

Abstract
Proteomics is a powerful tool for the identification of proteins, which provides a basis for rational vaccine design. However, it is still a highly technical and time-consuming task to examine a protein’s immunogenicity utilizing traditional approaches. Here, we present a platform for effectively evaluating protein immunogenicity and antibody detection. A tetanus toxin C fragment (Tet-c) was used as a representative antigen to establish this platform. A cell wall-anchoring sialidase-like protein (SLP) of Propionibacterium acnes was utilized to assess the efficacy of this platform. We constructed an Escherichia coli vector-based vaccine by overexpressing Tet-c or SLP in E. coli and utilized an intact particle of E. coli itself as a vaccine (E. coli Tet-c or SLP vector). After ultraviolet (UV) irradiation, the E. coli vector-based vaccines were administered intranasally into imprinting control region mice without adding exogenous adjuvants. For antibody detection, we fabricated antigen microarrays by printing with purified recombinant proteins including Tet-c and SLP. Our results demonstrated that detectable antibodies were elicited in mice 6 weeks after intranasal administration of UV-irradiated E. coli vector-based vaccines. The antibody production of Tet-c and SLP was significantly elevated after boosting. Notably, the platform with main benefits of using E. coli itself as a vaccine carrier provides a critical template for applied proteomics aimed at screening novel vaccine targets. In addition, the novel immunogenic SLP potentially serves as an antigen candidate for the development of vaccines targeting P. acnes-associated diseases.
Keywords: Antigen microarray, Immunogenicity, Propionibacterium acnes, Sialidase-like protein, Vector-based vaccine
1 Introduction
The technologies of genomics and proteomics have offered high-throughput approaches for rapid and global identification of proteins. Functional proteomics, which emphasizes the biological activity of each identified protein, is facilitating novel drug and vaccine development [1]. However, the transition from protein target selection to drug/vaccine creation is still moving slowly partly due to lack of an simple platform that can expedite drug or vaccine evaluation. Traditional immunoproteomic approaches mainly utilized circulating antibodies to probe the proteins in 2-DE gels for immunogenic detection. However, detected proteins, particularly low abundance and membrane antigens, often remain unidentifiable due to proteome complexity and limiting sample amounts [2]. Computer-assisted techniques provided a means to predict the protein antigenicity, but the probability of a correct prediction was extremely low in comparison with information obtained from vaccinated recipients [3, 4]. Recently, many novel immunoproteomic strategies have been proposed to enhance the selection of proteins as vaccine candidates [5]. However, once proteins are chosen, vaccine designers may face difficulties trying to decide which immunization modalities, adjuvants, animal models and antibody detection systems are most appropriate to use. Each option involves complicated procedures; therefore, it may take months to years to appraise the immunogenicities of protein antigens.
Here, we streamline this process by taking advantage of some recently developed approaches to accelerate the detection of protein immunogenicity. First, we used an E. coli vector-based vaccine system that utilizes an intact particle of E. coli as a natural adjuvant [6, 7]. It has been reported that recombinant proteins or vaccines derived from killed microbes often induce weaker cellular immunity than live vaccines [8]. However, it is not ideal to use live pathogens as vaccines due to safety concerns. Alternatively, killed but metabolically active (KBMA) microbes generated via ultraviolet (UV) or gamma irradiation have been reported to have the benefit of vaccines to induce an appropriate immune response without the issue of pathogen replication in the host [9, 10]. Here, we used an intact E. coli particle (vector) as a vaccine vehicle to carry a non-toxic tetanus toxin C fragment (Tet-c). Secondly, we choose an easier immunization route through intranasal administration of the vector. Such approach has been used for the Food and Drug Administration (FDA) approved influenza vaccine [11]. We observed that UV-irradiated E. coli Tet-c vector elicited a detectable humoral immune response without the assistance of exogenous adjuvants. In addition, the use of E. coli vector-based vaccines eliminates the time-consuming steps required for antigen purification. Furthermore, intranasal immunization circumvents the intrinsic problems associated with multiple needle injections. Thus, E. coli vector-based vaccines are beneficial for antigen screening as well as for large-scale and rapid vaccine production. Thirdly, we fabricated an antigen microarray by printing purified recombinant or gel-eluted proteins on glass slides for detection of antibody production. Unlike ELISA, protein-based microarrays potentially can rapidly characterize antibody against thousands of proteins simultaneously in one single glass slide [12]. The platform reported here thus benefits proteomics research intended for selecting protein target(s) for vaccine development.
To evaluate the efficiency of this platform in detecting novel immunogenic proteins, we chose a surface sialidase-like protein (SLP) (accession number: Q67A7F3) of Propionibacterium acnes. SLP contains an LPXTG cell wall-anchoring motif in the C-terminal domain [13]. Although it has been known that SLP shares identities (~30%) with sialidase (EC 3.2.1.18) (accession number: Q02834) of Micro-monospora viridifaciens and a cell wall surface anchor family protein (accession number: Q04M99) of Streptococcus pneumoniae serotype 2 [13], the immunogenicity of SLP remains unexplored. There has been an increase in evidence showing that P. acnes has cytotoxic properties and is involved in many human diseases including acne vulgaris, late-stage prosthetic joint infections, endocarditis, endophthalmitis, osteomyelitis, shunt-associated CNS infections, cranial neurosurgery infection, and biofilm formation on implanted biomaterials [14]. In particular, acne vulgaris, the most common human skin disease, affects 85–100% of people at some time during their lives.
Sialidase has been selected as a vaccine target for various diseases including influenza [15, 16]. Thus, establishing a facile platform to examine the SLP immunogenicity may accelerate the development of vaccines against P. acnes-associated diseases. In this study, we used Tet-c antigen to create an immunological platform that is capable of facilitating the production and detection of protein immunogenicity. The competence of this platform was evaluated by inducing the immunogenicity of SLP, a cell wall-anchoring protein of P. acnes.
2 Materials and methods
2.1 Construction, expression and purification of Tet-c and SLP overexpressed in an E. coli vector carrier
The codon-optimized Tet-c [17] was PCR amplified and cloned into pCAL-n-FLAG (Stratagene, La Jolla, CA) into BamHI sites. The correct orientation of the insert was confirmed by restriction enzyme mapping as well as DNA sequencing. The pCAL-n-FLAG which contains T7/LacO promoter is derived from a T7 RNA polymerase-based pET-11 series vector to achieve exceptionally high levels of protein expression in E. coli BL21 (DE3) [18]. For Tet-c expression, the E. coli vector-based Tet-c carrier [E. coli BL21 (DE3) T7/lacO Tet-c], grew to OD600/0.3, was induced with 1 mM IPTG for 4 h. An E. coli pellet was formed by centrifugation at 1000×g and resuspended in a lysis buffer containing 100 mM PMSF, PBS, 40 μg/mL lysozyme and 0.1 % Triton-X 100. After keeping on ice for 30 min and sonicating three times (10 s/time), the E. coli lysate was centrifuged for 10 min at 15 000×g to remove the cellular debris, and supernatant was then collected. For Tet-c purification, a cal-modulin affinity resin (Stratagene, La Jolla, CA) with high affinity to calmodulin-binding peptide (CBP)-tagged fusion proteins was added into the supernatant. After mixing gently for 1 h at 4°C, a resin pellet was obtained by centrifuging for 10 s at 1000×g. The Tet-c-FLAG fusion protein bound to CBP tag-binding resin was washed with threefold volumes of binding buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM β-mercaptoethanol, 1.0 mM magnesium acetate, 1.0 mM imidazole, and 2 mM CaCl2 and eluted with elution buffer containing 50 mM Tris-HCl (pH 8.0), 10 mM β-mercaptoethanol, 2 mM EGTA, 150 mM NaCl. Purified Tet-c was obtained by removing FLAG fusion protein and a CBP tag with enterokinase as well as a CBP-affinity resin. For construction of a SLP vector carrier, a gene encoding SLP was PCR amplified from template DNA prepared from P. acnes (ATCC 6919). A vector encoding SLP was constructed by inserting PCR products of SLP into the pEcoli-Nterm 6xHN vector (Clontech, Mountain View, CA) at the SaII and HindIII restriction sites. Specific primers including the sense (5′-ATGTCGCGAACACCACGG-3′) and anti-sense primers (5′-GAGACCCGGTACCGCCAGA-3′) were designed to clone SLP from the P. acnes. The construct was confirmed by restriction enzyme digestions and DNA sequencing (data not shown). The pEcoli-Nterm 6xHN vector containing T7/LacO promoter is derived from the pET system developed by William Studier and colleagues [19] to achieve exceptionally high levels of protein expression in the E. coli BL21 (DE3). For the SLP expression, E. coli vector-based SLP carrier [E. coli BL21 (DE3) T7/lacO SLP] was induced with IPTG for 4 h. The SLP-6xNH tag fusion protein [19] was obtained via In-Fusion Ready TALON Express Bacterial Expression and Purification kit (Clontech). The concentration of purified Tet-c and a SLP-6xNH fusion protein was determined by BCA Protein Assay Kit (Pierce, Rockford, IL). The E. coli lysate, Tet-c-FLAG fusion protein, Tet-c as well as SLP-6xNH fusion protein were subjected to 10% SDS-PAGE gels, then stained with CBB R250 (Bio-Rad, Hercules, CA).
2.2 MALDI-TOF MS and NanoLC-MS/MS analysis
Proteins in SDS-PAGE gels were extracted by in-gel trypsin digestion. Tryptic digests of Tet-c and SLP were analyzed by PerSeptive Voyager-DE MALDI-TOF MS (PerSeptive Biosys-tems, Framingham, MA) [20, 21] and HCTultra PTM NanoLC-MS/MS IT mass spectrometer (Bruker Daltonik, Bremen, Germany), respectively. Peptides in the tryptic digests were eluted from ZipTips with 75% ACN/0.1% TFA and air-dried. Peptide fragments were then mixed with a matrix solution containing CHCA dissolved in 50% ACN/0.1% TFA and analyzed with a MALDI-TOF MS. Peptides were laser-evaporated at 337 nm, and each spectrum was the cumulative average of 50–100 laser pulses. All peptides were measured as mono-isotopic masses, and a trypsin autolytic peak at 2164.0 m/z was used for internal calibration. Up to one missed trypsin cleavage was allowed, although most matches did not contain any missed cleavages. This procedure resulted in mass accuracies of 100 ppm. Peptide mass spectra above 5% of full scale were analyzed, interpreted, and matched to Swiss-Prot database using MASCOT, a searching algorithm available at the Matrix Science Homepage, http:/www.matrixscience.com. Matches were computed using a probability-based MOWSE score defined as −10 × log p, where p is the probability that the observed match was a random event [21] MOWSE scores greater than 70 were considered significant (p ≤0.05).
The automated NanoLC-MS/MS setup for SLP identity consisted of an Agilent 1200 2D LC system, a 10-port switch valve, a C18 trap column (Agilent, Santa Clara, CA), and a Pepmap™ 100 reversed phased column (15 cm in length, 75-μm id, Dionex, Sunnyvale, CA) with an integral spray tip (Picofrit, 15-μm tip, New Objective, Woburn, MA). RP LC directly coupled to HCTultra PTM system IT mass spectrometer (Bruker Daltonik, Bremen, Germany) was performed using linear gradient elution from buffer A (98% H2O, 2% ACN plus 0.1% formic acid) to 50% buffer A plus 50% buffer B (20% H2O, 80% ACN plus 0.1% formic acid) for 45 min. This instrument was operated in the data-dependent mode. MS/MS on the four strongest ions above intensity of 5 × 104 was collected with dynamic exclusion enabled. The mass tolerances of a peptide ion and a fragment ion were set as 1.2 and 0.8 Da, respectively. Maximum missed cleavage was set at 2. Peptide sequences derived from SLP were processed and searched using in-house MASCOT 2.0 server against a P. acnes database obtained from http://expasy.org/sprot/hamap/PROAC.html. Thirty-six internal peptides (37% coverage; MOWSE scores greater than 27) of SLP were fully sequenced (Supporting Information Table I).
2.3 Western blot
Proteins (5 μg) were subjected to 10% SDS-PAGE gels and transferred to NC membranes at 80 mV for 1 h. The membranes were incubated overnight at 4°C with blocking buffer containing 5% skim milk in Tween-Tris buffered saline (TTBS), then were washed for 5 min three times with TTBS and incubated with diluted mouse serum or a commercial goat anti-Tet-c antibody (Fitzgerald Industries International, Concord, MA) in TTBS buffer containing 2% BSA at room temperature overnight at 4°C. After that, the membranes were washed three times and incubated with HRP-conjugated secondary antibody at room temperature for 1 h. Finally, the membranes were washed with TTBS and detected by the ECL kit (Pierce, Rock-ford, IL).
2.4 UV-irradiation of E. coli vector-based vaccines
E. coli harboring expression vectors with or without inserted Tet-c and SLP genes were grown to logarithmic phase and induced with IPTG as described above. CFU (colony forming units) of E. coli (109) were UV irradiated at a total energy of 4500 J/m2 by a Spectrolinker (Spectronics, Westbury, NY). The viability of UV-irradiated E. coli was determined by counting the bacterium colonies on Lauria-Bertani (LB) agar plates. The changes in protein amounts after UV irradiation were examined by SDS-PAGE.
2.5 Intranasal immunization with UV-irradiated E. coli vector-based vaccines
Female imprinting control region (ICR) mice approximately 3-month-old (Harlan, Indianapolis, IN) were intranasally vaccinated by applying 109 CFU/25 μL of an UV-irradiated E. coli vector-based vaccine [E. coli BL21 (DE3) T7/lacO Tet-c or E. coli BL21 (DE3) T7/lacO SLP] into the nasal cavity of each mouse. Mice immunized with E. coli BL21 (DE3) T7/lacO Tet-c were boosted with the same amount of vaccine 3 and 6 weeks after vaccination. Serum was harvested for antigen microarray analysis and Western blot at 3, 6, and 9 weeks post vaccination. Mice immunized with E. coli BL21 (DE3) T7/lacO SLP were boosted with the same amount of vaccine 3 weeks after vaccination. Serum was harvested for antigen microarray analysis and Western blot at 3 and 6 weeks post vaccination. Mice immunized by applying an empty vector without Tet-c or SLP insertion served as a control group. To immunize mice with whole bacteria, P. acnes was killed by heat at 65°C for 30 min. ICR mice were immunized intranasally with heat-killed P. acnes (25 μL; 108 CFU) and boosted with the same amount of vaccine 3 and 6 weeks after vaccination. Serum was harvested for Western blot analysis at 3, 6, and 9 weeks post vaccination. Each group and each experiment was performed in triplicate. Each group contained four mice. All experiments using mice were conducted according to institutional guidelines. A pooled serum sample from four mice was used for antibody detection.
2.6 Elution of Tet-c from SDS-PAGE gels
The protein bands corresponding to Tet-c-FLAG and Tet-c in 10% CBB R250 stained SDS-PAGE gels were excised with razor blades and soaked into 1% SDS sealed with a dialysis membrane (Pierce). The proteins sealed in dialysis membranes were placed in a Mini-Sub horizontal electrophoresis cell (Bio-Rad). The protein elution was run at 100 mV for 2 h. The concentration of eluted proteins was determined by BCA Protein Assay Kit (Pierce). Eluted proteins were printed on antigen microarrays according to the protocols described below.
2.7 Antigen microarray printing and hybridization
Proteins including recombinant Tet-c-FLAG, Tet-c and gel-eluted Tet-c were spotted on poly-L-lysine slides to fabricate the antigen microarrays. For the fabrication of P. acnes antigen microarrays (Supporting Information Fig. 1), five recombinant P. acnes proteins with 6xNH tags were expressed and purified according to the same procedures that were used to obtain an SLP-6xNH fusion protein. Sixty spots in a P. acnes antigen microarray included five 6xNH tagged P. acnes proteins (a secreted sialidase B, SLP, a cell wall anchored sialidase, CAMP factor and lipase), H2O, saline sodium citrate (SCC) buffer, goat IgG and mouse IgG. Spotting an IgG served as a positive control. All proteins were diluted in 3x SSC before spotting. The purified Tet-c (0.35 and 0.7 μg) and various concentrations of P. acnes proteins including a SLP-6xNH fusion protein were printed on antigen microarrays based on the procedure described in the manual of the DeRisi arrayer with silicon microcontact printing pins (Parallel Synthesis Technologies, Santa Clara, CA) [22, 23]. Micro-arrays were UV cross-linked with a total energy of 700 J/m2, then blocked with 0.2% BSA in PBS for 1 h at room temperature, and hybridized with diluted mouse serum or a commercial anti-Tet-c antibody at 4°C overnight. After secondary antibody (Cy-3-labeled goat-anti-mouse IgG) reaction for 1 h at room temperature, microarrays were washed three times in PBS and finally rinsed with water and dried before scanning. Microarrays were scanned by GenePix 4000B scanner and analyzed by GenePix Pro 6.0 software (Molecular Devices Corporation, Sunnyvale, CA).
3 Results
3.1 Tet-c identity confirmed by MALID-TOF MS and Western blot
It has been known that the non-toxic Tet-c is immunogenic [6]. We thus selected Tet-c as a model antigen to establish a platform that can simplify and accelerate the vaccine development and antibody detection. The Tet-c coding sequence was inserted into a pCAL-n-FLAG plasmid and expressed in E. coli [E. coli BL21 (DE3)]. After IPTG induction, overexpressed Tet-c fusion protein from E. coli was detected in a CBB-stained SDS-PAGE gel at approximately 55-kDa molecular weight (Supporting Information Fig. 2A, lanes 1 and 2). The Tet-c fusion protein (Supporting Information Fig. 2A, lane 3) was purified using a calmodulin affinity resin. The fusion tags were removed by enterokinase (Supporting Information Fig. 2A, lane 4).
Figure 2.
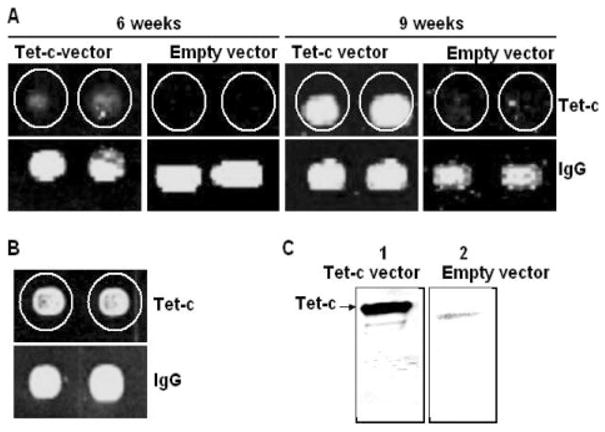
Detection of antibodies elicited by UV-irradiated E. coli vector-based vaccine via antigen microarray and Western blot. Mouse sera were collected 6 and 9 weeks after intranasal immunization with an E. coli vector-based vaccine [E. coli BL21 (DE3) T7/lacO Tet-c] (Tet-c vector) or an empty vector without Tet-c insertion (Empty vector). Antigen microarrays were in-house fabricated as described in Section 2. Briefly, two duplicated purified Tet-c (0.7 μg) and IgG (0.7 μg; a positive control) were spotted on an individual poly-L-lysine slide. Antigen arrays were hybridized with mouse sera (A) and commercial Tet-c antibody (B) (100-fold dilutions). The production of Tet-c antibody was also confirmed by Western blot (C). Purified Tet-c (5 μg) was loaded into 10% SDS-PAGE. Tet-c transferred nitro-cellulose membranes were reacted with mouse sera, which were collected 9 weeks post-immunization with a Tet-c vector (lane 1) and an empty vector (lane 2). Data are representative of three separate experiments with similar results.
The Tet-c expression and purification were confirmed by MALDI-TOF MS and Western blot. Purified Tet-c (Supporting Information Fig. 2A, lane 4) was excised and in-gel digested with trypsin. Eight tryptic digest fragments (1004.4, 1135.5, 1264.6, 1476.8, 1582.7, 1788.9, 1909.9, and 2034.0 m/z) identified in the MALDI-TOF MS spectrum matched with eight internal sequences of Tet-c (accession number: Q9LA13). MASCOT, a searching algorithm, was used. A MOWSE score of 132 was obtained that confirmed the protein identity. Western blot (Supporting Information Fig. 1C) using a commercial anti-Tet-c antibody was also performed to confirm the results from MALDI-TOF MS analysis. These results indicated that Tet-c was expressed in E. coli BL21 (DE3). More importantly, our results suggested that MS is an alternative method to confirm the protein expression and purification, which is particularly beneficial when an antibody to the protein of interest is not available.
Figure 1.
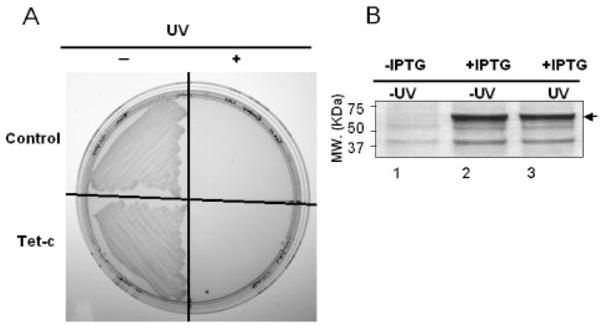
UV-irradiation of an E. coli vector-based vaccine overexpressed Tet-c. Prior to immunization, the E. coli vector-based vaccine was irradiated by UV. E. coli was plated on a LB-agar plate as indicated (A). The upper half of the plate contains control bacteria that were transformed with empty expression vectors. The lower half is the colonies of Tet-c overexpressed E. coli. The left and right halves of the plate are bacteria without (− UV) and with UV irradiation (+ UV), respectively. The protein profile in bacteria transformed by a Tet-c-FLAG fusion protein expression plasmid before (lane 1; − IPTG) and after (lanes 2 and 3: + IPTG) IPTG induction is shown on a CBB R250-stained gel (B). There was no significant change in the protein amount of Tet-c-FLAG (arrow) before (lane 2) and after (lane 3) UV-irradiation.
3.2 Intranasal immunization using an E. coli vector-based vaccine
The use of an intact microbe carrying the target antigen as a vaccine is an advantageous strategy [6, 24, 25]. We thus used an intact particle of E. coli as an E. coli vector-based vaccine [E. coli BL21 (DE3) T7/lacO Tet-c]. In an attempt to avoid the detrimental effects caused by live E. coli, we used UV to irradiate the E. coli vector-based vaccine prior to immunization. Total energy of UV ranging from 200 to 4500 J/m2 was utilized. A dose of 4500 J/m2 is capable of killing all E. coli, either expressing (Tet-c vector) or not expressing Tet-c genes (empty vector), as demonstrated by the inability to form colonies on LB agar plates (Fig. 1A). Although the primary effect of UV irradiation is DNA damage, changes in the level of Tet-c in E. coli vector before and after UV irradiation were detected via SDS-PAGE gels. The amounts of Tet-c in E. coli vectors were not altered by UV-irradiation (Fig. 1B), suggesting the UV-irradiated E. coli vector was a replication incompetent vehicle but preserved the overexpressed Tet-c.
3.3 Detection of Tet-c antibody production by antigen microarrays
It has been revealed that intranasal immunization offers a non-invasive, needle- and pain-free vaccination modality [26]. Furthermore, immunization with an E. coli vector-based vaccine may eliminate the necessity of protein/antigen purification [27]. We thus tested if the E. coli-based vector vaccine serves as a natural adjuvant that can elicit adaptive immunity. ELISA and Western blot are two most common methods for antibody detection [28]. However, these two methods are tedious and inappropriate for high-throughput analysis. Additionally, Western blot is not appropriate for quantitatively determining the antibody titers. In contrast, micro-array is a more high-throughput and quantitative method [29]. We thus fabricated an antigen microarray by printing Tet-c-FLAG, Tet-c and mouse IgG (a positive control) on glass slides. The antigen microarray was hybridized with mouse sera to generate fluorescent signals (green dots) that indicate Tet-c antibody production. There was no detectable Tet-c antibody in the serum harvested from mice 3 weeks after immunization with UV-irradiated E. coli-based vector vaccines [E. coli BL21 (DE3) T7/lacO Tet-c] without adding any exogenous adjuvants (data not shown). After boosting at the third week, we were able to obtain detectable Tet-c antibody in the serum harvested from mice 6 week after immunization (Fig. 2A). No green dots were found on antigen micro-arrays when serum from empty vector-immunized mice was used (Fig. 2A), indicating that generation of green dots is a specific signal for the interaction of Tet-c to Tet-c antibody. More importantly, this result demonstrated that intranasal immunization using an UV-irradiated E. coli-based vector vaccine [E. coli BL21 (DE3) T7/lacO Tet-c] for 6 weeks was sufficient for producing detectable levels of Tet-c antibody. The production of Tet-c antibody was significantly increased after a second boosting was performed at the sixth week and antibody was detected at the ninth week after immunization (Figs. 2A and 3A). To confirm that the green dots in the antigen microarray analysis were a result of interaction of Tet-c with Tet-c antibody in mouse serum, we hybridized antigen microarrays with a commercial Tet-c antibody (Fig. 2B). The intensities of green dots generated by the commercial antibody were equivalent to those generated by mouse serum obtained from mice 9 weeks post-immunization. The antibody detection by antigen microarrays was also validated by traditional Western blot analysis (Fig. 2C). A strong band generated by Tet-c and antibody interaction was detected when a Tet-c transferred membrane was probed with mouse serum harvested 9 weeks after immunization. Consistent with the antigen microarray results, Western blot data indicated that Tet-c antibody was produced only in mice immunized with a Tet-c vector, not an empty vector.
Figure 3.
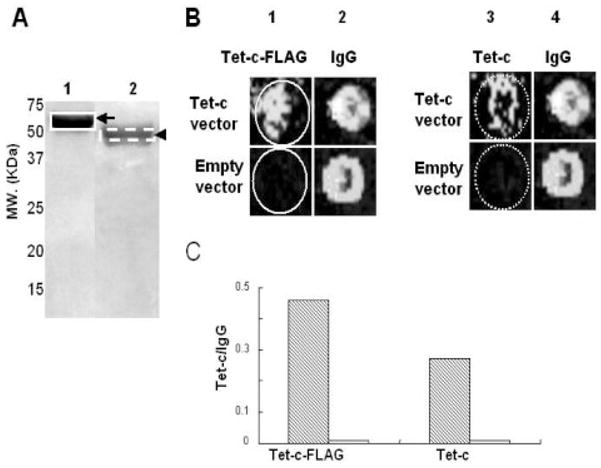
Printing microarrays with SDS-PAGE gel eluted Tet-c. Purified Tet-c-FLAG (A, lane 1) and Tet-c (A, lane 2) (50 μg) were loaded onto a 10% SDS-PAGE gel and stained with CBB R250. Tet-c-FLAG and Tet-c were then extracted by gel elution as described in Section 2. The gel-eluted Tet-c-FLAG (B, lane 1) and Tet-c (B, lane 3) (0.35 μg) were spotted on a microarray. Spotting IgG (0.7 μg) (B, lanes 2 and 4) serves as a positive control. The antigen microarrays printed with IgG as well as gel-eluted Tet-c-FLAG and Tet-c were incubated with mouse sera (100-fold dilution), which were collected 9 weeks post-immunization with a Tet-c vector (Tet-c vector) and an empty vector (empty vector). The intensities of fluorescent signals (green dots) reflecting the level of Tet-c antibodies are represented by a ratio (Tet-c/IgG) of those of Tet-c (hatched bar) to IgG (open bar) (C). Data are representative of three independent experiments with similar results.
3.4 Printing antigen microarrays with SDS-PAGE gel eluted Tet-c
We demonstrated that antigen microarrays printed with purified recombinant Tet-c can be used to detect antibody production (Fig. 2). We also printed antigen microarrays by spotting SDS-PAGE gel-eluted Tet-c on glass slides. Proteins eluted from gels will eliminate the complicated procedures for protein purification using affinity columns. Furthermore, protein eluted from a specific band of a SDS-PAGE gel instead of purified from protein-expressed E. coli will remove other contaminants from the E. coli protein lysate. Protein bands corresponding to Tet-c-FLAG and Tet-c in SDS-PAGE gels (Fig. 3A) were excised and eluted in dialysis membranes via electrophoresis. Eluted Tet-c-FLAG and Tet-c as well as mouse IgG (a positive control) were printed on antigen mciroarrays. The antigen microarrays were then incubated with mouse sera (100-fold dilution), which were collected 9 weeks post immunization with a Tet-c vector (Tet-c vector) and an empty vector (empty vector). No green dots appeared when antigen microarrays were incubated with sera from empty vector-immunized mice. In contrast, both eluted Tet-c-FLAG and Tet-c are capable of reacting to mouse serum collected from Tet-c vector-immunized mice (Fig. 3B). Printing with eluted Tet-c-FLAG generates a significantly higher intensity (Tet-c/IgG ratio) than printing eluted Tet-c (Fig. 3C). This may simply reflect more epitopes were recognized by a bigger Tet-c-FLAG fusion protein than Tet-c alone.
3.5 Quantitative antigen microarray analysis
For quantitative antigen microarray analysis, the Tet-c printed antigen microarrays were hybridized with diluted mouse sera (100-, 250-, and 1000-fold dilutions) (Fig. A). The intensities of green dots generated from Tet-c spots were divided by those generated from mouse IgG spots and presented as Tet-c/IgG ratios. The Tet-c/IgG ratio is extremely low (<0.1) when antigen microarrays were hybridized with empty vector-immunized mouse sera (data not shown). To detect the level of anti-Tet-c antibody at 6 weeks after immunization, antigen microarrays were hybridized with 250-, and 100-fold diluted mouse sera and Tet-c/IgG ratios at 0.3 and 0.7, respectively, were obtained (Fig. 4A; open bars). The Tet-c/IgG ratio was less than 0.1 when the serum was diluted 1000-fold. The level of anti-Tet-c antibody detected at 9 weeks after immunization was significantly elevated. The Tet-c/IgG ratios for 1,000-, 250-, and 100-fold diluted sera were 0.7, 0.8, and 1.7, respectively (Fig. 4A; solid bars). We also examined the sensitivity of antigen microarrays by spotting two different amounts (0.35 and 0.7 μg) of purified Tet-c (Fig. 4B). Antigen microarrays were hybridized with 1000-fold diluted sera obtained from mice 6 and 9 weeks post immunization. Green dots were detectable when antigen microarrays were spotted with 0.7 μg of Tet-c. Spotting 0.35 μg of Tet-c was not sensitive enough to detect the level of Tet-c antibody in the 6-week-immunized mice. However, green dots were evident when antigen microarrays (0.35 μg of Tet-c spotted) were hybridized with serum obtained from 9-week-immunized mice. These results illustrate that vaccination boost enhances the production of Tet-c antibody and that antigen micro-arrays are capable of quantitatively detecting antibody production.
Figure 4.
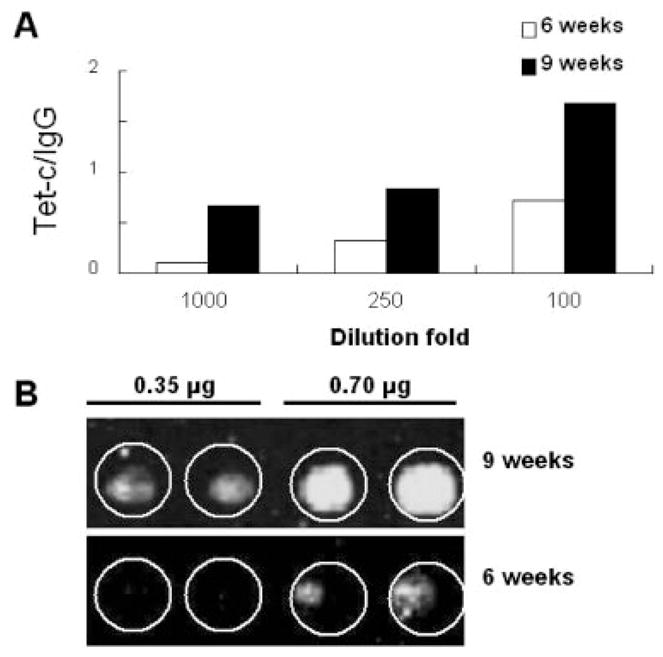
The quantitative detection of Tet-c antibody production via antigen microarray analysis. Tet-c and IgG (0.7 μg)-spotted antigen microarrays were incubated with mouse sera obtained from 6 and 9 weeks post-immunization with a Tet-c vector (solid bar) and an empty vector (open bar). Mouse sera were serially diluted (100-, 250- and 1000-fold) (A). Green dots indicating the binding of Tet-C to serum antibody were visualized by incubating antigen microarrays with Cy-3 labeled goat-anti-mouse IgG (Fig. 2). The level of Tet-c antibody production is represented by the ratio of green dot intensities of Tet-c to IgG (Tet-c/IgG). Two concentrations (0.35 and 0.7 μg) of purified Tet-c were spotted on microarrays in duplicate (B). Tet-c-spotted mciroarrays were reacted with sera (1000-fold dilution) obtained from mice immunized with Tet-c vectors for 6 (lower panel) and 9 (upper panel) weeks. Experiments were performed in triplicate. Representative data are selected for presentation from other similar results.
3.6 Selection of a surface protein to evaluate the competence of platform that facilitates the detection of protein immunogenicity
Surface proteins of pathogens are critical targets for vaccine development. To validate the efficiency of platform in detecting a novel immunogenic protein, we selected a cell wall-anchoring SLP (accession number: Q67A7F3) of P. acnes [30]. The rationale of this selection is primarily based on the lack of vaccines that can effectively combat P. acnes. Secondly, sialidases have been chosen as vaccine targets to counteract pathogens [15, 16]. The Dense Alignment Surface method (www.sbc.su.se/~miklos/DAS/) indicated that N terminus (14–24 amino acid residues) of SLP is a potential transmembrane segment. Although the sialidase activity of SLP has been not yet validated, it has been known that SLP shares identities (~ 30%) with sialidase (accession number: Q02834) of Micromonospora viridifaciens and exo-alpha-sialidase (accession number: Q8XMY5) of Clostridium perfringens [31]. A real-time PCR analysis confirmed that the P. acnes expressed SLP (Supporting Information Fig. 3).
3.7 SLP expression and MS identification
The SLP coding sequence was inserted into a pEcoli-Nterm 6xHN plasmid and expressed in E. coli [E. coli BL21 (DE3)]. After IPTG induction, overexpressed SLP-6xNH fusion protein from E. coli was detected in a CBB-stained SDS-PAGE gel at approximately 142-kDa molecular weight (Fig. 5A, lanes 1 and 2). The SLP-6xNH fusion protein (Fig. 5A, lane 3) was purified using a TALON resin column. The SLP expression was confirmed by MALDI-TOF MS (data not shown) as well as NanoLC-MS/MS sequencing (Fig. 5B, Supporting Information Table 1). Purified SLP-6xNH fusion protein (Figs. 5A, and 3) was in-gel digested with trypsin prior to NanoLC-MS/MS analysis. Thirty-six internal peptides (37% coverage; Supporting Information Table I) derived from SLP were fully sequenced by NanoLC-MS/MS analysis via an HCTultra PTM system IT mass spectrometer. The MS/MS spectra of sequenced peptides matched with those of SLP (accession number: Q67A7F3) of P. acnes KPA171202. An internal peptide (TGADGAGLGFSSSEAA-SATAAVGVSR; 1293–1318 amino acid residues) of SLP with a MOWSE score of 124 was presented (Fig. 5B). These results indicated that SLP was expressed in E. coli BL21 (DE3) and suggested that MS provided a powerful modality to confirm the expression of a protein that has no antibody available for Western blot analysis.
Figure 5.
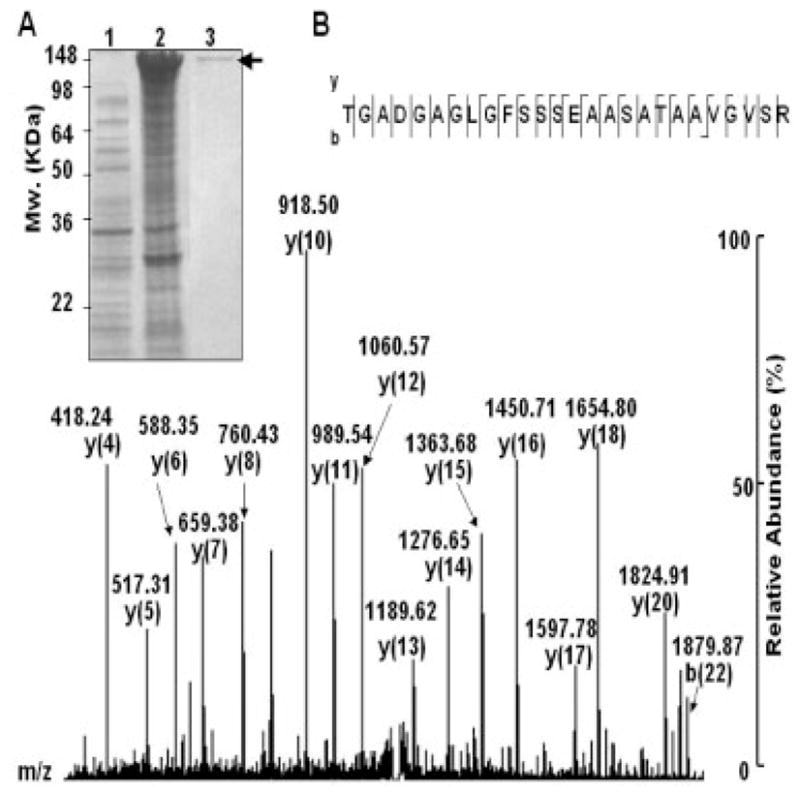
Expression and confirmation of SLP. A vector encoding SLP was constructed by inserting PCR products of SLP into the pEcoli-Nterm 6xHN vector containing a T7/LacO promoter to control protein expression as described in Section 2. The vector with SLP insertion was then transfected into E. coli BL21 (DE3) to construct an E. coli vector-based carrier [E. coli BL21 (DE3) T7/lacO SLP]. For protein expression, E. coli BL21 (DE3) T7/lacO SLP was induced with (A, lane 2) or without (A, lane 1) 1 mM IPTG. SLP-6xNH fusion protein with an approximately 142-kDa molecular weight was overexpressed and detected on a 10% SDS-PAGE. A SLP-6xNH fusion protein was obtained from the elution of a TALON resin column (A, lane 3, arrow). The expression of SLP was confirmed by NanoLC-MS/MS analysis. An internal peptide (TGADGAGLGFSSSEAASATAAVGVSR) of SLP is illustrated (B). The characteristic “y” and “b” series ions were indicated in a MS/MS spectrum.
3.8 The production of SLP antibody by intranasal immunization with an E. coli vector-based vaccine
A UV-irradiated E. coli vector-based vaccine [E. coli BL21 (DE3) T7/lacO SLP] was used to test the SLP immunogenicity. A dose of UV (4500 J/m2) was given to irradiate all E. coli, both expressing (SLP-vector) and not expressing SLP genes (empty vector), as demonstrated by the inability to form colonies on LB agar plates (Fig. 6A). Figure 6B indicates that the amount of SLP in E. coli vectors was not changed after UV irradiation. ICR mice were intranasally immunized with UV-irradiated E. coli BL21 (DE3) T7/lacO SLP and boosted 3 weeks after the first nasal inoculation. The production of antibody (IgG) in mouse sera was detected 3 and 6 weeks after immunization. A sixty-spot antigen micro-array printed with five P. acnes proteins including an SLP-6xNH fusion protein (Supporting Information Fig. 1) and mouse IgG (a positive control) was used for antibody detection. After hybridization with mouse sera, fluorescent signals (green dots) displayed in antigen microarrays indicated SLP antibody production. There was no detectable SLP antibody in the serum collected from mice 3 weeks after immunization (data not shown). After boosting once, the SLP antibody was detectable in the serum harvested from mice 6 weeks after immunization (Fig. 6C). The antibody against SLP did not cross-react with other P. acnes proteins (Supporting Information Fig. 1). No antibody production was found in empty vector-immunized mice. The antibody production was also confirmed by Western blot analysis (Fig. 6D). A strong band appearing at approximately 142 kDa was visualized when an SLP-6xNH fusion protein transferred membrane was reacted with mouse serum harvested 6 weeks after immunization. Antigen microarray and Western blot data both indicated that SLP was immunogenic when mice were intranasally immunized an E. coli vector-based vaccine [E. coli BL21 (DE3) T7/lacO SLP].
Figure 6.
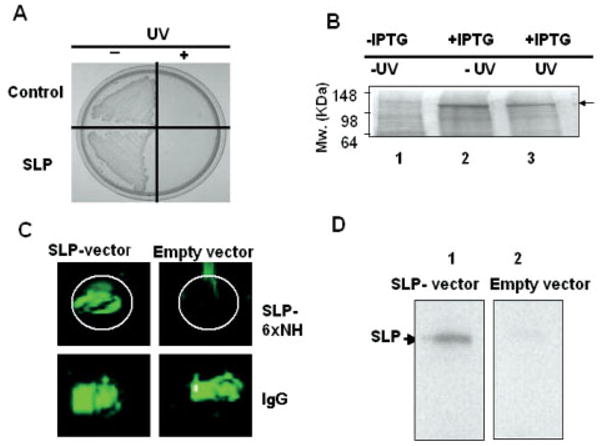
Production of SLP antibody by intranasal vaccination with an UV-irradiated E. coli vector and detection via antigen micro-array and Western blot. The E. coli vector-based vaccine overexpressed SLP [E. coli BL21 (DE3) T7/lacO SLP] was irradiated by UV. E. coli was plated on a LB-agar plate as indicated (A). The upper half of the plate is a control bacterium that was transformed with an empty expression vector. The lower half is the colonies of SLP overexpressed E. coli. The left and right halves of the plate are bacteria without (− UV) and with UV irradiation (+ UV), respectively. The protein profile in bacteria transformed by a SLP-6xNH fusion protein expression plasmid before (lane 1; − IPTG) and after (lane 2 and 3: + IPTG) IPTG induction is shown on a CBB R250-stained gel (B). Mouse sera were collected 6 weeks after intranasal vaccination with an E. coli vector-based vaccine [E. coli BL21 (DE3) T7/lacO SLP] (SLP-vector) or an empty vector without SLP insertion (Empty vector). A SLP-6xNH fusion protein (0.35 μg) and IgG (0.35 μg; a positive control) were spotted on an individual poly-L-lysine slide to create an antigen microarray for hybridization with mouse sera (C). The production of SLP antibody was also confirmed by Western blot (D). SLP-6xNH fusion protein (5 μg) was loaded into 10% SDS-PAGE, transferred to nitrocellulose membranes and reacted with mouse sera (1000-fold dilution), which were collected 6 weeks post-immunization with a SLP vector (lane 1) and an empty vector (lane 2). Data are representative of three separate experiments with similar results.
Among P. acnes-associated diseases, acne vulgaris is highly related to P. acnes infection. Patients with acne vulgaris are likely to produce anti-P. acnes antibodies [32], however, acne vulgaris still recurs in these patients. This suggests that patients infected with P. acnes may develop insufficient immunity to prevent subsequent P. acnes infection and acne recurrence. We thus examined whether SLP antibody can be produced in the P. acnes immunized mice. ICR mice were immunized intranasally with heat-killed P. acnes. P. acnes lysates and SLP-6xNH fusion protein were subjected to 10% SDS-PAGE and reacted with mouse serum which was obtained 6 (data not shown) and 9 weeks post immunization (Fig. 7). Many proteins with molecular weights greater than 50 kDa, but not SLP, were immunoreactive with mouse serum obtained from the heat-killed P. acnes-immunized mice, indicating that mice immunized with whole organism P. acnes do not produce antibodies to SLP. More importantly the results also implicated the benefit of using the E. coli vector since SLP became immunogenic in mice immunized with an E. coli vector-based vaccine [E. coli BL21 (DE3) T7/lacO SLP] (Fig. 6). In addition, insufficient or lack of SLP antibody production after P. acnes infection may provide one explanation for the recurrence of acne vulgaris. Thus, the E. coli vector-based vaccine targeting SLP may serve as a vaccine candidate to combat acne vulgaris.
Figure 7.
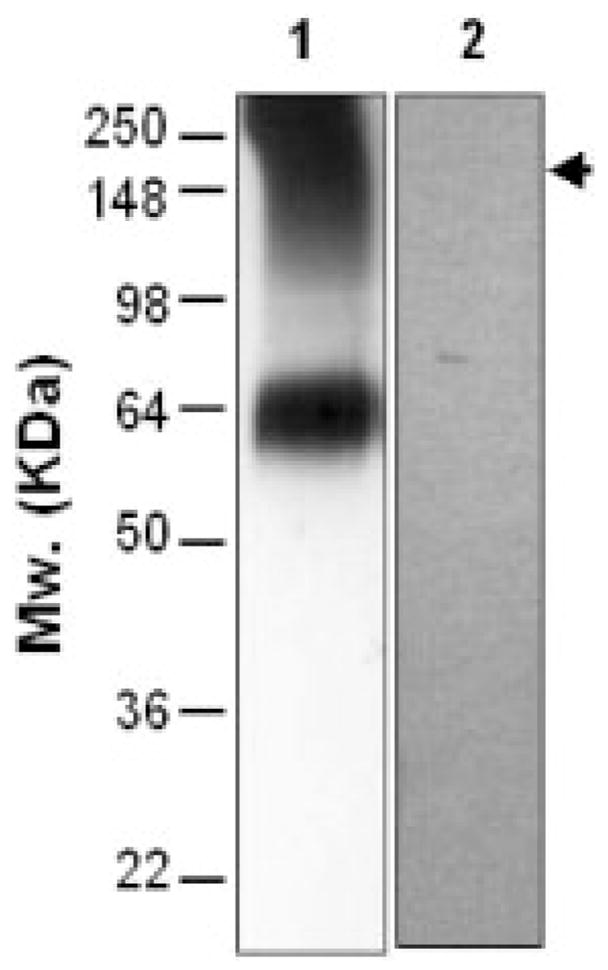
The SLP antibody was not produced in mice immunized with whole organism P. acnes. ICR mice were vaccinated intrana-sally with heat-killed P. acnes. Serum (1000-fold dilution), harvested 9 weeks post vaccination, was reacted with 5 μg of P. acnes lysates (lane 1) and SLP-6xNH fusion protein (lane 2) that had been run on a 10% SDS-PAGE. Although heat-killed P. acnes-vaccinated mice produced anti-sera against several P. acnes proteins (>50 kDa), an SLP specific antibody was not detected (arrow). Data are representative of three separate experiments with similar results.
In summary, we emphasized the advantages of an E. coli vector-based vaccine system for proteomics community (Fig. 8). A variety of pathogen proteomes with hundreds of identified proteins have been established in many proteomic laboratories. However, the validation of antigens selected from identified proteins is generally time-consuming due to the lack of a rapid antigen screening system. Our data demonstrated that the use of E. coli particles as vaccine vehicles to carry over-expressed antigens simplified the vaccination procedures. More importantly, the E. coli vector-based vaccine system could effectively elicit the protein immunogenicity, which may fail if whole pathogens as vaccines were used (Fig. 7). The E. coli vectors can be easily accessed in the laboratories. The use of intact E. coli particles with the benefits of elimination of the deleterious requirement for the antigen purification and the hazard of contemporary adjuvants will thus speed up the screening of immunogenicities of proteins selected from various pathogen genomes and/or proteomes.
Figure 8.
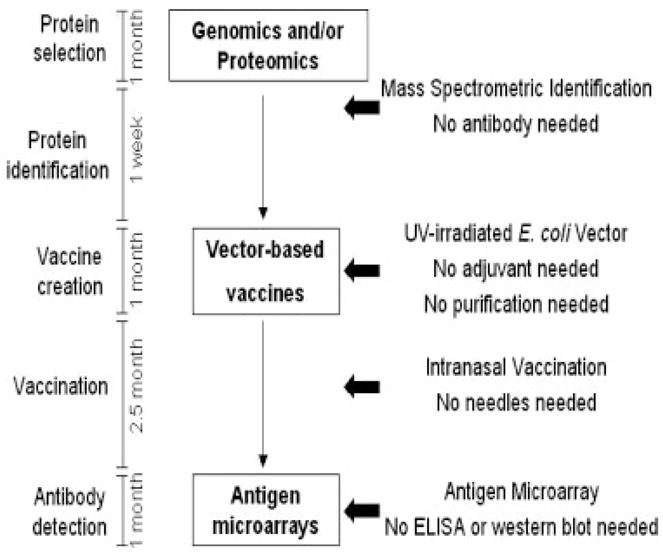
A summarized scheme highlights the benefits of a platform to vaccine and proteomics research. Once proteins are selected from genomics and/or proteomics, the vector-based vaccine system coupled to proteomic approaches and antigen microarrays expedites the detection of protein immunogenicities. Intranasal immunization with the vector-based vaccines liberates the dependence on adjuvants, protein purification and needles. The effectiveness of platform was demonstrated in this study by using two proteins (Tet-c and SLP), an E. coli vector (two different plasmids, pCAL-n-FLAG and pEcoli-Nterm 6xHN) and two mass spectrometers (MALDI-TOF MS and NanoLC-MS/MS).
4 Discussion
The identity of a protein purified from ectopic expression systems, such as E. coli, is always questionable if simply based on the predicted size on SDS-PAGE. It is important to confirm the correct protein is purified, particularly when a novel protein is cloned since no specific antibody is available. Immunoblotting with antibodies to tags fused to the protein is a common but indirect method for confirmation. Here we propose to use mass-spectrometry for direct confirmation to avoid a mistake that may be realized only months later after immunization. Additionally, MS can also provide information about the post-translational patterns of protein expression [33].
To simplify the immunization protocol, we employed an E. coli vector-based vaccine system using whole E. coli that expresses Tet-c as a vaccine without adding other adjuvants. It has been demonstrated that E. coli components such as lipopolysaccharide and the CpG DNA of E. coli, exert excellent adjuvant properties [10, 34]. The E. coli particles can trigger a specific gene expression profile in antigen-presenting cells such as dendritic cells and are associated with the activation of the Toll-like receptor signaling pathway [35, 36]. Thus, the E. coli particle itself may be a natural adjuvant to enhance the immunity of proteins that have been expressed in E. coli.
UV-irradiation was used to kill E. coli before vaccination. Unlike heat that can damage protein structures [37], UV irradiation may be a safer method that has less adverse effects on proteins. UV-irradiation equipment is readily available in most laboratories and it has been well documented that the major cause of UV inactivation of bacteria is nucleic acid damage [38]. Indeed, our data showed that UV irradiation did not significantly change the protein profile in E. coli (Fig. 1).
Previously, an intact particle of a live or gamma-irradiated E. coli over-expressing Tet-c has been used for epicutaneous vaccination [6]. Topical application of E. coli-vector-based vaccines protected mice against tetanus challenge. It was also found that cutaneous γδT cells were rapidly recruited to the site where the E. coli-vector-based vaccine was applied, suggesting that intact E. coli particles in the outer layer of skin may be disrupted by a γδ T cell-mediated innate defense mechanism. In this study, we immunized mice intranasally with UV-irradiated E. coli-vector-based vaccines. Intranasal immunization offers easy access and only a simple administration device is required. Intranasal immunization acts through the mucosal immune system and can trigger better immune responses than oral or parenteral administration [39]. It can induce mucosal cytotoxic T cell responses and activate the regulatory T cell (CD4+ and CD8+ T cells) to produce cytokines, such as interleukin (IL)-4 and IL-10 [40, 41]. Flumist, an intranasal influenza vaccine, has already been approved by the US-FDA for a few years [42]. In our experiments, UV-irradiation killed E. coli overexpressing Tet-c was intranasally inoculated into mice. Anti-Tet-c antibody (IgG) was detectable 6 weeks after immunization and was significantly elevated after boosting. It has been known that intranasal vaccination can elicit both a local and systemic immune response [43]. Although systemic IgG against Tet-C or SLP was detected in this study, it is worth measuring a local IgA mucosal response after intranasal vaccination with E. coil vector-based vaccines.
For antibody detection, ELISA has been most commonly used. However, there are many limitations, such as they are time-consuming, require large quantities of both reagents and biological samples, and barely achieve high-throughput screening [44]. It has been reported that antigen arrays have higher sensitivity than ELISA has and can be used to map epi-topes [45, 46]. We created antigen microarrays by printing purified recombinant Tet-c as well as SDS-PAGE gel-eluted Tet-c. Performance of antigen microarrays printed either with gel-eluted Tet-c or recombinant Tet-c was comparable (Figs. 3 and 4). Printing gel-eluted proteins on microarrays can be applied for detecting thousands of proteins obtained from spots on 2-DE gels even before the identities of those proteins are known. Recombinant Tet-c and SLP became detectable when they were spotted on antigen microarrays at the concentration higher than 0.35 μg (Fig. 4B, Supporting Information Fig. 1). Performing both antigen microarrays and ELISA side-by-side with the positive correlation of antigen concentration may be needed to quantitatively determine which assay can display higher sensitivity for detection of both Tet-c and SLP.
P. acnes is involved in many human diseases including acne vulgaris, a disease that affects more than fifty million people in the US. In addition, acne vulgaris is the most common skin disease, affecting 85–100% of people at some time during their lives. Systemic antibiotic therapy for acne lesions nonspecifically kills the majority of skin bacteria, which affects the homeostasis of skin resident flora. Although the genome of P. acnes has been completely sequenced [13] and has therefore created an excellent source for antigen selection, vaccines against acne vulgaris and other P. acnes-associated diseases are not currently available. A variety of sialidases has been uncovered in the genome of P. acnes. Since Tet-c has been used to establish a platform (Supporting Information Fig. 1 and Figs. 1–4) that can simplify the procedures of antibody production and detection, we thus selected a cell wall-anchoring sialidase (SLP) of P. acnes to validate this platform. Our data demonstrate for the first time that SLP is an immunogenic protein that can evoke antibody production when mice are immunized with an SLP-overexpressed E. coli-vector-based vaccine. It has been demonstrated that sialidase is involved in the adhesion of pathogens to host cells [30], and that treatment of host cells with sialidase changes the susceptibility of host cells to pathogens [47]. More importantly, SLP is a surface protein carrying a cell wall-anchoring LPXTG motif. Thus, SLP may be a valuable target to create vaccine against P. acnes-associated diseases.
In summary, we developed a platform that integrates proteomics technology, intranasal immunization with E. coli vector-based vaccines, and gel-eluted antigen microarrays (Fig. 7). The E. coli vector-based vaccine system is a key component of this platform that provides a simple modality produce antibodies in mice. Antigen microarrays spotted with either purified recombinant or gel-eluted antigens provide an alternative method for antibody detection. This platform was compared with traditional methods that used available antibody/Western blot for protein confirmation, purified proteins with adjuvants plus needles for vaccination, and ELISA for antibody detection (Fig. 8). More importantly, producing a detectable antibody of a cell wall-anchoring sialidase (SLP) of P. acnes strongly validated the competence of this platform. The antibody against SLP was detectable in E. coli vector-immunized mice, but not in killed P. acnes-immunized mice, which demonstrates the benefits of an E. coli vector in evoking the protein immunogenicity and providing an explanation for the recurrence of acne vulgaris in human. Accordingly, this cell wall-anchoring sialidase is a potential antigen candidate for vaccine development targeting acne vulgaris and other P. acnes-associated diseases [48].
Acknowledgments
This work was supported by National Institutes of Health Grants [R01-AI067395-01, R21-R022754-01, R21-I58002-01, R41AR056169-01 (C.-M. H.) and P30-AI36214-12S1 (Y.-T. L.)] and an award from National Taiwan University for C.-P. Huang’s research in the University of California, San Diego, USA. We thank A. J. Livengood for critical reading of the manuscript.
Abbreviations
- CBP
calmodulin-binding peptide
- CFU
colony forming units
- ICR
imprinting control region
- LB
Lauria-Bertani
- SLP
sialidase-like protein
- Tet-c
tetanus toxin C fragment
- TTBS
tween-tris buffered saline
Footnotes
The authors have declared no conflict of interest.
References
- 1.Chich JF, Schaeffer B, Bouin AP, Mouthon F, et al. Prion infection-impaired functional blocks identified by proteomics enlighten the targets and the curing pathways of an anti-prion drug. Biochim Biophys Acta. 2007;1774:154–167. doi: 10.1016/j.bbapap.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 2.Hess JL, Blazer L, Romer T, Faber L, et al. Immunoproteomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;815:65–75. doi: 10.1016/j.jchromb.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 3.Zenteno-Cuevas R, Huerta-Yepez S, Reyes-Leyva J, Hernández-Jáuregui P, et al. Identification of potential B cell epitope determinants by computer techniques, in hemagglutinin-neuraminidase from the porcine rubulavirus La Piedad Michoacan. Viral Immunol. 2007;20:250–260. doi: 10.1089/vim.2006.0066. [DOI] [PubMed] [Google Scholar]
- 4.Ferreira-da-Cruz Mde F, Giovanni-de-Simone S, Banic DM, Canto-Cavalheiro M, et al. Can software be used to predict antigenic regions in Plasmodium falciparum peptides? Parasite Immunol. 1996;18:159–161. doi: 10.1046/j.1365-3024.1996.d01-62.x. [DOI] [PubMed] [Google Scholar]
- 5.Wilson RA, Curwen RS, Braschi S, Hall SL, et al. From genomes to vaccines via the proteome. Mem Inst Oswaldo Cruz. 2004;99:45–50. doi: 10.1590/s0074-02762004000900008. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Shi Z, Kong FK, Jex E, et al. Topical application of Escherichia coli-vectored vaccine as a simple method for eliciting protective immunity. Infect Immun. 2006;74:3607–3617. doi: 10.1128/IAI.01836-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu YT, Lin SB, Huang CP, Huang CM. A novel immunogenic spore coat-associated protein in Bacillus anthracis: characterization via proteomics approaches and a vector-based vaccine system. Protein Expr Purif. 2008;57:72–80. doi: 10.1016/j.pep.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lauvau G, Vijh S, Kong P, Horng T, et al. Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science. 2001;294:1735–1739. doi: 10.1126/science.1064571. [DOI] [PubMed] [Google Scholar]
- 9.Brockstedt DG, Bahjat KS, Giedlin MA, Liu W, et al. Killed but metabolically active microbes: a new vaccine paradigm for eliciting effector T-cell responses and protective immunity. Nat Med. 2005;11:853–860. doi: 10.1038/nm1276. [DOI] [PubMed] [Google Scholar]
- 10.Datta SK, Okamoto S, Hayashi T, Shin SS, et al. Vaccination with irradiated Listeria induces protective T cell immunity. Immunity. 2006;25:143–152. doi: 10.1016/j.immuni.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 11.Meadows M. Strategies to reduce medication errors. How the FDA is working to improve medication safety and what you can do to help. FDA Consum. 2003;37:20–27. [PubMed] [Google Scholar]
- 12.Predki PF, Mattoon D, Bangham R, Schweitzer B, Michaud G. Protein microarrays: a new tool for profiling antibody cross-reactivity. Hum Antibodies. 2005;14:7–15. [PubMed] [Google Scholar]
- 13.Bruggemann H, Henne A, Hoster F, Liesegang H, et al. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science. 2004;305:671–673. doi: 10.1126/science.1100330. [DOI] [PubMed] [Google Scholar]
- 14.Perry AL, Lambert PA. Propionibacterium acnes. Lett Appl Microbiol. 2006;42:185–188. doi: 10.1111/j.1472-765X.2006.01866.x. [DOI] [PubMed] [Google Scholar]
- 15.Johansson BE, Brett IC. Changing perspective on immunization against influenza. Vaccine. 2007;25:3062–3065. doi: 10.1016/j.vaccine.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 16.Tai SS. Streptococcus pneumoniae protein vaccine candidates: properties, activities and animal studies. Crit Rev Microbiol. 2006;32:39–53. doi: 10.1080/10408410600822942. [DOI] [PubMed] [Google Scholar]
- 17.Makoff AJ, Oxer MD, Romanos MA, Fairweather NF, Ballantine S. Expression of tetanus toxin fragment C in E. coli: high level expression by removing rare codons. Nucleic Acids Res. 1989;17:10191–10202. doi: 10.1093/nar/17.24.10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 19.Zhou JC, Yi XY, Kalinna BH, McManus DP. Expression and purification of recombinant Schistosoma japonicum paramyosin. Hunan Yi Ke Da Xue Xue Bao. 2000;25:106–108. [PubMed] [Google Scholar]
- 20.Huang CM, Ananthaswamy HN, Barnes S, Ma Y, et al. Mass spectrometric proteomics profiles of in vivo tumor secretomes: capillary ultrafiltration sampling of regressive tumor masses. Proteomic. 2006;6:6107–6116. doi: 10.1002/pmic.200600287. [DOI] [PubMed] [Google Scholar]
- 21.Huang CM, Wang CC, Kawai M, Barnes S, Elmets CA. Surfactant sodium lauryl sulfate enhances skin vaccination: molecular characterization via a novel technique using ultrafiltration capillaries and mass spectrometric proteomics. Mol Cell Proteomics. 2006;5:523–532. doi: 10.1074/mcp.M500259-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Eisen MB, Brown PO. DNA arrays for analysis of gene expression. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang D, Urisman A, Liu YT, Springer M, et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003;1:E2. doi: 10.1371/journal.pbio.0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dentovskaia SV, Shaihutdinov RZ, Knirel’ I, Ivanov SA, Anisimov AP. Generation of vaccine strains of gram-negative bacteria with reduced adverse reactions. Gen Mikrobiol Virusol. 2006;2:3–8. [PubMed] [Google Scholar]
- 25.Detmer A, Glenting J. Live bacterial vaccines – a review and identification of potential hazards. Microb Cell Fact. 2006;5:23–24. doi: 10.1186/1475-2859-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 27.Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10:7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsang VC, Wilkins PP. Immunodiagnosis of schistosomiasis. Screen with FAST-ELISA and confirm with immunoblot. Clin Lab Med. 1991;11:1029–1039. [PubMed] [Google Scholar]
- 29.Shomaker TS, Ward K. Microarray technology in biomedical research. Hawaii Med J. 2006;65:253–256. [PubMed] [Google Scholar]
- 30.Kharat AS, Tomasz A. Inactivation of the srtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect Immun. 2003;71:2758–2765. doi: 10.1128/IAI.71.5.2758-2765.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleineidam RG, Kruse S, Roggentin P, Schauer R. Elucidation of the role of functional amino acid residues of the small sialidase from Clostridium perfringens by site-directed mutagenesis. Biol Chem. 2001;382:313–319. doi: 10.1515/BC.2001.038. [DOI] [PubMed] [Google Scholar]
- 32.Basal E, Jain A, Kaushal GP. Antibody response to crude cell lysate of Propionibacterium acnes and induction of pro-inflammatory cytokines in patients with acne and normal healthy subjects. J Microbiol. 2004;42:117–125. [PubMed] [Google Scholar]
- 33.Larsen MR, Trelle MB, Thingholm TE, Jensen ON. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Biotechniques. 2006;40:790–798. doi: 10.2144/000112201. [DOI] [PubMed] [Google Scholar]
- 34.Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005;11:S45–S53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 35.Singh M, Srivastava I. Advances in vaccine adjuvants for infectious diseases. Curr HIV Res. 2003;1:309–320. doi: 10.2174/1570162033485195. [DOI] [PubMed] [Google Scholar]
- 36.O’Hagan DT, MacKichan ML, Singh M. Recent developments in adjuvants for vaccines against infectious diseases. Biomol Eng. 2001;18:69–85. doi: 10.1016/s1389-0344(01)00101-0. [DOI] [PubMed] [Google Scholar]
- 37.Burgio MR, Bennett PM, Koretz JF. Heat-induced quaternary transitions in hetero- and homo-polymers of alpha-crystallin. Mol Vis. 2001;7:228–233. [PubMed] [Google Scholar]
- 38.Hijnen WA, Beerendonk EF, Medema GJ. Inactivation credit of UV radiation for viruses, bacteria and protozoan (oo) cysts in water: a review. Water Res. 2006;40:3–22. doi: 10.1016/j.watres.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 39.Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports–principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 40.Every AL, Kramer DR, Mannering SI, Lew AM, Harrison LC. Intranasal vaccination with proinsulin DNA induces regulatory CD4+ T cells that prevent experimental autoimmune diabetes. J Immunol. 2006;176:4608–4615. doi: 10.4049/jimmunol.176.8.4608. [DOI] [PubMed] [Google Scholar]
- 41.Debin A, Kravtzoff R, Santiago JV, Cazales L, et al. Intranasal immunization with recombinant antigens associated with new cationic particles induces strong mucosal as well as systemic antibody and CTL responses. Vaccine. 2002;20:2752–2763. doi: 10.1016/s0264-410x(02)00191-3. [DOI] [PubMed] [Google Scholar]
- 42.Meissner HC. Influenza vaccines: a pediatric perspective. Curr Opin Pediatr. 2007;19:58–63. doi: 10.1097/MOP.0b013e328012999e. [DOI] [PubMed] [Google Scholar]
- 43.Garmise RJ, Staats HF, Hickey AJ. Novel dry powder preparations of whole inactivated influenza virus for nasal vaccination. AAPS PharmSciTech. 2007;8:E81. doi: 10.1208/pt0804081. [DOI] [PubMed] [Google Scholar]
- 44.Tylewska-Wierzbanowska S, Chmielewski T. Limitation of serological testing for Lyme borreliosis: evaluation of ELISA and Western blot in comparison with PCR and culture methods. Wien Klin Wochenschr. 2002;114:601–605. [PubMed] [Google Scholar]
- 45.Neuman DV, Amara RR, Steinman L, Utz PJ, et al. Microarray profiling of antibody responses against simian-human immunodeficiency virus: postchallenge convergence of reactivities independent of host histocompatibility type and vaccine regimen. J Virol. 2003;77:11125–11138. doi: 10.1128/JVI.77.20.11125-11138.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson WH, DiGennaro C, Hueber W, Haab BB, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 47.Winter C, Schwegmann-Wessels C, Cavanagh D, Neumann U, Herrler G. Sialic acid is a receptor determinant for infection of cells by avian infectious bronchitis virus. J Gen Virol. 2006;87:1209–1216. doi: 10.1099/vir.0.81651-0. [DOI] [PubMed] [Google Scholar]
- 48.Nakatsuji T, Liu YT, Huang CP, Gallo RL, Huang CM. Vaccination targeting a surface sialidase of P. acnes: Implication for new treatment of acne vulgaris. PLoS ONE. 2008;3:e1551. doi: 10.1371/journal.pone.0001551. [DOI] [PMC free article] [PubMed] [Google Scholar]