

Abstract
CpG islands mark CpG-enriched regions in otherwise CpG-depleted vertebrate genomes. While the regulatory importance of CpG islands is widely accepted, it is little appreciated that CpG islands vary greatly in lengths. For example, CpG islands in the human genome vary ∼30-fold in their lengths. Here we report findings suggesting that the lengths of CpG islands have functional consequences. Specifically, we show that promoters associated with long CpG islands (long-CGI promoters) are distinct from other promoters. First, long-CGI promoters are uniquely associated with genes with an intermediate level of gene expression breadths. Notably, intermediate expression breadths require the most complex mode of gene regulation, from the standpoint of information content. Second, long-CGI promoters encode more RNA polymerase II (Polr2a) binding sites than other promoters. Third, the actual binding patterns of Polr2a occur in a more tissue-specific manner in long-CGI promoters compared to other CGI promoters. Moreover, long-CGI promoters contain the largest numbers of experimentally characterized transcription start sites compared to other promoters, and the types of transcription start sites in them are biased toward tissue-specific patterns of gene expression. Finally, long-CGI promoters are preferentially associated with genes involved in development and regulation. Together, these findings indicate that functionally relevant variations of CpG islands exist. By investigating consequences of certain CpG island traits, we can gain additional insights into the mechanism and evolution of regulatory complexity of gene expression.
CpG islands are genomic regions unusually enriched in CpG dinucleotides in otherwise CpG-depleted vertebrate genomes (Bird 1986). The general lack of CpGs in vertebrate genomes is a likely consequence of DNA methylation, which occurs exclusively at cytosines in CpG contexts. Because methylated cytosines in CpGs are highly vulnerable to spontaneous mutations to thymines, DNA methylation effectively reduces CpG dinucleotide contents (Coulondre et al. 1978; Bird 1980). The human genome, for example, contains only ∼20% of CpGs compared to what is expected from its G + C content (Bird 1980; Cooper and Krawczak 1989; Elango et al. 2008).
CpG islands, however, avoid DNA methylation and preserve their CpGs (Antequera and Bird 1993). The avoidance of DNA methylation by CpG islands appears to critically rest on their ability to encode various regulatory sequences (Illingworth and Bird 2009). CpG islands may avoid DNA methylation by directly encoding demethylation signals. Alternatively, they may escape DNA methylation machinery altogether by hosting DNA-binding proteins, notably transcription factors. As such, CpG islands play important regulatory roles. Aberrant methylation of CpG islands manifests in serious disease phenotypes, including cancer (Robertson and Wolffe 2000). Revealing the nature of regulatory mechanisms of CpG islands remains an important topic in epigenetic studies (Mohn and Schübeler 2009).
Interestingly, the lengths of CpG islands in mammalian genomes exhibit substantial variations. For example, in the human genome, CpG islands vary ∼30-fold in their lengths, according to the annotations in the University of California, Santa Cruz (UCSC) genome browser. We hypothesize that such variation may reflect functional differences.
In this study, we focus on the relationship between CpG island lengths and the expression of associated genes. For this purpose, we analyze CpG islands overlapping with promoter regions. Previous studies revealed that the presence/absence of CpG islands in promoters is tightly linked to patterns of downstream gene expression. Specifically, CpG islands in promoters are linked to broad expression of housekeeping genes (e.g., Carninci et al. 2006; Saxonov et al. 2006; Elango and Yi 2008; Illingworth and Bird 2009). Here, we demonstrate that the lengths of CpG islands provide additional layers of complexity. Specifically, we show that we can further divide promoters according to the lengths of CpG islands and that this distinction coincides with different patterns of gene expression and promoter characteristics.
MATERIALS AND METHODS
Genome sequences and promoter-associated CpG islands annotation:
The human genome (version hg 18), the mouse genome (version mm 9), and CpG islands annotations were downloaded from the UCSC genome database (Kent et al. 2002; Karolchik et al. 2008). Briefly, the CpG islands annotation algorithm at UCSC searches genome sequences one base at a time, scoring each dinucleotide (+17 for CpG and −1 for others). Next, it finds maximally scoring segments and annotates the segment as a CpG island if it satisfies the following criteria: (1) G + C content >50%, (2) length >200, and (3) the ratio of observed to expected number of CpG dinucleotides (“CpG O/E”) >0.6. In the human genome, Alu elements occupy substantial portions (International Human Genome Sequencing Consortium 2001). Because Alu elements are short and G + C-rich, many genomic regions identified by the above criteria are Alu elements rather than bona fide CpG islands (Takai and Jones 2002). Therefore, to avoid false positives due to Alu elements, only the CpG islands >500 bp in length were used in this study.
To find promoter-associated CpG islands, we first investigated the distribution of CpG O/E values of putative promoter regions, defined as nucleotides straddling the transcription start site (TSS). Saxonov et al. (2006) showed that the average CpG O/E of human promoters peaks at the TSS and decays gradually as the distance from the TSS increases. We found a similar pattern. Notably, CpG O/E reached the genomic background at ∼4000–5000 bp distance from the TSS in both directions. Thus we defined promoters as the 5000-bp region on each side of TSSs. CpG islands are annotated as promoter associated if they lie within a distance of 5000 bp in each direction around the TSS.
To identify one-to-one correspondence between a promoter region and expression traits of a gene, we further performed the following filtering steps: first, promoters that overlapped with promoters from other genes were removed from all the analyses. Second, for genes containing alternate TSSs, one representative TSS was randomly chosen.
Gene expression data:
We used three types of expression data. First, we used the EST counts in the Unigene database (Wheeler et al. 2008). Genes with EST count ≥1 in a tissue were considered to be expressed in that tissue. The expression breadth of a gene is the number of tissues in which it is expressed. The total numbers of tissues analyzed in human and mouse are 49 and 47, respectively.
Second, we analyzed exon microarray expression data from six tissues (heart, kidney, liver, muscle, spleen, and testis) (Xing et al. 2007). We used the tissue specificity index as a measure of expression pattern. Tissue specificity index of a gene is defined as
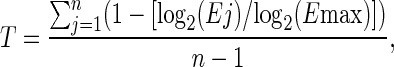 |
where n is the number of tissues analyzed, Ej is the expression level of the gene in the jth tissue, and E max is the maximum expression level of the gene across the n tissues (Yanai et al. 2005; Liao et al. 2006). The higher the tissue specificity index of a gene is, the more tissue-specific it is. A major advantage of the tissue specificity index is that it measures the tissue specificity of genes without imposing thresholds on expression levels (e.g., EST count ≥1 that we have used in expression breadth analyses).
Finally, we analyzed the Gene Atlas data for human and mouse, which are obtained via oligonucleotide microarray hybridizations (Su et al. 2004). We removed cancerous tissues from our analyses.
RNA polymerase II occupancy and cap analysis of gene expression data:
RNA polymerase II (Polr2a) occupancy data were obtained from Barrera
et al. (2008). Briefly, Barrera
et al. (2008) produced a genome-wide map of Polr2a occupancy in five mouse tissue types (brain, heart, kidney, liver, and embryonic stem cells), using the ChIP-chip method. Approximately 24,000 Polr2a binding sites are mapped in this study. The relative Polr2a occupancy at each binding site across the five tissues was characterized using the Shannon entropy 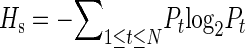 , where
, where 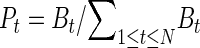 .
.
Bt is the average ChIP-chip log2 ratio in the 1-kb region centered at the midpoint of the binding site. A high entropy value means that the Polr2a is bound to that site uniformly across all tissues, whereas a low value of entropy means a more tissue-specific binding pattern.
Cap analysis of gene expression data:
To investigate the relationship between promoter CpG island lengths and experimentally determined characteristics of TSSs, we analyzed the cap analysis of gene expression (CAGE) tag data from Carninci et al. (2006). These data provided lists of experimentally verified TSSs (identified using tag clusters) from large numbers of different libraries from the human and mouse genomes (41 and 145 different libraries from human and mouse, respectively). The tag clusters were further divided into four types (Carninci et al. 2006). These include “broad” (BR), “single dominant peak” (SP), “bi- or multimodal” (MU), and “broad with dominant peak” (PB). We mapped the coordinates of tag clusters to the human and mouse genomes and analyzed the relationship between the frequencies of tag clusters and the lengths of promoter CpG islands.
Gene ontology analysis:
Gene ontology analyses were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (Dennis et al. 2003). To find the GO terms overrepresented in genes associated with long CpG islands, all genes with CpG islands were used as the background and Fisher's exact test was performed. Analyses were performed on the molecular function and the biological processes domains.
RESULTS
Long CpG islands are associated with intermediate tissue specificity:
We first examine the relationship between the lengths of promoter CpG islands and the breadths of gene expression. According to the information theory, genes that need to be expressed in an intermediate number of tissues require the most complex choice of switch on/off transition (Vinogradov 2006). Following this logic, we hypothesized that promoters associated with long CpG islands are capable of more complex regulation of gene expression than other promoters, because CpG islands may contain sequences involved in gene regulation (see Introduction).
Promoter CpG islands in the human and mouse genomes exhibit a large variation in their sizes (supporting information, Figure S1). To investigate the relationship between lengths of CpG islands and breadths of gene expression, we first divided human genes into several equal-sized bins on the basis of the lengths of the associated CpG islands. We then examined the mean expression breadths of each bin using EST data.
Following several experiments using different cutoff values, we observe that genes associated with CpG islands >2000 bp are expressed in significantly fewer tissues than others (Figure 1A and Figure S2). We thus refer to the human promoters harboring CpG islands (CGIs) that are >2000 bp as “long-CGI promoters” (LCGI promoters), as opposed to “short-CGI promoters” (SCGI promoters; CpG island lengths <2000 bp). For the sake of brevity, in the rest of this section, we use “LCGI genes” to refer to genes harboring LCGI promoters, and SCGI and NCGI genes to refer to those with SCGI promoters and those without CpG islands, respectively.
Figure 1.—

Long CpG island promoters are associated with genes expressed in fewer numbers of tissues compared to genes with short CpG island promoters. (A) Human and (B) mouse promoters are divided into eight equal-sized bins. Mean expression breadths of the genes in each bin, measured using EST data, are shown, along with their confidence intervals.
Genes associated with promoter CpG islands are generally more broadly expressed than those without CpG islands (Antequera 2003; Elango and Yi 2008; Saxonov et al. 2006; Weber et al. 2007). We find that when those genes are divided into LCGI and SCGI genes, expression breadths of LCGI genes are distinctively intermediate between those of NCGI and SCGI genes (Figure 2A).
Figure 2.—

Long CpG island promoters are associated with intermediate tissue specificity of downstream genes. (A) Human and mouse genes are divided into the following three types: no CpG island (NCGI), short CpG island (SCGI), and long CpG island (LCGI) promoter genes. See text for the exact definitions. The mean expression breadth of genes within each group measured using EST data is plotted with its confidence interval. (B) The mean tissue specificity indexes of NCGI, SCGI, and LCGI genes in the human and mouse genomes, determined by exon array data. (C) The mean tissue specificity indexes of NCGI, SCGI, and LCGI genes in the human and mouse genomes determined by the Gene Atlas oligonucleotide microarray data.
Mouse CpG islands are on average shorter than human CpG islands (Figure 1B). Despite this difference, the relationship between lengths of CpG islands and the breadths of gene expression is conserved between the two mammals (Figure 1B). Similar to the results from the human genome, LCGI genes in mouse (defined as CpG island lengths >1400 bp, Figure S3 and Figure S4) are expressed in significantly fewer tissues than SCGI genes are, but in a significantly greater number of tissues than NCGI genes are (Figure 2A, Figure S3, and Figure S4).
We observe the same trends using data from exon microarrays (Xing et al. 2007). We use the metric “tissue specificity index” for comparing gene expression breadths from exon microarrays (i.e., Liao et al. 2006). The tissue specificity index is inversely correlated with the number of tissues a gene is expressed in. NCGI and SCGI genes exhibit the highest and the lowest mean tissue specificity indexes, confirming their highly tissue-specific and broad expression, respectively (Figure 2B). Tissue specificities of LCGI genes are in between those of NCGI and SCGI genes (Figure 2B).
Analyses of oligonucleotide microarray data (Su et al. 2004) provide the same results. LCGI genes exhibit intermediate levels of tissue specificities (Figure 2C), while NCGI and SCGI genes represent high and low ends of tissue specificities. These results all support the idea that LCGI promoters are associated with intermediate tissue specificity, which typically require complex gene regulation strategies (Vinogradov 2006).
LCGI promoters are complex in terms of Polr2a occupancy:
In this section, we investigate characteristics of promoters themselves, analyzing genome-wide maps of Polr2a binding from five mouse tissues (brain, heart, kidney, liver, and embryonic stem cells) (Barrera et al. 2008). We mapped experimentally characterized Polr2a binding sites onto promoter regions and asked whether the numbers of Polr2a binding sites differ between the three promoter types (namely, LCGI, SCGI, and NCGI promoters). Since more binding sites likely suggest more complex regulatory mechanisms, we hypothesize that LCGI promoters encode greater numbers of Polr2a binding sites than SCGI or NCGI promoters do.
Indeed, the numbers of Polr2a binding sites in LCGI promoters are significantly greater than those of SCGI or NCGI promoters (P < 0.001, Mann–Whitney test for both comparisons). Figure 3A illustrates distinctive distributions of the numbers of encoded Polr2a binding sites in the NCGI, SCGI, and LCGI promoters. While the majority of NCGI and SCGI promoters contain a single Polr2a binding site, a large number of LCGI promoters contain two Polr2a binding sites. LCGI promoters also contain a greater proportion of promoters with ≥3 binding sites than NCGI or SCGI promoters do.
Figure 3.—

Long CpG island promoters exhibit a more complex Polr2a occupancy pattern. (A) Mouse promoters are divided into NCGI, SCGI, and LCGI promoters. Within each promoter type, the proportion of promoters containing a certain number (1–7) of experimentally verified Polr2a binding sites is plotted. (B) The average Shannon entropy of Polr2a occupancy in binding sites that overlap with the three types of promoters.
Furthermore, we measured the average entropy of Polr2a binding sites overlapping with the three promoter types (Figure 3B). For a binding site, a high entropy means that the Polr2a is bound to that site uniformly across all tissues. A low entropy indicates a more tissue-specific binding pattern. We show that the average entropy of binding sites in NCGI promoters is the lowest, which is in accord with the highly tissue-specific expression pattern of NCGI genes (see above). Binding sites in SCGI promoters have the highest entropies, reflecting their association with genes with “housekeeping” functions. The mean entropy of binding sites in LCGI promoters is lower than that of SCGI promoters and larger than that of NCGI promoters. This reaffirms the pattern that LCGI genes exhibit intermediate tissue specificity.
LCGI promoters contain a large number of TSSs biased toward tissue-specific expression:
We further investigated the relationship between CpG island lengths and the numbers and characteristics of experimentally characterized transcription start sites, using tag data obtained from the CAGE experiments from 145 and 41 mouse and human libraries (Carninci et al. 2006).
LCGI promoters on average contain significantly larger numbers of TSSs compared to SCGI and NCGI promoters. In the human genome, the median number of transcription start sites, defined as the number of tag clusters from CAGE, in LCGI promoters is 17.5 compared to 9 in SCGI promoters (P < 10−15, Mann–Whitney test). In comparison, the median number of TSSs in NCGI promoters is 4. A similar pattern is found in the mouse genome: the median number of TSSs in the LCGI promoters is 19 compared to 11 in SCGI promoters (P < 10−15, Mann–Whitney test). The median number of TSSs in NCGI promoters is 5.
CAGE data also provide a rare opportunity to assess detailed functional landscapes of TSSs in promoters. Carninci et al. (2006) demonstrated that tag clusters can be divided into four distinctive types. These include BR, SP, MU, and PB. Among these, the SP type TSSs are known to be tissue specific, while the BR type TSSs are broadly expressed (Carninci et al. 2006).
We hypothesize that LCGI promoters may contain a larger proportion of SP types compared to SCGI promoters. Indeed, the patterns in both human and mouse genomes fit this prediction (Figure S5). The frequency of the SP TSS in LCGI promoters is 1.7-fold higher than that in SCGI promoters in the human genome (P < 0.001). In the mouse genome, the difference is 2-fold (P < 0.001). In comparison, approximately half of TSSs in NCGI promoters belong to the SP type in human and mouse genomes, respectively (Figure S5). Thus, similar to the pattern found in distributions of tissue specificity (Figure 2) and Pol2ra binding sites (Figure 3), the proportion of SP TSSs in LCGI promoters is intermediate between those of NCGI and SCGI promoters. These findings indicate that regulatory complexities of LCGI promoter gene expression are at least partially mediated by the presence of large numbers of transcription start sites, each attuned to a tissue-specific pattern of gene expression.
LCGI promoters are preferentially associated with development and regulation:
We have demonstrated that LCGI genes are conspicuously different from SCGI and NCGI genes in terms of gene regulation complexity. We further ask whether LCGI genes are associated with certain gene functions, by examining gene overrepresentation of gene ontology (GO) terms. The top10 overrepresented GO terms are shown in Table 1. In the case of biological processes, we find that LCGI genes are associated mainly with ontology terms involved in development and gene regulation. The top two biological processes GO terms overrepresented in LCGI genes are “development” (Fisher's exact test, P < 10−14) and “regulation of transcription DNA dependent” (Fisher's exact test, P < 10−11). In terms of molecular function, LCGI genes are primarily associated with gene regulation and signaling. The top two molecular function GO terms overrepresented in the LCGI genes are “transcription factor activity” (Fisher's exact test, P < 10−15) and “transcription regulator activity” (Fisher's exact test, P < 10−14).
TABLE 1.
GO terms enriched in genes with long CpG island promoters
| Biological processes |
Molecular function |
||
|---|---|---|---|
| Overrepresented GO term | Fisher's exact test P-value | Overrepresented GO term | Fisher's exact test P-value |
| Development | 1.0 × 10−15 | Transcription factor activity | 1.8 × 10−16 |
| Regulation of transcription, DNA dependent | 7.1 × 10−12 | Transcription regulation activity | 2.8 × 10−15 |
| Regulation of nucleobase, nucleoside, nucleotide, and nucleic acid metabolism | 1.1 × 10−11 | Sequence-specific DNA binding | 9.3 × 10−11 |
| Regulation of transcription | 1.4 × 10−11 | DNA binding | 1.7 × 10−10 |
| Transcription, DNA dependent | 4.4 × 10−11 | Transmembrane receptor activity | 1.0 × 10−5 |
| Regulation of cellular metabolism | 1.5 × 10−10 | Signal transducer activity | 2.3 × 10−5 |
| Transcription | 1.6 × 10−10 | G-protein–coupled receptor activity | 4.1 × 10−5 |
| Regulation of cellular physiological processes | 2.1 × 10−10 | Nucleic acid binding | 5.3 × 10−5 |
| Regulation of biological processes | 2.4 × 10−10 | Receptor activity | 9.8 × 10−5 |
| Regulation of metabolism | 4.9 × 10−10 | Binding | 1.9 × 10−4 |
DISCUSSION
Since coined in 1980s (Bird 1986), the term “CpG island” is widely used to refer to genomic regions with high G + C content and unusual clusters of CpG dinucleotides. CpG islands are deeply involved in regulatory processes. In particular, the presence and absence of CpG islands in promoters clearly distinguish genes into housekeeping vs. tissue-specific patterns of expression (Antequera 2003; Saxonov et al. 2006; Weber et al. 2007) across distantly related vertebrate taxa (Elango and Yi 2008).
While these previous studies classified promoters into two groups with respect to their associations with CpG islands, our work demonstrates that not all CpG islands are equal. Specifically, we show that CpG island promoters in mammalian genomes consist of functionally distinctive groups according to CpG island lengths. Long CpG islands, defined as those >2000 bp in humans and >1400 bp in mouse, are associated with distinctively “intermediate” levels of tissue specificity (Figures 1 and 2). Furthermore, LCGI promoters have a larger number of Polr2a binding sites compared to SCGI and NCGI promoters, and the binding sites that overlap with LCGI promoters exhibit intermediate tissue specificity (Figure 3).
To confirm that our results are not a consequence of the annotation method used, we performed the same analysis using a different annotation method that depends solely on the clustering property of CpG dinucleotides, the threshold for which is chosen objectively (Glass et al. 2007). We observed the same patterns (results not shown).
Detailed analyses of experimentally characterized TSS positions and types reveal that LCGI promoters tend to house a large number of TSSs that are enriched with the SP type tags, typically associated with tissue-specific genes. Thus, the ability of LCGI promoters to regulate complex modes of gene expression may be directly attributable to the large number of TSSs, each attuned for tissue-specific expression. Alternatively, LCGI promoters could represent chromatin states that are more permissive to attracting specific regulatory machineries. CpG islands themselves may provide specific sequence context for such a purpose. For example, the Polycomb group protein complex targets a subset of CpG islands (Tanay et al. 2007), which tend to be long and are often found in genomic regions enriched with conserved noncoding elements (Akalin et al. 2009).
Our study begins to address the potential functional significance of within-genome variation of CpG islands and demonstrates that grouping CpG islands altogether can obscure the functional importance of certain characteristics of CpG islands. In light of this link between CpG island lengths and gene regulation, it is interesting to note that the lengths of CpG islands vary greatly across taxa (Glass et al. 2007). CpG islands in fish are much smaller than those in mammals (Aerts et al. 2004). The lengths of CpG islands within mammals also exhibit intriguing variation (Jiang et al. 2007). Comparative studies of CpG island traits, paired with functional data, may provide new insights into the regulatory role and evolution of CpG islands.
Caveats and future directions:
One caveat of our analyses is uncertainties in identifying promoters. We have designated genomic regions 5 kb on either side of the TSS as putative promoters, on the basis of the profile of degradation of CpG O/E values (materials and methods). Even though it is a common practice to define regions straddling the TSS as putative promoters (e.g., Aerts et al. 2004; Schug et al. 2005), this method is prone to errors. For example, the lengths of the promoter region may not be exactly 5 kb on either side of the TSS for all genes. To gauge if our definition is biased, we examined a subset of CpG islands that directly overlap with TSSs and hence have higher probabilities of being associated with true promoters. We found similar results (Figure S6). The pattern we discovered is likely to reflect a true relationship between CGI lengths and expression complexity.
Another potential issue is the notion of “tissue-specific” patterns of gene expression. While several studies converged on characterizing patterns of tissue-specific gene expression (e.g., Mortazavi et al. 2008), recent RNA-seq data demonstrate that typically many more genes than previously appreciated are expressed in a ubiquitous manner (e.g., Blencowe et al. 2009; Ramsköld et al. 2009). However, RNA-seq studies still find qualitative differences between transcriptomes of different tissues and that the expression levels of ubiquitously expressed genes are not uniform across tissues (Ramsköld et al. 2009). Our findings may well be the first step toward elucidating the properties of CpG islands and their importance in regulation of genes that are not expressed uniformly across tissues.
A puzzling phenomenon in the eukaryotic transcriptome is the ubiquitous presence of transcription outside of well-annotated protein-coding genes (Jacquier 2009; Mercer et al. 2009). It is of interest whether some CpG islands, especially long-CpG islands currently annotated as “intergenic,” facilitate tissue- and developmental stage-specific expression of “noncoding” transcripts. Overall, analyses of different CpG island traits, combined with newly emerging functional genomics data, have a potential to become a powerful tool to reveal hidden mechanisms of complexity of transcriptomes.
Acknowledgments
We thank Pierre Carninci for sharing the CAGE data and the editor and anonymous reviewers for comments on the previous versions of the manuscript. This study was supported by funds from the Blanchard–Milliken Fellowship, the Alfred P. Sloan Foundation, and a National Science Foundation grant (MCB-0950896) to S. Yi.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.126094/DC1.
References
- Aerts, S., G. Thijs, M. Dabrowski, Y. Moreau and B. De Moor, 2004. Comprehensive analysis of the base composition around the transcription start site in Metazoa. BMC Genomics 5 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin, A., D. Fredman, E. Arner, X. Dong, J. Bryne et al., 2009. Transcriptional features of genomic regulatory blocks. Genome Biol. 10 R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera, F., 2003. Structure, function and evolution of CpG island promoters. Cell Mol. Life Sci. 60 1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera, F., and A. Bird, 1993. Number of CpG islands and genes in the human and mouse. Proc. Natl. Acad. Sci. USA 90 11995–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera, L. O., Z. Li, A. D. Smith, K. C. Arden, W. K. Cavenee et al., 2008. Genome-wide mapping and analysis of active promoters in mouse embryonic stem cells and adult organs. Genome Res. 18 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, A., 1980. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, A., 1986. CpG-rich islands and the function of DNA methylation. Nature 321 209–213. [DOI] [PubMed] [Google Scholar]
- Blencowe, B. J., S. Ahmad and L. J. Lee, 2009. Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev. 23 1379–1386. [DOI] [PubMed] [Google Scholar]
- Carninci, P., A. Sandelin, B. Lenhard, S. Katayama, K. Shimokawa et al., 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38 626–635. [DOI] [PubMed] [Google Scholar]
- Cooper, D. N., and M. Krawczak, 1989. Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum. Genet. 83 181–188. [DOI] [PubMed] [Google Scholar]
- Coulondre, C., J. H. Miller, P. J. Farabaugh and W. Gilbert, 1978. Molecular basis of base substitution hotspots in Escherichia coli. Nature 274 775–780. [DOI] [PubMed] [Google Scholar]
- Dennis, G., B. Sherman, D. Hosack, J. Yang, W. Gao et al., 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4 R60. [PubMed] [Google Scholar]
- Elango, N., and S. Yi, 2008. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol. Biol. Evol. 25 1602–1608. [DOI] [PubMed] [Google Scholar]
- Elango, N., S.-H. Kim, NISC Comparative Sequencing Program, E. Vigoda and S. Yi, 2008. Mutations of different molecular origins exhibit contrasting patterns of regional substitution rate variation. PLoS Comput. Biol. 4 e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, J. L., R. F. Thompson, B. Khulan, M. E. Figueroa, E. N. Olivier et al., 2007. CG dinucleotide clustering is a species-specific property of the genome. Nucleic Acids Res. 35 6798–6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth, R. S., and A. P. Bird, 2009. CpG islands—‘a rough guide’. FEBS Lett. 583 1713–1720. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium, 2001. Initial sequencing and analysis of the human genome. Nature 409 860–921. [DOI] [PubMed] [Google Scholar]
- Jacquier, A., 2009. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 10 833–844. [DOI] [PubMed] [Google Scholar]
- Jiang, C., L. Han, B. Su, W.-H. Li and Z. Zhao, 2007. Features and trend of loss of promoter-associated CpG islands in the human and mouse genomes. Mol. Biol. Evol. 24 1991–2000. [DOI] [PubMed] [Google Scholar]
- Karolchik, D., R. M. Kuhn, R. Baertsch, G. P. Barber, H. Clawson et al., 2008. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 36 D773–D779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. J., C. W. Sugnet, T. S. Furey, K. M. Roskin, T. H. Pringle et al., 2002. The human genome browser at UCSC. Genome Res. 12 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, B.-Y., N. M. Scott and J. Zhang, 2006. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol. Biol. Evol. 23 2072–2080. [DOI] [PubMed] [Google Scholar]
- Mercer, T. R., M. E. Dinger and J. S. Mattick, 2009. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10 155–159. [DOI] [PubMed] [Google Scholar]
- Mohn, F., and D. Schübeler, 2009. Genetics and epigenetics: stability and plasticity during celluar differentiation. Trends Genet. 25 129–136. [DOI] [PubMed] [Google Scholar]
- Mortazavi, A., B. A. Williams, K. McCue, L. Schaeffer and B. Wold, 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5 621–628. [DOI] [PubMed] [Google Scholar]
- Ramsköld, D., E. T. Wang, C. B. Burge and R. Sandberg, 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 5 e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, K. D., and A. P. Wolffe, 2000. DNA methylation in health and disease. Nat. Rev. Genet. 1 11–19. [DOI] [PubMed] [Google Scholar]
- Saxonov, S., P. Berg and D. L. Brutlag, 2006. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 103 1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug, J., W.-P. Schuller, C. Kappen, J. M. Salbaum, M. Bucan et al., 2005. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 6 R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, A. I., T. Wiltshire, S. Batalov, H. Lapp, K. A. Ching et al., 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 101 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai, D., and P. A. Jones, 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 99 3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay, A., A. H. O'Donnell, M. Damelin and T. H. Bestor, 2007. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci. USA 104 5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov, A. E., 2006. ‘Genome design’ model and multicellular complexity: golden middle. Nucleic Acids Res. 34 5906–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, M., I. Hellmann, M. B. Stadler, L. Ramos, S. Pääbo et al., 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39 457–466. [DOI] [PubMed] [Google Scholar]
- Wheeler, D. L., T. Barrett, D. A. Benson, S. H. Bryant, K. Canese et al., 2008. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 36 D13–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, Y., Z. Ouyang, K. Kapur, M. P. Scott and W. H. Wong, 2007. Assessing the conservation of mammalian gene expression using high-density exon arrays. Mol. Biol. Evol. 24 1283–1285. [DOI] [PubMed] [Google Scholar]
- Yanai, I., H. Benjamin, M. Shmoish, V. Chalifa-Caspi, M. Shklar et al., 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21 650–659. [DOI] [PubMed] [Google Scholar]