficacy compounds (20b and 15c). Again 15b stood out for having a substantially different profile to the other ligands, it did not stimulate MOR and was in fact a high potency MOR antagonist.
The unsubstituted 6β-cinnamoylnaltrexamine (3) has previously been evaluated in vivo in the warm water tail withdrawal and acetic acid induced abdominal stretch assays and found to be a potent and efficacious KOR agonist.12 For comparison, two of the current series of ligands were chosen for preliminary evaluation in the similar paraphenylquinone (PPQ) induced abdominal stretch assay,21 an assay in which MOR and KOR agonists of even relatively low efficacy would be expected to show activity. 12b was chosen as it has a very similar profile to 3 in vitro, while the second choice, 12a, had lower efficacy in vitro at both MOR and KOR. 12a was found to be an agonist of quite low potency (ED50 18.5 (13.2 – 26.1) mg/kg, s.c.) and the effect of an ED80 concentration of 12a could be blocked by norBNI (AD50 5.28 mg/kg, s.c.), a selective KOR antagonist but not by the MOR antagonist β-FNA (up to 30 microg icv) or the DOR antagonist naltrindole (NTI) up to 30 mg/kg sc. In contrast 12b was significantly more potent (ED50 3.5 (1.6 – 7.7) mg/kg, s.c.) and the effect of an ED80 dose was blocked by β-FNA (AD50 7.1 μg/icv), inconsistently by norBNI (38% at 1, 13% at 3 μg/brain) and partially NTI (26% at 10, 23% at 30 mg/kg, s.c.) indicating a predominantly MOR mediated mode of action for 12b.
Discussion
The current series of 6β-naltrexamine derived ligands confirms our earlier finding that having a secondary amide at C6 results in compounds with no selectivity for any particular opioid receptor. This effect is in contrast to the tertiary amides such as TRK-820 (2), which are selective towards the KOR. The low nanomolar to subnanomolar affinities seen here are entirely in keeping with those observed with earlier members of the series.11,12 The predominant activity in the [35S]GTPγS assay of these new ligands is of high efficacy agonist activity at the KOR. Again this is in keeping with our earlier work in which 3 and p-substituted analogues were KOR agonists in the electrically stimulated guinea pig ileal longitudinal muscle (GPI)12 and the work of others.7,10 Compared to the p-methyl and p-chloro derivatives, previously reported as partial agonists11 the o-analogues (12c, 12d) reported here seem to be of higher efficacy, certainly equivalent to the unsubstituted parent compound (3).
Our previous studies with 3 indicated that it had little efficacy at MOR in either the GPI (no reversal by the MOR antagonist CTAP) or in vivo (antinociceptive effect not reversed by β-FNA).12 The new results in the [35S]GTPγS assay confirm its low MOR efficacy. Of the substituted analogues (12a – 12d) the o-Cl, o-Me and m-NO2 substituted ligands (12b – 12d) all have similar low efficacy at MOR of around 20% of DAMGO’s effect, whereas the MOR efficacy of 12a (o-NO2) is even lower (7% of DAMGO). This is reminiscent of the effect of aryl substitution in the cinnamoyl derivatives of the 14-aminomorphinones where the effect of orientation with nitro substitution was different to that with chloro and methyl.14
Substituents were added to the C14-oxygen in expectation of increasing the MOR efficacy of the ligands. This approach has shown partial success with both the allyl ether (20b) and the phenylacetate (15c) having double the MOR efficacy of 3. However the effect was not consistent, both the benzyl ether (20a) and the acetate (15a) displayed no change in efficacy compared to 3, while the benzoate (15b) showed no stimulation of [35S]GTPγS binding at MOR. Certainly the effect of introducing a substituent at this position is much less dramatic than seen in the somewhat related series of substituted 14-amino analogues of oxymorphindole where significant changes in potency and efficacy could be seen.16 The effect of benzyl substitution in this series can also be compared with the effect of 14-O benzylation of naltrexone.22 With naltrexone, this modification lead to an increase in affinity and antagonist potency at KOR compared to naltrexone itself, which is in contrast to the current findings where 20a has somewhat lower affinity and potency at KOR compared to 3.
The activity of the two nitro-substituted cinnamoyl-6β-naltrexamines (12a and 12b) in vivo is worthy of note. In the [35S]GTPγS binding assay both appeared to be high efficacy KOR agonists with some low level efficacy at MOR, with the o-substituted analogue (12a) having somewhat lower efficacy at both receptors compared to 12b. In the PPQ induced abdominal stretch assay 12a was found to be a KOR agonist, correlating well with the in vitro data, while 12b, in the same assay, had predominant MOR activity which would not have been predicted from the in vitro data. Either the [35S]GTPγS binding assay has overestimated efficacy at KOR or underestimated efficacy at MOR. The latter has been observed and discussed previously in a related series of 14-cinnamoylaminomorphinones and may be related to the slower rate and extent of receptor occupation by the ligands in vivo compared to in the in vitro assays.14,23
Experimental
Reagents and solvents were purchased from Aldrich or Lancaster and used as received. M.p.: Gallenkamp MFB-595 melting point apparatus; uncorrected. NMR Spectra: Jeol Delta-270-MHz instrument: 1H at 270 MHz, and Varian Mercury-400-MHz instrument: 1H at 400 MHz, 13C at 100 MHz; δ in ppm, J in Hz with TMS as an internal standard. ESIMS: micrOTOF (BRUKER). Microanalysis: Perkin-Elmer 240C analyser. Ligands were tested as their hydrochloride or oxalate salts, prepared by adding one equivalent of (5N hydrochloric acid in isopropanol) to an ether solution and one equivalent oxalic acid to an ethanolic solution of the compound. Synthetic methods are detailed for one example in each series. Full details for all experiments can be found in the Supporting Information.
General procedure (A): Acylation of Naltrexone
To a stirring mixture of the appropriate cinnamic acid (2.7 mmol) in PhMe (10 mL), was added oxalyl chloride (14 mmol), followed by DMF (4 drop) at 0 °C. The reaction mixture was stirred for 1 h, and concentrated under reduced pressure to yield the appropriate cinnamoyl chloride. The crude cinnamoyl chloride (2.2 mmol), was added to a solution of 6-β-naltrexamine (2.2 mmol) in dry CH2Cl2 (15 mL) at 25 °C. Et3N (11.2 mmol) was added and the reaction mixture stirred for 1 h before diluting with H2O (20 mL), and extraction with CH2Cl2 (3 × 20 mL). The combined organic layers washed with (20 mL) brine, dried over MgSO4 and concentrated under reduced pressure. Flash-chromatography eluent (0.5/0.5/99 MeOH/Et3N/CH2Cl2) afforded the corresponding cinnamoyl naltrexamine.
General procedure (B): 14-O-acylation
To a stirred solution of 3-O- tetrabutyldimethylsilyl-6-β-cinnamoyl-naltrexamine (13) (0.25 mmol), in PhMe (14 mL), was added the appropriate anhydride. The reaction mixture was stirred at reflux for 24 h. After cooling to room temperature the suspension was diluted with (15mL), of saturated NaHCO3, and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried over anhydrous MgSO4 filtered and concentrated under reduced pressure. Flash-chromatography (10/90 EtOAc/Hexane) gave the appropriate protected-6-β-cinnamoyl-14-acyl naltrexamine (14).
General procedure (C): TBDMS deprotection
To a stirred solution of the appropriate 3-O-tetrabutyldimethylsilyl-6-β-cinnamoyl-14-acyl-naltrexamine (14) (0.28mmol), in THF (3 mL), was added TBAF (0.18 mmol) and the suspension stirred for 2 h. Flash-chromatography (2/98 MeOH/CH2Cl2) afforded the appropriate 6-β-cinnamoyl-14-acyl-naltrexamine derivatives (15).
General procedure (D): 14-O-alkylation
Sodium hydride (5.28 mg 0.22 mmol), was added to a solution of 3-O-methyl-6-β-cinnamoyl naltrexamine (18) in DMF (anhydrous, 15mL) at 0 °C. The reaction mixture stirred for 5 min and then allowed to warm to r.t and stirred for a further 20 minutes before adding alkyl bromide (0.41 mmol) and stirring at r.t for 6 h. Excess NaH was destroyed carefully by addition of small pieces of ice and diluted with H2O (50 mL), and the mixture extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Flash-chromatography (1/99 MeOH/CH2Cl2) afforded the products (19).
General procedure (E): 3-O-demethylation
To a solution of the appropriate (19) (0.019 mmol) in CH2Cl2 (61 μl) was added a 1M solution of BBr3 in CH2Cl2 (0.12 mL) over 10 min at −15 °C. After 5 minutes ice (0.20 g) and concentrated NH3 (94 μl) were added and the resulting mixture was stirred at 0 °C for 30 minutes. The organic layers was separated and the aqueous layers extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4 and concentrated under reduced pressure. Flash-chromatography (5/95 MeOH/CH2Cl2) afforded pure material.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(cinnamoylamino)morphinan (3)
3 was prepared using the general method A described above, to obtain 675 mg (63%) of pale white solid. Rf 0.40 (1/5/94 NH3/MeOH/CH2Cl2); 1H-NMR (270 MHz, CDCl3): δ 7.59 (1H, d, J = 15.6 Hz, CH = CH), 7.48 – 7.47 (2H, m, CHAr), 7.34-7.32 (3H, m, CHAr), 6.67 (1H, d, J = 8 Hz, H1), 6.55 (1H, d, J = 8 Hz, H2), 6.39 (1H, d, J = 15.6 Hz, CH = CH), 5.43 (1H, brs, OH), 5.28 (1H, s, OH), 4.43 (1H, d, J = 6.8 Hz, H5), 4.10 – 4.10 (1H, brm, H6), 3.09 (1H, d, J = 5.6 Hz, H9), 3.00 (1H, d, J = 18.0 Hz H10β), 2.66 – 2.63 (2H, m), 2.33 (2H, d, J = 6.6 Hz, cyclopropylmethyl CH2), 1.83 – 1.79 (2H, m), 1.65 – 1.60 (2H, m), 1.50 – 1.47 (2H, m), 1.28 – 1.23 (1H, m), 0.84 – 0.82 (1H, m, cyclopropyl CH), 0.51 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.12 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); 13C NMR (CDCl3): δ 166.5, 145.8, 141.6, 139.0, 135.3, 130.8, 128.6, 128.4, 127.6, 127.1, 120.9, 118.0, 77.2, 71.8. 71.0, 61.9, 60.5, 48.4, 46.4, 32.5, 27.1, 22.2, 19.4, 9.2, 3.8, 3.7; ESIMS m/z: 473 [M + 1]+; Anal. (C29H32N2O4.HCl.2H2O) CHN.
6β-naltrexamine (10)
To ZrCl4 (392 mg, 1.6 mmol) in dry THF (15 mL) was added portion wise NaBH4 (212 mg, 5.6 mmol) at 0 °C. After the mixture had been stirred for 30 minutes naltrexene O-benzyl oxime (11c) (500 mg, 1.1 mmol) was added portion wise. The reaction mixture was warmed to r.t and then refluxed for 24 h under N2. The suspension was concentrated to dryness, before adding 2M HCl at 0 °C (slowly), and then heated at 80 °C for at least 1 hour. The aqueous layer was basified with Na2CO3 and extracted with EtOAc (3 × 30 mL), washed with brine (30 mL), dried with MgSO4, and concentrated under reduced pressure. Flash-chromatography eluent (5/95 MeOH/CH2Cl2) afforded 10 as a white solid 161 mg, (42%). Rf 0.24 (1/5/94 NH3/MeOH/CH2Cl2); 1H-NMR (270 MHz, CDCl3): δ 6.60 (1H, d, J = 8.0 Hz, H1), 6.51 (1H, d, J = 8.0 Hz, H2), 5.21 (3H brs, OH, NH2), 4.23 (1H, d, J = 6.8 Hz, H5), 3.04 (1H, d, J = 5.5 Hz, H9), 2.96 (1H, d, J = 18.0 Hz H10β), 2.60 – 2.56 (4H, m), 2.33 (2H, d, J = 6.6 Hz, cyclopropylmethyl CH2), 2.22 – 2.08 (2H, m), 1.80 – 1.72 (2H, m), 1.65 – 1.35 (2H, m), 0.80 – 0.79 (1H, m, cyclopropyl CH), 0.48 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.10 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); ESIMS m/z: 343 [M + 1]+.
Naltrexone O-benzyl oxime (11c)
To a solution of naltrexone benzoate (3.02 g, 6.5 mmol), and sodium acetate (1.58 g, 19.2 mmol) in EtOH/H2O (80 mL:5 mL), was added O-benzyl hydroxylamine hydrochloride (1.57 g, 9.8 mmol). The suspension of white solid was heated to reflux for 24 h. After evaporation to dryness the residue was diluted with H2O (20 mL), basified with Na2CO3 (pH 10), extracted into CHCl3 (3 × 20 mL), washed with brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure. Flash-chromatography eluent (5/95 MeOH/CH2Cl2) afforded 11c as a pale cream foam (2.6 g, 89%); Rf 0.72; 1H-NMR (270 MHz, CDCl3): δ 7.55 – 7.28 (5H, m, CHAr), 6.60 (1H, d, J = 8.0 Hz, H1), 6.51 (1H, d, J = 8.0 Hz, H2), 5.35 (2H s, Ph-CH2-O), 5.00 (1H, brs, OH14), 4.68 (1H, s, H5), 3.09 (1H, d, J = 5.5 Hz, H9), 3.05 (1H, d, J = 18.0 Hz H10β), 2.58 – 2.24 (6H, m), 1.87 (2H, d, J = 6.6 Hz, cyclopropylmethyl CH2), 1.59 – 1.58 (2H, m), 1.24 – 1.20 (1H, m), 0.86 – 0.83 (1H, m, cyclopropyl CH), 0.54 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.13 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); ESIMS m/z: 447 [M + 1]+.
3-t-Butyldimethylsilyloxy-17-cyclopropylmethyl-14β-hydroxy-4,5α-epoxy-6β-(cinnamoylamino)morphinan (13)
To a stirred solution of 3 (0.25 mmol), in CH2Cl2 (5 mL) imidazole (0.67 mmol), and t-butyldimethylsilylchloride (0.25 mmol) were added at 0 °C. The suspension was stirred at r.t for 24 h. The reaction mixture then was diluted with H2O (10 mL), and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with H2O, (20 mL) brine, (20 mL) and then dried over anhydrous MgSO4 filtered and concentrated under reduced pressure. Flash-chromatography (10/90 EtOAc/Hexane) gave 13 (206 mg, 86%) of white powder. Rf 0.86 (5/95 MeOH/CH2Cl2); 1H-NMR (270 MHz, CDCl3): δ 8.03 (1H, brs, NH), 7.55 (1H, d, J = 15.6 Hz, CH = CH), 7.48 – 7.47 (2H, m, CHAr), 7.33-7.32 (3H, m, CHAr), 6.68 (1H, d, J = 8 Hz, H1), 6.57 (1H, d, J = 8 Hz, H2), 6.43 (1H, d, J = 15.6 Hz, CH = CH), 4.40 (1H, d, J = 6.8 Hz, H5), 4.07 – 4.03 (1H, m, H6), 3.10 (1H, d, J = 5.5 Hz, H9), 3.03 (1H, d, J = 18.0 Hz H10β), 2.65 – 2.60 (2H, m), 2.35 (2H, d, J = 6.6 Hz, cyclopropylmethyl CH2), 2.34 – 2.18 (2H, m), 1.72 – 1.46 (5H, m), 0.92 (9H, s, Si-(CH3)3) 0.92 – 0.89 (1H, m, cyclopropyl CH), 0.51 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.14 (6H, s, Si-(CH3)2), 0.12 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); ESIMS m/z: 587 [M + 1]+.
14β-Acetoyloxy-3–t-butyldimethylsilyloxy-17-cyclopropylmethyl-4,5α-epoxy-6β-(cinnamoylamino)morphinan (14a)
14a was prepared using the general method B described above, to obtain 90 mg (56%) of white solid. Rf 0.33 (5/95 MeOH/CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.56 (1H, d, J = 15.6 Hz, CH = CH), 7.49 – 7.46 (2H, m, CHAr), 7.36-7.34 (3H, m, CHAr), 6.62 (1H, d, J = 8 Hz, H1), 6.53 (1H, d, J = 8 Hz, H2), 6.36 (1H, d, J = 15.6 Hz, CH = CH), 4.46 (1H, d, J = 6.8 Hz, H5), 4.33. (1H, d, J = 5.5 Hz, H9), 3.78 – 3.66 (1H, m, H6), 3.02 (1H, d, J = 18.0 Hz H10β), 2.70 – 2.40 (2H, m), 2.36 – 2.23 (4H, m), 2.11 (3H, s CO-CH3), 2.06 – 1.36 (5H, m), 0.92 (9H, s, Si-(CH3)3) 0.75 – 0.68 (1H, m, cyclopropyl CH), 0.43 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.12 (6H, s, Si-(CH3)2), 0.05 – 0.04 (2H, m, cyclopropyl CH2 trans); 13C-NMR (CDCl3): δ 169.9, 140.9, 129.6, 128.8, 127.7, 122.2, 120.6, 118,8, 91.3, 83.0, 77.3, 77.0, 76.6, 59.3, 55.7, 52.3, 48.1, 44.1, 30.0, 25.6, 24.1, 23.1, 22.3, 9.6, 3.82, 3.5, −4.5, −4.6; ESIMS m/z: 629 [M + 1]+.
14β-Acetoyloxy-17-cyclopropylmethyl-4,5α-epoxy-3-hydroxy-6β-(cinnamoylamino)morphinan (15a)
15a was prepared using the general method C described above, to obtain 60 mg (93%) of white solid. Rf 0.27 (5/95 MeOH/CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 8.03 (1H, brs, NH), 7.56 (1H, d, J = 15.6 Hz, CH = CH), 7.49 – 7.46 (2H, m, CHAr), 7.36-7.34 (3H, m, CHAr), 6.62 (1H, d, J = 8 Hz, H1), 6.53 (1H, d, J = 8 Hz, H2), 6.36 (1H, d, J = 15.6 Hz, CH = CH), 4.77 (1H, d, J = 6.8 Hz, H5), 4.25 (1H, d, J = 4.2 Hz, H9), 3.62 – 4.03 (1H, m, H6), 2.93 (1H, d, J = 18.0 Hz H10β), 2.62 – 2.61 (1H, m), 2.51 – 2.44 (1H m), 2.38 – 2.23 (3H, m), 2.107 (3H, s, CO-CH3) 2.07 – 1.86 (2H, m), 1.78 – 1.72 (1H, m), 1.28 -1.18 (2H, m), 0.92 (1H, t, J = 5.0 Hz),0.76– 068 (1H, m, cyclopropyl CH), 0.44 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.6 – 0.2 (2H, m, cyclopropyl CH2 trans); 13C-NMR (CDCl3): δ 170.8, 166.4, 141.2, 140.9, 139.9, 134.4, 129.7, 128.6, 127.7, 120.7, 119.6, 82.7, 59.3, 55.7, 52.3, 48.1, 44.1, 29.7, 26.1, 24.0, 22.9, 22.2, 9.5, 3.8, 3.5; ESIMS m/z: 515 [M + 1]+; Anal. (C31H34N2O5.HCl) CHN.
β-Naltrexamine-3-O-methyl ether (17)
17 was prepared from 3-O-methylnaltrexone following the same procedure for formation of 6β-naltrexamine 10, to obtain 2.41 g (69%) of white solid. Rf 0.31 (1/5/94 NH3/MeOH/CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 6.73 (1H, d, J = 8.0 Hz, H1), 6.60 (1H, d, J = 8.0 Hz, H2), 5.56 (2H brs, NH2), 4.48 (1H, d, J = 6.8 Hz, H5), 3.88 (3H, s, OCH3), 3.49 (1H, d, J = 5.5 Hz, H9), 3.23 -3.20 (1H, m, H6), 3.02 (1H, d, J = 18.0 Hz H10β), 2.77 – 1.38 (11H, m), 0.86– 0.83 (1H, m, cyclopropyl CH), 0.54 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.19 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); ESIMS m/z: 357 [M + 1]+.
17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxy-3-methoxy-6β-(cinnamoylamino)morphinan (18)
18 was prepared using the general method A described above, to obtain 31 mg (18%) of white powder. Rf 0.66 (1/5/94 NH3/MeOH/CH2Cl2); 1H NMR (400 MHz, CDCl3):8.03 (1H, brs, NH), 7.41 (1H, d, J = 15.6 Hz, CH = CH), 7.47 – 7.35 (2H, m, CHAr), 7.34-7.33 (3H, m, CHAr), 6.70 (1H, d, J = 8 Hz, H1), 6.60 (1H, d, J = 8 Hz, H2), 6.41 (1H, d, J = 15.6 Hz, CH = CH), 5.29 (1H, brs, OH14), 4.45 (1H, d, J = 6.8 Hz, H5), 4.14 – 4.09 (1H, m, H6), 3.83 (3H, s, OCH3), 3.11 (1H, d, J = 5.5 Hz, H9), 3.00 (1H, d, J = 18.0 Hz H10β), 2.68 – 2.61 (2H, m), 2.35 (2H, d, J = 6.6 Hz, cyclopropylmethyl CH2), 2.19 – 2.03 (2H, m), 1.73 – 1.24 (5H, m), 0.85 – 0.83 (1H, m, cyclopropyl CH), 0.52 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.12 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); ESIMS m/z: 487 [M + 1]+.
14β-Allyloxy-17-cyclopropylmethyl-4,5α-epoxy-3-methoxy-6β-cinnamoylamino)morphinan (19b)
19b was prepared using the general method D described above, to obtain 2.19 g (83%) of white powder. Rf 0.46 (1/5/94 NH3/MeOH/CH2Cl2); 1H-NMR (270 MHz, CDCl3): δ 8.16 (1H, brs, NH), 7.66 (1H, d, J = 15.6 Hz, CH = CH), 7.45 – 7.31 (5H, m, CHAr), 6.77 – 6.67 (3H, m, H1, H2, CH = CH), 6.04 – 5.92 (1H, m, allyl CH = CH2) 5.44 – 5.13 (1H, m, allyl CH2 cis), 4.79 – 4.65 (1H, m, allyl CH2 trans), 4.24 – 4.20 (2H, q, H5 H6), 3.85 (3H, s, OCH3), 3.70 – 3.68 (2H, s, CH2-CH=CH2), 3.09 – 3.05 (2H, m, H9 H10β), 2.65 – 2.58 (2H, m, cyclopropylmethyl CH2), 2.37 – 2.22 (3H, m), 1.63 – 1.42 (5H, m), 1.27 – 1.26 (1H, m), 0.87 - 0.83 (1H, m, cyclopropyl CH), 0.51 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.14 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); ESIMS m/z: 527 [M + 1]+.
14β-Allyloxy-17-cyclopropylmethyl-4,5α-epoxy-3-hydroxy-6β-(cinnamoylamino)morphinan (20b)
20b was prepared using the general method E described above, to obtain 76 mg (39%) of white powder. Rf 0.61 (1/5/94 NH3/MeOH/CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 8.08 (1H, brs, NH), 7.57 (1H, d, J = 15.6 Hz, CH = CH), 7.57 – 7.54 (1H, m, CHAr), 7.45-7.43 (1H, m, CHAr), 7.41 – 7.32 (3H, m, CHAr), 6.95 (1H, d, J = 15.6 Hz, CH = CH), 6.75 (1H, d, J = 8 Hz, H1), 6.65 (1H, d, J = 8 Hz, H2), 6.09 – 5.94 (1H, m, allyl CH = CH2) 5.41 (1H, d, J = 17.0 Hz, allyl CH2 cis), 5.32 – 5.14 (1H, m, allyl CH2 trans), 4.68 (1H, d, J = 7 Hz, H5), 4.32 – 4.00 (3H, m, H6, CH2-CH=CH2), 3.14 (1H, d, J = 5.5 Hz, H9), 3.08 (1H, d, J = 18.7 Hz H10β), 2.72 – 2.62 (2H, m), 2.42 (2H, d, J = 6.6 Hz, cyclopropylmethyl CH2), 2.31 – 2.12 (2H, m), 1.65 – 1.24 (5H, m), 0.93 - 0.88 (1H, m, cyclopropyl CH), 0.54 (2H, d, J = 8.0 Hz, cyclopropyl CH2 cis), 0.16 (2H, d, J = 8.0 Hz, cyclopropyl CH2 trans); 13C-NMR (CDCl3): δ 168.2, 143.4, 143.0, 142.5, 140.3, 139.9, 135.1, 134.8, 129.7, 129.3, 128.8, 127.9, 127.8, 119.0, 118.7, 118.4, 117.2, 70.0, 62.3, 62.1, 59.1, 47.8, 47.7, 43.9, 43.8, 30.6, 30.3, 22.7, 22.6, 9.4, 3.8, 3.7; ESIMS m/z: 513 [M + 1]+; Anal. (C31H35N2O4.HCl.H2O) CHN.
Biology
Materials
[3H]diprenorphine and [35S]GTPγS were from Perkin Elmer Life and Analytical Sciences (Boston, MA). BCA protein assay reagents were from Pierce (Rockford, IL). Cell culture reagents were from Invitrogen (Carlsbad, CA). Sigma Cote, U69593, DAMGO and GDP were from Sigma-Aldrich (St. Louis, MO). EcoLume scintillation cocktail was from ICN (Aurora, OH). Whatman GF/C glass-fiber filtermats were from Brandel (Gaithersburg, MD).
Cell Culture
C6 glioma cells stably expressing the rat mu-opioid receptor (C6μ)25 were grown in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 90 units/ml penicillin, 90 μg/ml streptomycin, and 0.5 mg/ml geneticin. Chinese hamster ovary cells stably expressing the human kappa-opioid receptor (CHOκ)24 were maintained similarly, except that DMEM-F12 was used. Both cell lines were grown under 5% CO2 at 37 °C.
Preparation of membranes from C6μ or CHOκ cells
Cells were washed three times with 1X PBS and detached from plates using 37 °C harvesting buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, and 0.68 mM EDTA). After centrifugation at 200 × g for 3 min, the cell pellet was resuspended in 50 mM Tris-HCl, pH 7.4 and homogenized with a Tissue Tearor (Biospec Products, Inc.) 10 times at setting 4. The homogenate was pelleted at 20,000 × g for 20 min at 4 °C and the pellet was resuspended in 50 mM Tris-HCl, pH 7.4 and homogenized with a Tissue Tearor 5 times at setting 2. The final pellet was resuspended in 50 mM Tris-HCl, pH 7.4 and frozen in aliquots at −80 °C. The protein concentrations of the aliquots were determined using the BCA protein assay.
[3H]diprenorphine competitive binding assay
Due to the stickiness of the compounds plasticware was precoated with Sigma Cote. Membranes (20 μg) were incubated in 50 mM Tris-HCl, pH 7.4 with 0.2 nM [3H]diprenorphine in the absence or presence of varying concentrations of unlabeled compound for 60 min in a shaking water bath at 25 °C. Nonspecific binding was measured using 10 μM naloxone. Samples were filtered through GF/C glass-fiber filtermats mounted on a Brandel cell harvester and rinsed four times with 4 °C 50 mM Tris-HCl, pH 7.4 buffer. Filtermats were dried and 0.1 ml EcoLume scintillation cocktail was added to each sample area to soak the filter. Each filtermat was placed in a polyethylene bag, heat sealed, and radioactivity retained in each sample area was counted in a Wallac 1450 MicroBeta Liquid Scintillation and Luminescence Counter. Ki values were determined using GraphPad Prism.
[35S]GTPγS Binding Assay
Due to the stickiness of the compounds plasticware was precoated with Sigma Cote. As described previously,20 membranes (20 μg) were incubated in 20 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 100 mM NaCl, 2.2 mM dithiothreitol (prepared freshly), 30 μM GDP, 0.1 nM [35S]GTPγS, and 10 μM compound or Super Q H2O. The membranes were incubated for 60 min in a shaking water bath at 25 °C. Samples were filtered through GF/C glass-fiber filtermats mounted on a Brandel cell harvester and rinsed four times with 4 °C 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, and 100 mM NaCl. Filtermats were then processed as described above. EC50 and relative efficacy values were calculated using GraphPad Prism.
Supplementary Material
Acknowledgments
This work was funded by NIDA Grant # DA07315
Abbreviations
- β-FNA
β-funaltrexamine
- β-NTA
β-naltrexamine
Footnotes
Supporting Information Available: 1H NMR, 13C NMR, mass spectra and microanalysis data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Heidbreder C, Shippenberg TS. U69,593 prevents cocaine sensitization by normalizing basal accumbens dopamine. NeuroReport. 1994;5:1797–1800. doi: 10.1097/00001756-199409080-00028. [DOI] [PubMed] [Google Scholar]
- 2.Crawford CA, McDougall SA, Bolanos CA, Hall S, Berger SP. The effects of the κ agonist U-50,488 on cocaine-induced conditioned and unconditioned behaviours and Fos immunoreactivity. Psychopharmacology. 1995;120:392–399. doi: 10.1007/BF02245810. [DOI] [PubMed] [Google Scholar]
- 3.Shippenberg TS, LeFevour A, Heidbreder CH. κ-Opioid receptor agonists prevent sensitization to the conditioned rewarding effects of cocaine. J Pharmacol Exp Ther. 1996;276:545–554. [PubMed] [Google Scholar]
- 4.Mello NK, Negus SS. Interactions between kappa opioid agonists and cocaine – preclinical studies. New medications for drug abuse. Ann N Y Acad Sci. 2000;909:104–132. doi: 10.1111/j.1749-6632.2000.tb06678.x. [DOI] [PubMed] [Google Scholar]
- 5.Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: Comparison with butorphanol and hydromorphone in humans. Psychopharmacology. 2001;157:151–162. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- 6.Negus SS, Mello NK. Effects of kappa opioid agonists on cocaine self-administration under a progressive ratio schedule in rhesus monkeys. Drug Alcohol Depend. 2001b;63(Suppl 1):S113. [Google Scholar]
- 7.Takemori AE, Larson DL, Portoghese PS. The irreversible narcotic antagonistic and reversible agonistic properties of the fumaramate methyl-ester derivative of naltrexone. Eur J Pharmacol. 1981;70:445–451. doi: 10.1016/0014-2999(81)90355-1. [DOI] [PubMed] [Google Scholar]
- 8.Takemori AE, Portoghese PS. Affinity labels for opioid receptors. Ann Rev Pharmacol Toxicol. 1985;25:193–223. doi: 10.1146/annurev.pa.25.040185.001205. [DOI] [PubMed] [Google Scholar]
- 9.Mjanger E, Yaksh TL. Characteristics of dose-dependent antagonism by beta-funaltrexamine of the antinociceptive effects of intrathecal-mu agonists. J Pharmacol Exp Ther. 1991;258:544–550. [PubMed] [Google Scholar]
- 10.Nagase H, Hayakawa J, Kawamura K, Kawai K, Takezawa Y, Matsuura H, Tajima C, Endo T. Discovery of structurally novel opioid κ-agonist derived from 4,5-epoxymorphinan. Chem Pharm Bull. 1998;46:366–369. doi: 10.1248/cpb.46.366. [DOI] [PubMed] [Google Scholar]
- 11.Derrick I, Lewis JW, Moynihan HA, Broadbear J, Woods JH. Potential Irreversible ligands for opioid receptors. Cinnamoyl derivatives of β-naltrexamine. J Pharm Pharmacol. 1996;48:192–196. doi: 10.1111/j.2042-7158.1996.tb07121.x. [DOI] [PubMed] [Google Scholar]
- 12.Derrick I, Moynihan HA, Broadbear J, Woods JH, Lewis JW. 6N-Cinnamoyl-β-naltrexamine and its p-nitro derivative. High efficacy κ-opioid agonists with weak antagonist actions. Bioorg Med Chem Lett. 1996;6:167–172. [Google Scholar]
- 13.Halfpenny PR, Hill RG, Horwell DC, Hughes J, Hunter JC, Johnson S, Rees DC. Highly selective κ-opioid analgesics. 2. Synthesis and structure-activity relationships of novel N-[(2-aminocyclohexyl)aryl]acetamide derivatives. J Med Chem. 1989;32:1620–1626. doi: 10.1021/jm00127a036. [DOI] [PubMed] [Google Scholar]
- 14.Nieland NPR, Moynihan H, Carrington S, Broadbear J, Woods JH, Traynor JR, Husbands SM, Lewis JW. Structural Determinants of Opioid Activity in Derivatives of 14-Aminomorphinones: Effect of Substitution in the Aromatic Ring of Cinnamoylaminomorphinones and codeinones. J Med Chem. 2006;49:5333–5338. doi: 10.1021/jm0604777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greiner E, Spetea M, Krassnig R, Schuller F, Aceto MD, Harris LS, Traynor JR, Woods JH, Coop A, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 18. N-substituted 14-phenylpropyloxymorphinan-6-ones with unanticipated agonist properties: Extending the scope of common structure-activity relationships. J Med Chem. 2003;46:1758–1763. doi: 10.1021/jm021118o. [DOI] [PubMed] [Google Scholar]
- 16.Grundt P, Jales AJ, Traynor JR, Lewis JW, Husbands SM. 14-Amino, 14-alkylamino- and 14-acylamino analogs of oxymorphindole. Differential effects on opioid receptor binding and functional profiles. J Med Chem. 2003;43:1563–1566. doi: 10.1021/jm021073r. [DOI] [PubMed] [Google Scholar]
- 17.Sayre LM, Portoghese PS. Stereospecific synthesis of the 6α- and 6β-amino derivatives of naltrexone and oxymorphone. J Org Chem. 1980;45:3366–3368. [Google Scholar]
- 18.Mohamed MS, Portoghese PS. Stereoselectivity of the Reduction of Naltrexone Oxime with Borane. J Org Chem. 1986;51:105–106. [Google Scholar]
- 19.Lee KO, Akil H, Woods JH, Traynor JR. Differential binding properties of oripavines at cloned μ- and δ-opioid receptors. Eur J Pharmacol. 1999;378:323–330. doi: 10.1016/s0014-2999(99)00460-4. [DOI] [PubMed] [Google Scholar]
- 20.Traynor JR, Nahorski SR. Modulation by opioid agonists of guanine-5′-O-(3-[35]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol. 1995;47:848–854. [PubMed] [Google Scholar]
- 21.Aceto MD, Harris LS, Bowman ER. Etorphines: μ-opioid receptor-selective antinociception and low physical dependence capacity. Eur J Pharmacol. 1997;338:215–223. doi: 10.1016/s0014-2999(97)81924-3. [DOI] [PubMed] [Google Scholar]
- 22.Schullner F, Meditz R, Krassnig R, Morandell G, Kalinin VN, Sandler E, Spetea M, White A, Schmidhammer H, Berzetei-Gurske IP. Synthesis and biological evaluation of 14-alkoxymorphinans. 19. Effect of 14-O-benzylation on the opioid receptor affinity and antagonist potency of naltrexone. Helv Chim Acta. 2003;86:2335–2341. [Google Scholar]
- 23.McLaughlin JP, Hill KP, Jiang Q, Sebastian A, Archer S, Bidlack JM. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: in vivo and in vitro characterization of μ-selective agonist and antagonist activity. J Pharmacol Exp Ther. 1999;289:304–311. [PubMed] [Google Scholar]
- 24.Zhu J, Luo LY, Li JG, Chen C, Liu-Chen LY. Activation of the cloned human kappa opioid receptor by agonists enhances [35S]GTPgammaS binding to membranes: determination of potencies and efficacies of ligands. J Pharmacol Exp Ther. 1997;282:676–684. [PubMed] [Google Scholar]
- 25.Emmerson PJ, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the mu opioid receptor. J Pharmacol Exp Ther. 1996;278:1121–1127. [PubMed] [Google Scholar]
- 26.Rennison D, Moynihan H, Traynor JR, Lewis JW, Husbands SM. Structural Determinants of Opioid Activity in Derivatives of 14-Aminomorphinones: Effects of Changes to the C14-amino to Aryl Ring Linker Chain. J Med Chem. 2006;49:6104–6110. doi: 10.1021/jm060595u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.