

Abstract
Microvascular pericytes and perivascular fibroblasts have recently been identified as the source of scar-producing myofibroblasts that appear after injury of the kidney. We show that cross talk between pericytes and endothelial cells concomitantly dictates development of fibrosis and loss of microvasculature after injury. When either platelet-derived growth factor receptor (R)-β signaling in pericytes or vascular endothelial growth factor (VEGF)R2 signaling in endothelial cells was blocked by circulating soluble receptor ectodomains, both fibrosis and capillary rarefaction were markedly attenuated during progressive kidney injury. Blockade of either receptor-mediated signaling pathway prevented pericyte differentiation and proliferation, but VEGFR2 blockade also attenuated recruitment of inflammatory macrophages throughout disease progression. Whereas injury down-regulated angiogenic VEGF164, the dys-angiogenic isomers VEGF120 and VEGF188 were up-regulated, suggesting that pericyte-myofibroblast differentiation triggers endothelial loss by a switch in secretion of VEGF isomers. These findings link fibrogenesis inextricably with microvascular rarefaction for the first time, add new significance to fibrogenesis, and identify novel therapeutic targets.
Prompted by recent identification that kidney pericytes and to a lesser extent perivascular fibroblasts are the source of scar-forming myofibroblasts and previous studies in the eye and brain that identified pericytes as pivotal cells in developmental angiogenesis and vascular stabilization,1–4 we wanted to determine whether injury-stimulated pericyte-endothelial cross talk led not only to migration of pericytes from capillaries and differentiation into myofibroblasts but also to destabilization of capillaries in the kidney. Microvascular rarefaction after injury in the kidney and other organs is increasingly implicated in organ ischemia, chronic inflammation, and progressive loss of organ function.5–8 Therefore, strategies to prevent microvascular rarefaction are highly desirable. Regulated platelet-derived growth factor (PDGF) signaling from endothelial cells to pericyte PDGF receptor (R)-β (PDGFR-β) has been shown to be vital in vascular stabilization and in sprouting angiogenesis.9–11 PDGFs have also been implicated in fibrogenesis.12–15 PDGF over expression in endothelial cells, however, paradoxically promotes vascular rarefaction, suggesting that pro-apoptotic factors are released from, or survival factors are down-regulated in, expanded pericytes/fibroblasts.16 Although vascular endothelial growth factor (VEGF) signaling from pericytes to endothelial cells has been implicated in developmental angiogenesis,17–19 and more recently VEGF signaling from the specialized pericytes of the kidney glomerulus known as podocytes has been shown to be crucial for vascular stabilization,20 the presence of VEGF receptor 1 (VEGFR1) FMS-like tyrosine kinase 1 (Flt1) and receptor 2 (VEGFR2) VEGF receptor 3 and Neuropilin 1 on endothelial cells, the regulated expression of four distinct VEGF genes, and the regulated expression of multiple transcription splice variants render VEGF signaling more complicated than simple binary ligand-receptor interactions.21–26
Materials and Methods
Coll-GFP Mouse Model
Coll-GFP transgenic mice were generated and validated as previously described on the C57BL/6 background.1 All studies were carried out under protocols approved by the Institutional Animal Care and Use Committee, National Taiwan University College of Medicine; Animal Research and Comparative Medicine, Harvard Medical School; and the Office of Animal Welfare, University of Washington.
Mouse Models of Fibrosis
Unilateral ureteric obstruction (UUO) was performed in adult mice (8 to 12 weeks), as previously described.1 Briefly, under anesthesia by ketamine/xylazine (100/10 mg/kg i.p), the left ureter was exposed through flank incision in the prone position. The ureter was ligated twice using 4−0 nylon surgical sutures at the level of the lower pole of the kidney. In some experiments, sham operation was performed by flank incision only. The unilateral ischemia-reperfusion injury (IRI) model was performed, as previously described27 using a 30-minute ischemic time at 36.8°C to 37.3°C core temperature.
Construction of Adenovirus
Adenovirus expressing soluble (s) PDGFRβ and soluble (s) VEGFR2 were generated and purified as previously described.9 Briefly, PDGFRβ ectodomain cDNA (corresponding to amino acids 1 to 527) was amplified from embryonic day 12.5 mouse embryo cDNA with C-terminal His6 epitope tag and VEGFR2 cDNA sequence encoding signal peptide, and the ectodomain (to TIRRVRKEDGG, amino acid 731) was fused to murine IgG2a Fc (fragment of IgG after papain digesion with constant regions) fragment. Adenovirus was then generated by homologous recombination followed by production in 293 cells and CsCl gradient purification. Adenovirus titers were calculated as plaque-forming units on 293 cells. The sPDGFRβ and sVEGFR2 were purified from adenoviral supernatants using affinity chromatography (GE Healthcare, Pittsburgh, PA).
Adenovirus Administration and Detection of Plasma Transgene Expression
Adult Coll-GFP mice (8 to 12 weeks) received a single i.v. tail vein injection of 1 × 109 plaque-forming units of the indicated at the indicated time points (n = 6 for each group at each time point). At the indicated time points after injection, whole blood was obtained by retro-orbital phlebotomy followed by Western blot analysis of 1 μl of plasma using anti-His probe antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and then anti-rabbit IgG-HRP for PDGFRβ ectodomain, and anti-IgG2a Fc-HRP antibody (Thermo Scientific, Rockford, IL) for VEGFR2 ectodomain.
Cell Culture and Purification
Purification of Pericytes from the Normal Kidney
Pericyte purification from the normal kidney was previously described.1 Briefly the kidney was decapsulated, diced, and then incubated at 37°C for 30 minutes with liberase (0.5 mg/ml) (Roche) and DNase (100 U/ml, Roche) in HBSS. After centrifugation, cells were resuspended in 5 ml of PBS/1% bovine serum albumin, and filtered (40 μm). Pericytes were purified from the single cell suspension by isolating GFP+ cells using FACSAria cell sorting, and then total RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA). Note this purification does not distinguish pericytes from perivascular fibroblasts.
Purification and Culture of Myofibroblasts from the Kidney
Purification of kidney myofibroblasts from the kidney on day 7 after UUO was previously described.1 The kidney was decapsulated, diced, and then incubated at 37°C for 1 hour with liberase (0.5 mg/ml) (Roche) and DNase (100 U/ml, Roche) in HBSS. After centrifugation, cells were resuspended in 5 ml of PBS/1% bovine serum albumin, and filtered (40 μm). Myofibroblasts from the kidney after UUO were purified from single-cell suspension by isolating GFP+ cells using FACSAria cell sorting, and then the total RNA was isolated, or purified cells were cultured in Dulbecco's modified Eagle's medium with 20% fetal bovine serum (FBS). The primary cultured cells used in this study were between passages 4 and 6 and have been previously characterized.27 For transcriptional analysis, total RNA was isolated from sorted UUO kidney fibroblasts using RNeasy Mini Kit (Qiagen).
Preparation of Peripheral Blood Monocytes and Macrophages from the Kidney after UUO
Blood (500 μl) was collected from the inferior vena cava in sodium citrate (0.38%). Remaining blood was flushed out with ice-cold PBS; then the kidney on day 7 after UUO was harvested. Peripheral blood mononuclear cells were isolated from citrated whole blood using Ficoll-Paque PLUS (GE Healthcare). The kidney was decapsulated, diced, and incubated at 37°C for 1 hour with liberase/DNase as previously described. Peripheral blood mononuclear cells and digested kidney tissue were resuspended in Dulbecco's modified Eagle's medium/F12 medium with 10% FBS, plated onto cell culture dishes and incubated at 37°C for 1 hour. Nonadherent cells were washed away vigorously by ice-cold PBS five times; then the adherent peripheral blood monocytes or macrophages from UUO kidney (UUO macrophages) were lysed using RNeasy Mini Kit (Qiagen) for total RNA extraction.28 Bone marrow macrophages were prepared as previously described from whole bone marrow.27
Culture of Human Umbilical Vein Endothelial Cells
Human umbilical vein endothelial cells (HUVEC) were prepared from human umbilical cords by collagenase digestion and then cultured in M199 medium supplemented with 20% FBS. The primary cultured cells used in this study were between passages 4 and 7.7
In Vitro Analysis of Soluble Ectodomain Function
Myofibroblasts from UUO kidney were cultured in Dulbecco's modified Eagle's medium with 0.5% FBS for 16 hours and then incubated with platelet growth derived factor-BB (PDGF-BB) (10 ng/ml) (R&D Systems, Minneapolis, MN) in the presence or absence of affinity-purified sPDGFRβ (200 ng/ml) or sVEGFR2-Fc (5 μg/ml) for 15 minutes. The HUVEC were cultured in M199 with 0.5% FBS for 1 hour and then incubated with VEGF (20 ng/ml) (R&D Systems) in the presence or absence of sVEGFR2-Fc or sPDGFRβ for 15 minutes.9 The extent of PDGFRβ or VEGFR2 phosphorylation was determined by Western blot analysis of whole-cell extracts. Day 7 bone marrow macrophages were cultured in 12-well plates for 24 hours with supernatants containing sPDGFRβ, sVEGFR2, or sFc, and co-activated with LPS (Sigma) 100 ng/ml. Activation was determined by the extent of IL1β transcripts normalized to GAPDH transcripts.29
Tissue Preparation and Histology
Mouse tissues were prepared and stained as previously described.1 Primary antibodies against the following proteins were used for immunolabeling: αSMA-Cy3 (1:200, clone 1A4, Sigma), Ki-67, PDGF-B (1:200, Abcam, Cambridge, UK), F4/80, CD31 (1:200, eBioscience, San Diego, CA), VEGF (1:200, Santa Cruz Biotechnology), Tyr751 phospho-PDGFRβ, and Tyr1175 phospho-VEGFR2 (1:100, Cell Signaling Technology), Laminin (1:1000, Sigma). Fluorescent conjugated affinity purified secondary antibody labeling (1:400–1:800) (Jackson ImmunoResearch, West Grove, PA), co-labeled with DAPI, mounting with Vectashield (VectorLabs, Burlingam, CA) with DAPI, image capture, and processing were carried out as previously described. To study endothelial cells entering the cell cycle after UUO injury, cryosections were labeled with antibodies against Ki-67, followed by Cy3-conjugated secondary antibody, and then anti-CD31 antibody, followed by Alexa Fluor 350-conjugated secondary antibody (1:500) (Molecular Probes, Carlsbad, CA), and were mounted with ProLong Gold (Invitrogen, Carlsbad, CA). Quantification of specific cells in tissue sections was carried out as previously described. In brief, sections were co-labeled with DAPI; Coll-GFP+ cells were identified by blue and green nuclear co-localization; αSMA+, and F4/80+ cells were identified by greater than 75% of the cell area immediately surrounding nuclei (detected by DAPI) staining positive with Cy3 fluorescence indicative of the antigen expression; Ki-67+ cells were identified by positive nuclear staining for Cy3 fluorescence. Specific cells were counted in 10 cortical interstitial fields randomly selected at ×400 magnification per mouse. Vessel fluorescence was analyzed in images at ×400 magnification captured from CD31-stained sections of 10 different fields from six different animals. Based on fluorescence intensities ranging from 0 to 255, peritubular capillaries were distinguished from the background by empirically determining threshold values that marked only blood vessels in specimens from control kidney in sham-operated mice. The threshold was constant for all measurement.9 Alternatively, in IRI studies, vascular rarefaction was scored using a grid-based method.30 Interstitial fibrosis was quantified in Picrosirius red-stained paraffin sections. The morphometry of CD31+ peritubular capillary and Picrosirius red+ collagen was quantified using the FoveaPro4 program (Reindeer Graphics, Inc.) as described previously.1 Human kidney biopsy fresh tissues were obtained during routine biopsy collection for pathologic analysis during the course of routine clinical practice (IRB protocol 2007P000268). Samples were prepared for EM using standard methods for routine EM imaging.
RT-PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen). Purity was determined by A260 to A280. cDNA was synthesized using oligo(dT) and random primers. Conventional and quantitative PCR was performed using methods previously described.1 The specific primer pairs used in PCR are listed in Tables 1–3. The primer pairs for VEGF-A in Tables 1 and 2 were designed to amplify all isoforms including VEGF120, VEGF164, and VEGF188.31
Table 1.
Primer Sequences Used in RT-PCR
| Gene | Sequences |
|---|---|
| GAPDH | |
| Forward | 5′-ACTCCACTCACGGCAAATTC-3′ |
| Reverse | 5′-CACATTGGGGGTAGGAACAC-3′ |
| Coll1a1 | |
| Forward | 5′-CTGGTGAACAGGGTGTTCCT-3′ |
| Reverse | 5′-AGAACCATCAGCACCTTTGG-3′ |
| Kim1 | |
| Forward | 5′-ATGAATCAGATTCAAGTCTTC-3′ |
| Reverse | 5′-TCTGGTTTGTGAGTCCATGTG-3′ |
| F4/80 | |
| Forward | 5′-TTTTCAGATCCTTGGCCATC-3′ |
| Reverse | 5′-ACACTGGGGCACTTTTGTTC-3′ |
| VEGFR2 | |
| Forward | 5′-AGTGGCTCTGTCCTCCAAGA-3′ |
| Reverse | 5′-GCAAACCTTCCAAAACCAAA-3′ |
| VEGF-A | |
| Forward | 5′-ATCTTCAAGCCGTCCTGTGT-3′ |
| Reverse | 5′-GCATTCACATCTGCTGTGCT-3′ |
Table 2.
Primer Sequences Used in Quantitative RT-PCR
| Gene | Sequences |
|---|---|
| PDGF-B | |
| Forward | 5′-CCCACAGTGGCTTTTCATTT-3′ |
| Reverse | 5′-GTGAACGTAGGGGAAGTGGA-3′ |
| TGFβ1 | |
| Forward | 5′-GGACTCTCCACCTGCAAGAC-3′ |
| Reverse | 5′-GACTGGCGAGCCTTAGTTTG-3′ |
| FGF2 | |
| Forward | 5′-AGCGGCTCTACTGCAAGAAC-3′ |
| Reverse | 5′-TGGCACACACTCCCTTGATA-3′ |
| BMP7 | |
| Forward | 5′-GTACGTCAGCTTCCGAGACC-3′ |
| Reverse | 5′-GGTGGCGTTCATGTAGGAGT-3′ |
| TNFα | |
| Forward | 5′-TAGCCAGGAGGGAGAACAGA-3′ |
| Reverse | 5′-TTTTCTGGAGGGAGATGTGG-3′ |
| CCL2 | |
| Forward | 5′-AGCACCAGCCAACTCTCACT-3′ |
| Reverse | 5′-CGTTAACTGCATCTGGCTGA-3′ |
| CCR2 | |
| Forward | 5′-ATTCTCCACACCCTGTTTCG-3′ |
| Reverse | 5′-GATTCCTGGAAGGTGGTCAA-3′ |
| CX3CL1 | |
| Forward | 5′-TCGGACTTTGTTGGTTCCTC-3′ |
| Reverse | 5′-CAAAATGGCACAGACATTGG-3′ |
| CX3CR1 | |
| Forward | 5′-GGAGACTGGAGCCAACAGAG-3′ |
| Reverse | 5′-CCTGATCCAGGGAATGCTAA-3′ |
| VEGF-A | |
| Forward | 5′-ATCTTCAAGCCGTCCTGTGT-3′ |
| Reverse | 5′-GCATTCACATCTGCTGTGCT-3′ |
| VCAM1 | |
| Forward | 5′-TACCAGCTCCCAAAATCCTG-3′ |
| Reverse | 5′-TCTGCTAATTCCAGCCTCGT-3′ |
| ICAM1 | |
| Forward | 5′-AGCACCTCCCCACCTACTTT-3′ |
| Reverse | 5′-AGCTTGCACGACCCTTCTAA-3′ |
| GAPDH | |
| Forward | 5′-CTGGAGAAACCTGCCAAGTA-3′ |
| Reverse | 5′-AAGAGTGGGAGTTGCTGTTG-3′ |
Table 3.
Primer Sequences Used in Quantitative RT-PCR of VEGF Isoforms
| Gene | Sequences | |
|---|---|---|
| VEGF | Common forward | 5′-ACAGCAGATGTGAATGCAGACCAAAGAAAG-3′ |
| VEGF120 | Reverse | 5′-CGGCTTGTCACATTTTTCTGG-3′ |
| VEGF144 | Reverse | 5′-CAAGGCTCACAGTGATTTTCTGG-3′ |
| VEGF188 | Reverse | 5′-AACAAGGCTCACAGTGAACGCT-3′ |
Western Blot Analysis
Total cellular protein extracted using radioimmunoprecipitation assay buffer was subjected to Western blot analysis using methods previously described.32 The following primary antibodies were used to detect the specific protein: VCAM1, ICAM1, VEGF (Santa Cruz Biotechnology), Tyr751 phospho-PDGFRβ, Tyr1175 phospho-VEGFR2, VEGFR2 (Cell Signaling Technology, Beverley, MA), and PDGFRβ (gift from Dr. William Stallcup), and GFP (Medical & Biological Laboratories Co., Nagoya, Japan).
Fluorescence-Activated Cell Sorting (FACS) Analysis
Single cells (1 × 105) from cultured HUVEC and UUO kidney myofibroblasts were resuspended in FACS buffer and incubated with antibodies against VEGFR2 (1:400) (R&D Systems) for 30 minutes at 4°C in the presence of 1% mouse serum. After washing with FACS wash buffer, and resuspending in 200 μl FACS buffer, cells were analyzed using BD FACSCalibur flow cytometer.29
Statistical Analysis
Error bars are the SE of the mean. Statistical analyses were carried out using GraphPad Prizm (GraphPad Software, La Jolla, CA). The statistical significance was evaluated by one-way analysis of variance.
Results
Blockade of PDGFRβ or VEGFR2 Attenuates Fibrogenesis
Soluble ectodomains of PDGFRβ (sPDGFRβ) and VEGFR2 (sVEGFR2-Fc) effectively blocked tyrosine phosphorylation of native cognate receptors triggered by PDGF or VEGF in cultured kidney myofibroblasts or endothelial cells, respectively (Figure 1, A and B) indicating that these receptors can function as soluble inhibitors of endogenous receptor function. Mice treated with single IV injections of adenovirus led to stable and robust expression of soluble ectodomains of PDGFRβ (sPDGFRβ), VEGFR2 (sVEGFR2-Fc), or control protein (sFc) in the circulation (Figure 1C). Reporter mice expressing the transgene Coll1a1-GFP (Coll-GFP) were given adenovirus and were then subjected to progressive kidney injury induced by surgical UUO. Coll-GFP is detected in pericytes, podocytes, and perivascular fibroblasts of the kidney.1,2 Whereas fibroblasts are spindle-shaped cells of mesenchymal origin surrounded by collagen matrix, pericytes in the kidney are defined anatomically as extensively branched cells of mesenchymal origin (derived from FoxD1-expressing progenitors) that partially surround the endothelium of capillaries. The branched processes of the pericytes are sheathed within the capillary basement membrane and the basement membrane is often broken or incomplete between the endothelial cell and pericyte, allowing close appositions or interdigitations to occur (Supplemental Figure S1, see http://ajp.amjpathol.org).33–38 Perivascular fibroblasts, on the other hand, although of similar origin to kidney pericytes, surround arterioles within a collagenous matrix and have no close appositions with endothelial cells38 (Supplemental Figure S1, see http://ajp.amjpathol.org). In mice that produced circulating control sFc due to adenoviral injection, tyrosine phosphorylation of PDGFRβ and VEGFR2 was detected in pericytes and endothelial cells, respectively, focally, in UUO kidney as early as day 1 after UUO surgery (Figure 1, D and E). Tyrosine phosphorylation of both PDGFRβ and VEGFR2 was readily detectable by Western blot analysis of the whole kidney on day 4 after UUO (Figure 1F). Circulating sVEGFR2-Fc abolished phosphorylation of both PDGFRβ and VEGFR2, whereas sPDGFRβ only exerted potent inhibition on PDGFRβ (Figure 1F). In diseased mice producing circulating control sFc, there was an expected increase in Coll-GFP fluorescence in kidney on day 4 (Figure 1, G and H) and day 10 (Figure 1H), indicating increase of collagen 1 (α1) transcripts and expansion of pericyte-derived interstitial myofibroblasts. Increased Coll-GFP fluorescence was attenuated by either sPDGFRβ or sVEGFR2-Fc. These findings in tissue sections were supported by decreased GFP protein production and decreased collagen 1 (α1) transcripts in the kidneys after 4 days of injury (Figure 1L). The consequence of inhibition of collagen 1 (α1) transcription was marked attenuation of fibrosis (Figure 1, J and K). Notably downregulation of transcripts for pro-fibrotic effectors PDGF-B, TGFβ1, and FGF2 was achieved by either sPDGFRβ or sVEGFR2-Fc (Figure 1L). However, levels of transcripts for anti-fibrotic BMP7, which are down-regulated after kidney injury, were partially restored by sVEGFR2, and this difference was not significant when compared with kidneys exposed to sFc; therefore, it is unlikely to play a role mechanistically (Figure 1L). Thus, targeting receptors expressed exclusively by either pericytes (including perivascular fibroblasts) or endothelial cells prevents fibrogenesis.
Figure 1.
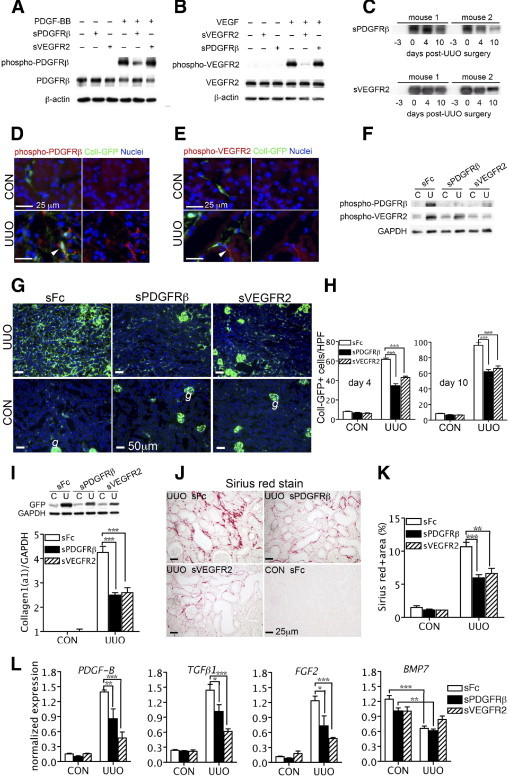
Selective inhibition of VEGFR2 or platelet-derived growth factor (PDGFR)β signaling by soluble circulating receptors attenuates kidney fibrosis after ureteral obstruction. A, B: Western blots of kidney myofibroblast phospho-PDGFRβ (A) or human umbilical vein endothelial cells phospho-VEGFR2 (B) stimulated by PDGF-BB or VEGF and inhibited by purified sPDGFRβ and sVEGFR2-Fc in vitro. C: Time course of circulating adenovirus-mediated sPDGFRβ and sVEGFR2-Fc production by Western blot analysis of 1 μl of plasma after i.v. administration of adenoviruses (1 × 109 plaque-forming units) to adult Coll-GFP mice 3 days before unilateral ureteral obstruction (UUO) surgery. D–F: Immunofluorescence images (D, E) of day 1 post-UUO Coll-GFP kidneys and Western blot (F) of day 4 post-UUO kidneys from mice with circulating adenovirus-mediated sPDGFRβ, sVEGFR2-Fc, or sFc, detecting phospho-PDGFRβ or phospho-VEGFR2 (arrowheads in D and E). G, H: Low-power fluorescence images of Coll-GFP in kidney sections in control (CON) or day 4 after UUO (g denotes glomerular podocytes) (G), indicating collagen 1 (α1) transcription and interstitial pericytes/perivascular fibroblasts (H), from mice-producing systemic sPDGFRβ, sVEGFR2-Fc, or sFc. I: Western blot of GFP protein or GAPDH levels (upper) or Q-PCR for collagen 1 (α1) transcripts (lower) in control [C] and day 4 after UUO [U] kidney cortex (n = 6/group). J, K: Images of picrosirius red-stained kidney sections for interstitial fibrillar collagens (red) (J), and morphometric quantification of fibrillar collagen from whole sagittal kidney sections (K). L: Q-PCR of transcripts of profibrotic effectors, in kidney cortex day 4 after UUO in mice-producing systemic sPDGFRβ, sVEGFR2-Fc, or sFc. *P < 0.05, **P < 0.01, ***P < 0.001 (n = 6/group).
To determine whether the anti-fibrotic properties of sPDGFRβ and sVEGFR2-Fc were generalizable to other models of injury to the cortex and medulla of kidney, we studied their properties in a second fibrogenic model, which we previously reported, called the unilateral IRI model.27 Single kidneys exposed to IRI develop progressive interstitial fibrosis by 15 days. Similar to the studies of UUO injury, mice received viruses 3 days before IRI and organs and blood were assessed on day 15 (Supplemental Figure S2A, see http://ajp.amjpathol.org). Mouse blood contained readily detectable receptor ectodomains on day 0 through day 15 after IRI surgery, and kidneys from mice with sPDGFRβ or sVEGFR2-Fc in blood experienced considerably less interstitial fibrosis on day 15 post IRI (Supplemental Figure S2, B and C, see http://ajp.amjpathol.org), which were similar to our observations in the UUO kidney injury model.
Blockade of PDGFRβ or VEGFR2 Limits Pericyte Proliferation and Detachment from Capillaries
To understand the mechanism of action further, proliferation of Coll-GFP+ interstitial cells was quantified on day 4 after injury onset in the UUO model. Either VEGFR2 or PDGFRβ blockade attenuated Coll-GFP+ cell proliferation (Figure 2, A and B). By contrast, proliferation in injured epithelial tubules was unaffected (Figure 2, A and C). By day 1 after kidney injury onset, pericytes could be detected detaching and moving away from capillaries, and by day 2, the pericyte population had increased. In mice expressing circulating sPDGFRβ or sVEGFR2-Fc, pericytes remained adherent to endothelial cells in diseased kidney and the pericyte population was not increased (Figure 2, D and E). Because kidney injury in this model is believed to initiate in epithelial tubules due to high back pressure and loss of tubular flow, the finding that epithelial proliferation is unaffected by receptor blockade serves as both an important control for epithelial injury, but it also localizes the effect of PDGFRβ blockade and VEGFR2 blockade to the perivascular signaling compartment. The αSMA is a robust marker of the differentiated myofibroblast cell in the adult kidney, but is not expressed by adult mouse pericytes.1,2 Not surprisingly, the number of αSMA-expressing cells decreased in response to VEGFR2 blockade or PDGFRβ blockade (Figure 2, F and G). However, in addition the proportion of Coll-GFP+ cells that co-expressed the myofibroblast marker αSMA also decreased from 99% to 94% and 89% in response to PDGFRβ and VEGFR2 blockade, respectively (Figure 2, F and H), providing further evidence that both administered proteins prevented pericyte migration and differentiation. Notably VEGFR2 blockade was most effective in blocking this differentiation. Similarly in the unilateral IRI model of kidney fibrosis, αSMA expression was also attenuated in mice with circulating sPDGFRβ or sVEGFR2-Fc (Supplemental Figure S2, D and F, see http://ajp.amjpathol.org) indicating a similar mechanism of action in that injury model.
Figure 2.

The platelet-derived growth factor (PDGFR)β or VEGFR2 blockade prevents pericyte proliferation, migration, and differentiation in kidneys after unilateral ureteral obstruction (UUO) injury. A–C: Split-image immunofluorescence micrographs of Coll-GFP+ pericytes/myofibroblasts in control or day 4 after UUO kidneys co-labeled with the pan-cell-cycle marker Ki-67 (A), and quantification of Coll-GFP+, Ki-67+ interstitial cells (arrowheads in A and B), or Ki-67+ tubule epithelial cells (C). D: Immunofluorescence micrographs of endothelial (CD31, red) cell, and Coll-GFP+ cell interactions in mouse kidneys on days 1 and 2 after UUO or control kidneys showing Coll-GFP+ pericyte detachment and migration from capillaries (arrowheads) by day 1 and pericyte population expansion by day 2, only in mice producing circulating sFc protein. E: Graph of Coll-GFP+ interstitial cell number at early timepoints after UUO. F–H: Immunofluorescence micrographs (F) of kidney interstitial αSMA expression (red) by Coll-GFP+ cells, 4 days after UUO or control (a denotes vascular smooth muscle cells of arterioles (arrowhead) Coll-GFP+ cell without αSMA staining). Quantification of αSMA+ interstitial cells (G) and % of αSMA+, Coll-GFP+ interstitial cells (H). *P < 0.05, ***P < 0.001 versus sFc-treated UUO kidney (n = 6/group).
Blockade of PDGFRβ or VEGFR2 Limits Innate Immune Responses
In recent studies, we identified recruited inflammatory macrophages as a potent leukocyte in immune-mediated fibrosis progression through cellular cross talk.27,29,39,40 Inflammatory macrophages likely play a direct role in pericyte differentiation. Both PDGFRβ and VEGFR2 blockade attenuated recruitment of inflammatory macrophages in the UUO model of fibrosis (Figure 3, A–C), but this was more sustained and greater when VEGFR2 signaling was blocked. Consistent with this, injury-stimulated expression of endothelial transcripts and protein levels for the monocyte adhesion molecules VCAM1 and to a lesser extent ICAM1, was inhibited by the blockade of VEGFR2 signaling (Figure 3, D and E). We calculated that the reduction in VCAM1 level reflected true downregulation of the endothelial cell adhesion molecule, not merely a reflection of VEGFR2-mediated effects on vascular density. This key finding in vivo was supported by findings that VEGF-induced VCAM1 upregulation in cultured HUVECs was inhibited by sVEGFR2-Fc (Supplemental Figure S3, see http://ajp.amjpathol.org). Furthermore, VEGFR2 blockade had the additive effect of inhibiting monocyte chemokines CCL2 (MCP1) and CX3CL1 (fractalkine) (Figure 3F), whereas PDGFRβ blockade less consistently inhibited these key regulators of monocyte recruitment (Figure 3, D–F). Therefore, one of the mechanisms by which VEGFR2 blockade inhibits fibrogenesis may be through inhibition of macrophage recruitment. Similar observations were made in the fibrosing IRI model (Supplemental Figure S4, A–C, see http://ajp.amjpathol.org). Although PDGFRβ may be expressed by some subpopulations of macrophages, we have not observed kidney macrophage expression of PDGFRβ and VEGFR2 expression is restricted to endothelium (see as follows) (Supplemental Figure S5, see http://ajp.amjpathol.org). Nevertheless, because receptor blockade limited inflammatory responses, then to exclude the possibility that VEGFR2 blockade or PDGFRβ blockade acted through inflammatory macrophages rather than through endothelial cells or pericytes, we tested the effect of soluble receptors in macrophage activation (Table 4). As expected the soluble receptors had no effect on M1 type activation of macrophages.29 Neither PDGF-BB nor VEGF-A activated macrophages (not shown).
Figure 3.

Platelet-derived growth factor (PDGFR)β or VEGFR2 blockade attenuates recruitment of inflammatory macrophages. A–C: Immunofluorescence micrographs showing interstitial F4/80+ macrophages in Coll-GFP control kidneys, or after unilateral ureteral obstruction (UUO) in mice with circulating sFc, sPDGFRβ, or sVEGFR2-Fc. Graphs (B, C) quantifying F4/80+ kidney interstitial macrophages. D–F: Western blot of VCAM1 and ICAM1 and GAPDH expression in whole kidney in control (C) or day 4 after UUO (U). E: Q-PCR of GAPDH-normalized whole kidney transcripts VCAM1, ICAM1, and TNFα from control and day 4 after UUO mice. F: Q-PCR of monocyte chemokine and chemokine receptor transcripts CCL2 (MCP1) and CX3CL1 (fractalkine) and receptors CCR2, CX3CR1. *P < 0.05, and ***P < 0.001 versus sFc-treated UUO kidney (n = 6/group).
Table 4.
The Effect of sPDGFRβ and sVEGFR2-Fc on Bone Marrow Derived Macrophage IL1β Transcripts After 24 Hours' Stimulation in Vitro
| Activation conditions | Vehicle | sFc | sPDGFRβ | sVEGFR2-Fc |
|---|---|---|---|---|
| Nil | 81 ± 8 | 36 ± 5 | 61 ± 10 | 39 ± 4 |
| LPS (10 ng/ml) | 414±103 | 247±85 | 485±94 | 261±42 |
Blockade of PDGFRβ or VEGFR2 Prevents Microvascular Rarefaction
We were surprised to find that the UUO model, which is triggered by obstruction of urine flow, causing back pressure in the epithelium of the nephrons, is also characterized by early microvascular leak, indicative of microvascular injury (Supplemental Figure S6A, see http://ajp.amjpathol.org). To understand the significance to the microvasculature of VEGFR2-mediated or PDGFRβ-mediated pericyte migration from the microvasculature, and pericyte differentiation into scar-forming myofibroblasts, we assessed the peritubular capillaries of the kidney after injury. The VEGFR2 or PDGFRβ blockade attenuated microvascular leak (Supplemental Figure S6B, see http://ajp.amjpathol.org). Endothelial cells of the peritubular capillaries entered the cell cycle from day 1 after UUO injury, which peaked at day 4 and then returned to baseline gradually (Figure 4, A and B). Morphometric quantification of the kidney microvasculature showed increased capillary density early after injury onset, reflecting the early angiogenic response to injury. This increased density was detectable from day 2 after injury onset and coincided with pericyte migration from endothelial cells (Figure 4C, and Figure 2D). The angiogenic response persisted through day 7 (Figure 4, A-C), but as the injury progressed, there was significant rarefaction of the microvasculature by day 10 after injury, suggesting that endothelial proliferation may be initially effective but later deleterious. The VEGFR2 blockade completely attenuated the early angiogenic response to injury when quantified on day 2 and day 4 after the injury onset and PDGFRβ blockade partially attenuated this response (Supplemental Figure S6, C and D, see http://ajp.amjpathol.org, and Figure 4, D, E, and G). Conversely, both VEGFR2 blockade and PDGFRβ blockade prevented the capillary rarefaction in the second week of injury (Figure 4, D and F). Thus, VEGFR2 blockade stabilizes the vasculature and although PDGFRβ blockade is partially permissive for angiogenesis, it nevertheless blocks rarefaction. To test whether the blockade of this initial phase of successful angiogenesis (day 2 to day 4) was necessary for the subsequent protection from microvascular rarefaction, UUO injury was performed on kidneys of Coll-GFP mice, but adenoviruses were administered on day 3 resulting in soluble VEGFR2-Fc or sPDGFRβ production in the circulation from day 4 onward. In these studies, although the receptor blockade was not initiated until after peak angiogenesis, blockade of both receptors was nevertheless highly effective in preventing microvascular rarefaction on day 10 of disease (Supplemental Figure S7, A–D, see http://ajp.amjpathol.org). These studies suggest that the early angiogenesis observed reflects stable angiogenesis and is in continuum with the angiogenesis after day 4, where it is dysfunctional, unstable, and drives capillary loss. We tested whether VEGFR2 blockade or PDGFRβ blockade also protected the microvasculature in the fibrosing IRI model, and similar to observations in the UUO model, blockade of both receptors significantly attenuated microvascular rarefaction (Supplemental Figure S4, D and E, see http://ajp.amjpathol.org).
Figure 4.

VEGFR2 blockade or PDGFRβ blockade prevent microvascular rarefaction but by different mechanisms. A, B: Split panel immunfluorescence micrographs (A) and quantification (B) of endothelial cells of peritubular capillaries expressing the pan-cell-cycle marker Ki-67 in kidneys after unilateral ureteral obstruction (UUO). C: Morphometric quantification of the kidney microvasculature showing angiogenic response to injury, followed by rarefaction of the microvasculature. *P < 0.05, **P < 0.01, ***P < 0.001 versus normal kidney on day (d) 0. D–F: Immunofluorescence micrographs showing kidney microvasculature (red) in Coll-GFP control kidneys or after UUO in mice with circulating sFc, sPDGFRβ, or sVEGFR2-Fc. E and F: Graphs showing morphometric quantification of kidney microvasculature. Note on day 4 sVEGFR2-Fc, but not sPDGRFβ, prevents angiogenesis. G: Graph quantifying peritubular endothelial (CD31+) cell proliferation in control kidneys or day 4 after UUO. **P < 0.01, ***P < 0.001 (n = 6/group).
VEGFA Isotype Switching May Contribute to Unstable Angiogenesis
To further dissect the mechanism by which blockade of these receptors could have two distinct beneficial roles for the kidney after injury, expression of the ligands and receptors of the VEGFR2 signaling pathway and PDGFRβ signaling pathways was assessed. The VEGF-A was widely expressed in the normal kidney (Supplementary Figure S8A, see http://ajp.amjpathol.org). It was strongly expressed by podocytes of the glomerulus and tubules of the kidney. However, it was weakly detectable in pericytes of the normal kidney (Supplemental Figure S8A, see http://ajp.amjpathol.org), and in some resident macrophages that lay along the microvasculature (Supplemental Figure S8B, see http://ajp.amjpathol.org). Pericytes form direct communications with endothelial cells at peg and socket and other junctions where direct cell-cell signaling, including VEGF signaling, has been reported to occur.18,19 After injury, there was increased expression of VEGF-A in pericyte-derived myofibroblasts (Supplemental Figure S8C, see http://ajp.amjpathol.org), and in inflammatory macrophages (Supplemental Figure S8D, see http://ajp.amjpathol.org), although total kidney VEGF-A was little affected by injury suggesting a compensatory decline in epithelial VEGF-A (Figure 5A), which was apparent in immunostained kidney sections (Supplemental Figure S8, B and D, see http://ajp.amjpathol.org). Transcripts for all three isoforms of VEGF-A were detected in whole kidney. Whereas injury triggered an increased expression of VEGF-A isoforms VEGF120 and VEGF188, it triggered decreased expression of VEGF164 in the UUO model (Figure 5A), and these observations were mirrored in the unilateral fibrosing IRI model (Supplemental Figure S4, F and G, see http://ajp.amjpathol.org). Furthermore, both PDGFRβ blockade and VEGFR2 blockade inhibited the injury induced increase in VEGF120 and VEGF188 transcripts (Figure 5B), suggesting that these isoforms were being generated in myofibroblasts or macrophages and also indicating that targeting the endothelial receptor alone was sufficient to regulate VEGF transcription in neighboring cells. In purified kidney pericytes VEGF164 was the predominant isoform, but in purified kidney myofibroblasts VEGF 120 and VEGF188 were expressed at higher levels (Supplemental Figure S8, F and I, see http://ajp.amjpathol.org). A similar transcriptional switch, especially the de novo expression of VEGF188, was observed in kidney macrophages compared with circulating monocytes (Supplementary Figure S8, H and I, see http://ajp.amjpathol.org). Moreover, the timing of switch of VEGF164 expression in favor of VEGF120 and VEGF188 (day 4 onward) implied an important role for this switch in disease progression (Figure 5C). As expected, VEGFR2 was expressed almost exclusively in the kidney endothelium (Figure 5D; Supplemental Figure S5A, see http://ajp.amjpathol.org), not in kidney pericytes or myofibroblasts (Figure 5D; Supplemental Figure S5, B and C, see http://ajp.amjpathol.org). Thus, in the healthy kidney, VEGF164 is the abundant isoform that binds to VEGFR2, but during injury, both recruited macrophages and pericyte-derived myofibroblasts switch to synthesis of VEGF120 and VEGF188, isoforms that are known to lead to dysangiogenesis in tumors and developing organs.25,26
Figure 5.

Kidney injury-mediated switch from VEGF164 to VEGF120 and VEGF188 isoforms is prevented by PDGFRβ blockade and VEGFR2 blockade. A: Q-PCR time course for whole kidney VEGFA transcript expression and the VEGFA splice variants VEGF164, VEGF120, and VEGF188, in days after the unilateral ureteral obstruction (UUO) surgery. *P < 0.05, **P < 0.01, ***P < 0.001 versus normal kidney at day (d) 0. B: Graphs of normalized Q-PCR for VEGF isomer transcripts showing day 10 after UUO, both PDGFRβ blockade (black bars) and VEGFR2 blockade (hatched bars) prevented the injury-induced increase in VEGF120 and VEGF188 transcripts compared to control Fc (white bars). **P < 0.01, and ***P < 0.001 versus sFc-treated UUO kidney; *P < 0.5, and ***P < 0.001 versus CON. C: Time course Western blot for VEGFA isoforms in kidneys after UUO. D: Immunofluorescence images of control and day 4 after UUO kidneys showing VEGFR2 (red) is not expressed by Coll-GFP+ cells. E: Immunofluorescence images of control and day 4 after UUO kidneys showing PDGFRβ is expressed exclusively in Coll-GFP+ pericytes/fibroblasts. F: Q-PCR time course of whole kidney for normalized PDGF-B transcripts in kidneys after UUO. *P < 0.05, **P < 0.01, ***P < 0.001 versus normal kidney at day 0 (n = 5/group).
The components of the PDGF-PDGFR system are expressed constitutively or are up-regulated in most renal cells and play important roles in kidney fibrosis.12,13,15 The PDGFRβ was notably expressed in pericytes/myofibroblasts (Figure 5E). The major ligand for PDGFRβ is PDGF-B, which was difficult to detect by immunofluorescence in normal kidney and whose transcript was relatively low (Figure 5F). Kidney injury triggered increased expression of PDGF-B in tubules, endothelial cells, and macrophages (Figure 5F, Supplemental Figure S8, J and K, see http://ajp.amjpathol.org). Together with the inhibitory effect of VEGFR2 blockade on PDGF-B transcript (Figure 1L), these data reveal that targeting the endothelial receptor alone is sufficient to regulate PDGF-B expression in both endothelial cells and neighboring cells, and may be a second mechanism by which expansion of the myofibroblast population is blocked.
Discussion
These studies are the first to show that targeting the kidney endothelial VEGFR2 receptor alone or PDGFRβ alone profoundly inhibits the triad of microvascular rarefaction, interstitial fibrosis, and inflammation in two models of non-glomerular kidney injury. These receptors represent potential novel therapeutic targets for kidney injuries leading to chronic kidney disease. Loss of pericytes from the capillaries of the kidney due to injury-induced detachment, migration, and differentiation into myofibroblasts is a major mechanism targeted by such receptor blockade. Moreover, targeting the endothelium-restricted VEGFR2 can prevent fibrosis, indicating that endothelial determinants regulate pericyte detachment and differentiation.
Increasingly, microvascular rarefaction is recognized as a central component of chronic kidney disease, the final common pathway in the kidney leading to organ failure. Rarefaction is always detected in chronic diseases of the kidney and leads to tubular and interstitial ischemia, which in turn drives activation of parenchymal cells, which recruits inflammatory cells, mainly macrophages, which become activated, hence promoting further vascular loss. The role of microvascular injury has not been previously described well in the UUO model, but the kidney IRI model, also used in these studies, has a well-defined microvascular injury component that is due to acute ischemic injury to the kidney similar to that seen in many forms of human native and transplanted kidney disease.
It is striking that VEGFR2 blockade prevents not only signaling at the VEGFR2, but also at the pericyte PDGFRβ. This observation, together with the findings that nonvascular kidney injury results in early capillary leak, places capillary injury at the initiation of processes that result in chronic kidney disease. It suggests that injured peritubular capillary endothelium becomes susceptible to VEGFA signaling (from pericytes), which triggers both angiogenesis and endothelial signals to pericytes (possibly by endothelial PDGF), which are now susceptible to PDGF-B signaling, resulting in phosphorylation of PDGFRβ on pericytes. This injury-stimulated cellular cross talk is central to the early events after kidney injury.
It is also striking that in the UUO model of progressive injury there is an early angiogenic response, but after day 4 this yields to rarefaction, although endothelial cells are still observed to proliferate at a similar rate. This suggests that while early endothelial proliferation results in increased capillary formation, after day 4, in the UUO model endothelial proliferation is dysfunctional and instead results in endothelial cell loss. One explanation for this is the loss of pericytes from peritubular capillaries.4,10,38,41 In other tissue beds and in developmental angiogenesis, pericytes are necessary for stable sprouting angiogenesis and vascular stabilization. Since by day 4, pericytes have detached from capillaries and migrated into the interstitial space it is likely that this process of pericyte detachment is a key component of the observed capillary loss. Although early injury-stimulated angiogenesis seems central to the disease process, when we blocked VEGFR2 or PDGFRβ from day 4 of the UUO model onward, we were nevertheless able to prevent rarefaction and fibrosis, indicating that the early successful angiogenesis is not crucial to subsequent events, and indicating that pericyte detachment may be reversible.
Although PDGFRβ and VEGFR2 have been reported in cell types other than pericytes/perivascular fibroblasts and endothelium, respectively, our studies have not identified significant other populations of myeloid-derived cells expressing these receptors in these models of kidney disease. Therefore, the effects on myeloid recruitment are secondary to endothelial and pericyte activation rather than direct effects of soluble PDGFRβ or soluble VEGFR2.
When pericytes differentiate into scar-forming myofibroblasts, VEGFA expression is increased less than twofold. However, myofibroblast differentiation results in an isotype switch from VEGF164 to VEGF120 and VEGF188 in the kidney interstitial milieu. Moreover, kidney inflammatory macrophages undergo a similar isotype switch. Although functional testing of each of these isoforms in kidney disease is beyond the scope of these studies, we speculate that isotype switching is central to the dysfunctional angiogenesis that characterizes UUO from day 4 onward and post-IRI kidney injury from day 7 onward. In tumor neovascularization, VEGF120 and the highly extracellular matrix bound VEGF188 are responsible for dysfunctional angiogenesis, which results in impaired tumor growth and vascular instability.25 Therefore, it is likely that VEGFR2 blockade or PDGFRβ blockade inhibit similar deleterious events in the kidney by preventing pericyte detachment and differentiation, and preventing inflammatory macrophage recruitment.
The effect of VEGFR2 blockade in short-term studies in kidney epithelial injury models presented here is contrary to observations of chronic VEGFA deletion in glomerular podocytes,20 in which VEGFA deletion is deleterious, resulting in thrombotic micro-angiopathy and severe kidney disease. The fact that our findings are reproduced in two models of kidney disease suggests to the authors that the peritubular capillary compartment of the kidney is quite different from the glomerular capillary compartment with respect to VEGFA signaling. The glomerulus is known as a vascular bed with high shear stresses and very high turnover of glomerular endothelial cells. It is likely that unlike peritubular capillaries, VEGFA signaling from podocytes to glomerular endothelial cells is active physiologically, and that a chronic angiogenic response is normal. Complete loss of the cross talk with endothelium in the glomerulus in physiological conditions destabilizes the glomerular endothelium. By contrast, the PTCs are nonangiogenic in physiological conditions, and injury-stimulated angiogenesis is at first adaptive but subsequently maladaptive. Moreover, unlike genetic deletion of VEGFA, sVEGFR2-Fc attenuates VEGF signaling, but does not completely ablate it. Clearly, the distinction between VEGFA-VEGFR2 signaling in the glomerulus and PTCs requires further study because it may be that short-term VEGFR2 blockade is beneficial in acute injury, whereas chronic blockade is deleterious.
Although the population of Foxd1 progenitor-derived, Coll-GFP+ and PDGFRβ+ cells in the kidney have been defined here as pericytes, this includes a small number of perivascular fibroblasts, and although this newly-described population of mesenchymal cells is likely to be heterogeneous, these studies highlight the fact that this population of cells as a whole exhibits clear pericyte functionality by having vascular-stabilizing properties.38,41 It is clear that anatomically, some of these pericytes have processes that abut tubular basement membrane in addition to processes with capillary basement membrane42; therefore, they may have been named as fibroblasts in earlier studies.43,44 Further studies will be required to determine whether kidney pericytes/fibroblasts can function as a cell-signaling bridge between the epithelium and endothelium. It is currently unclear whether all of the Foxd1 progenitor-derived, Coll-GFP+ and PDGFRβ+ interstitial cells have processes within the CBM. Currently, there are no molecular markers that distinguish cells that have pericyte projections from those that might not. Further studies are required to define molecular markers that will yield functional signficance to the heterogeneity of this cell population.
Although endothelium to pericyte signaling is necessary for pericyte detachment and differentiation into myofibroblasts, these studies also indicate that PDGFRβ ligation on pericytes, most likely by endothelial PDGF, is necessary for interstitial fibrosis, inflammation, and microvascular rarefaction. These studies place the vital cross talk between endothelial cells and pericytes at the center of injury responses in the kidney, regardless of epithelial events. Furthermore, they add new meaning to fibrosis, because for the first time, targeting fibrogenesis will have far greater impact than simply limiting interstitial matrix deposition.
Acknowledgments
We thank Dr. William Stallcup (Burnham Institute, La Jolla, CA) for anti-PDGFRβ antibody, Dr. David A. Brenner (University of California, San Diego, CA) for valuable input, Dr. Manjeri Venkatachalam (University of Texas Health Science Center at San Antonio, TX) for advice, Colleen Ford (Brigham & Women’s Hospital, Boston, MA) for assistance with electron microscopy, and Wei-Chun Chang, Menghua Tsai, Chen-Chih Chang, and Kuo-Tong Huang (National Taiwan University Hospital, Taipei, Taiwan) for technical assistance for equipment support.
Footnotes
This study was supported by grants from the National Science Council (97-2314-B-002-049 and 99-2628-B-002-013 to S.L.L.; 96-2314-B-002-059 to Y.M.C.), National Taiwan University Hospital (98N1264 to S.L.L.), Ta-Tung Kidney Foundation, Mrs. Hsiu-Chin Lee Kidney Research Foundation, and National Institutes of Health (DK73299, DK84077, DK87389 to J.S.D.; HL074267-01, NS052830-01 to C.J.K.).
Supplemental material for this article can be found on http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.10.012.
Supplemental data
Characterization of pericytes and perivascular fibroblasts in normal adult kidney. A: Confocal four-color images of normal kidney cortex showing pericytes (upper and middle panels) and perivascular fibroblasts (lower panel) and their relationship to endothelium (CD31) and basement membranes (laminin). Arrowheads show examples of areas of intimate connection between either pericyte body or pericyte processes and endothelium. In lower panels, g denotes a glomerulus, and the arrows show fibroblasts (green) surrounding an arteriole. B: Electron microscopy images of cortical peritubular capillaries (PTCs). a and b: Low- and high-power images of a PTC showing lumen (L) and endothelial cell body (E). Note clearly visible capillary basement membrane (CBM) and tubule basement membrane and interstitial space. Pericyte processes (Pp; arrows) are shown within the CBM. The pericyte process in b invaginates the endothelium, indicative of a “peg and socket” type junction (arrowhead). c: Image of PTC showing a pericyte body (P) within the CBM. d and e: Low- and high-power images of a PTC showing endothelial cell and three pericyte processes within the CBM. f: PTC lumen showing endothelial cell body and a pericyte process along the capillary. g–i: Low-power (g) and high-power (h and i) images of a PTC showing three pericyte processes on one side of the capillary and a single process (i) on the other side. Note this process overlies a junction between two endothelial cells. j–l: Low-power (j) and high-power (k and l) images of a PTC showing lumen and a long Pp within the CBM. Note that there are numerous points of intimate connection between the endothelial cells and Pp. An invagination of the Pp with a process from the endothelial cell can be seen (arrowhead).
Selective inhibition of PDGFRβ and VEGFR2 signaling by soluble circulating receptors attenuates kidney fibrosis after unilateral ischemia–reperfusion injury (IRI). (A) Time course of circulating adenovirus-mediated sPDGFRβ and sVEGFR2-Fc production by Western blot analysis of 1 μl of plasma following i.v. administration of adenoviruses (1 × 109 plaque-forming units) to adult C57BL/6 mice 3 days before unilateral ischemia–reperfusion injury surgery. B and C: Images of picrosirius red–stained kidney sections for interstitial fibrillar collagens (red) in control (CON) or d15 after IRI (B), and morphometric quantification of fibrillar collagen from whole sagittal kidney sections (C). D and E: Immunohistochemistry of kidney sections CON or d15 after IRI for α-smooth muscle actin (αSMA) (brown) (D), and morphometric quantification of αSMA from whole sagittal kidney sections (E). F: Quantitative PCR (Q-PCR) of transcripts of αSMA, type I collagen (α1) [Col1(α1)], and type 3 collagen (α1) [Col3(α1)] in kidneys of CON or d15 after IRI in mice producing systemic sPDGFRβ, sVEGFR2-Fc, or sFc. G: Q-PCR of transcripts of PDGF-B, TGFβ1, FGF2, and BMP7 in kidneys of CON or d15 after IRI in mice producing systemic sPDGFRβ, sVEGFR2-Fc, or sFc. Scale bar = 25 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 6/group).
Purified sVEGFR2-Fc down-regulates expression of VCAM1 and CX3CL1 in VEGF-stimulated human umbilical venous endothelial cells (HUVEC). HUVECs were treated with VEGF (10 ng/ml) for 6 hours in the presence or absence of affinity-purified sVEGFR2-Fc (5 µg/ml). A: VCAM1, CX3CL1, and GAPDH were analyzed by Western blot analysis. B: Western blot band density normalized to GAPDH. Three independent experiments were performed.
PDGFRβ and VEGFR2 blockade attenuates macrophage infiltration and microvascular rarefaction after ischemia–reperfusion injury (IRI). A and B: Immunofluorescence micrographs showing interstitial F4/80+ macrophages (red) in kidneys CON or d15 after IRI in mice with circulating sFc, sPDGFRβ, or sVEGFR2-Fc (A). Graphs (B) quantifying interstitial F4/80+ macrophages. C: Q-PCR of monocyte chemokine transcripts CCL2 (MCP1) and CX3CL1 (fractalkine) and receptor transcripts CCR2, CX3CR1 in kidneys of CON or d15 after IRI. *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 6/group). D: Immunofluorescence micrographs showing kidney microvasculature (red) in kidneys of CON or d15 after IRI in mice with circulating sFc, sPDGFRβ, or sVEGFR2-Fc. E: Morphometric quantification of peritubular capillary loss (PTC loss). ***P < 0.001 (n = 6/group). F: Quantitative PCR time course for normalized whole-kidney VEGFA transcript expression and the VEGFA splice variants VEGF164, -120, and -188, in days following IRI surgery. *P < 0.05, **P < 0.01, ***P < 0.001 versus normal kidney at day 0 (n = 3/group). G: Time-course Western blot for VEGFA isoforms in kidneys after IRI. Representative blot showed two mice in each time point.
Characterization of VEGFR2 expression in different cellular compartments of the kidney. A: Split-panel immunofluorescence images of control and kidneys 4 days after unilateral ureteral obstruction (UUO) showing VEGFR2 (red) exclusively expressed by CD31+ (green) peritubular capillary endothelial cells. B: Fluorescence-activated cell sorting (FACS) analysis showing cell surface VEGFR2 expression (white) in human umbilical venous endothelial cells (HUVEC) and UUO kidney myofibroblasts compared with isotype control (black). Note no expression of the receptor in myofibroblasts. C: RT-PCR detecting very weak expression of VEGFR2 (310 bp) in UUO kidney myofibroblasts.
Microvascular leak in ureteral obstruction model and VEGFR2-mediated attenuation of the early angiogenic response to unilateral ureteral obstruction (UUO) injury. A: Vascular permeability as detected by retention of Evans Blue (albumin-bound) dye in kidney 1 hour after injection i.v. B: Comparison of Evans Blue dye retention in d4 UUO kidney in mice with circulating receptors. (n = 4/group ***P < 0.001). C: Split-panel immunofluorescence images of kidneys d2 after UUO. Arrows indicate CD31+ endothelial cells that are also positive for pan-cell cycle marker Ki67. Note high blue fluorescent background in epithelial cells. D: Graph showing quantification of CD31+ endothelial cells coexpressing Ki67 in mice treated with different circulating soluble receptors. ***P < 0.001 versus sFc-treated mice.
Selective inhibition of PDGFRβ or VEGFR2 signaling by soluble circulating receptors following the initial angiogenic phase attenuates fibrosis and capillary rarefaction after unilateral ureteral obstruction (UUO). A: Circulating adenovirus-mediated sPDGFRβ and sVEGFR2-Fc production by Western blot analysis of 1 μl of plasma at days 3 and 10 after UUO surgery. Adenoviruses (1 × 109 plaque-forming units) were injected into adult Coll-GFP mice via the tail vein after plasma sampling at day 3 following UUO surgery. Representative images were from three mice in each group. B and C: Images of picrosirius red–stained kidney sections for interstitial fibrillar collagens (red) in control (CON) or d10 after UUO (B), and morphometric quantification of fibrillar collagen from whole sagittal kidney sections (C). Scale bar = 25 μm. ***P < 0.001 (n = 6/group). D: Immunofluorescence micrographs showing kidney microvasculature (red) and pericytes/myofibroblasts (green) in kidneys of d10 after UUO surgery in mice with adenoviruses administration 3 days after UUO surgery. Scale bar = 25 μm. E and F: Morphometric quantification of peritubular capillary loss (PTC loss) (E) and pericytes/fibroblasts (F). ***P < 0.001 (n = 6/group).
Characterization of VEGF and PDGF expression in different cellular compartments of the kidney. A and B: Split-panel immunofluorescence images of VEGF-A in the normal kidney of Coll-GFP mice (A) or wild-type (WT) mice (B). Note VEGF-A was strongly expressed in glomerular podocytes (*) and renal tubules, and weakly detectable in pericytes (arrowhead) and in some resident macrophages (arrow) that lay along the microvasculature. C and D: Split-panel immunofluorescence images of VEGF-A in the kidney d7 after unilateral ureteral obstruction (UUO) of Coll-GFP mice (C) or WT mice (D). In d7 UUO kidneys, there was increased expression of VEGF-A in myofibroblasts (C) and macrophages (D). Note decreased expression in tubular epithelia (indicated by TE). (E, F, and I) Semiquantitative RT-PCR for total VEGF-A (E) or VEGF-A isoforms VEGF120, VEGF 164, and VEGF188 (F), in purified pericytes from normal kidney and purified fibroblasts from d7 UUO kidney. Collagen I (α1) (Coll1a1) and F4/80 transcripts were amplified to assess purity. In purified pericytes, VEGF164 was the predominant isoform, but in cultured myofibroblasts, VEGF 120 and VEGF188 were expressed at higher levels, as assessed by quantitative PCR (Q-PCR) (I). G–I: Semiquantitative RT-PCR for total VEGF-A (G) or VEGF-A isoforms VEGF120, VEGF 164, and VEGF188 (H), in purified peripheral blood monocytes (PBMo) or macrophages from kidney d7 after UUO (UUOMϕ). F4/80, Coll1a1, and Kim1 were amplified to assess purity. Q-PCR for total VEGF-A, VEGF120, VEGF164, and VEGF188 transcripts normalized by GAPDH (I). J and K: Split-panel immunofluorescence images of kidney d7 after UUO, showing expression of PDGF-B in tubules (indicated by TE), F4/80+ Mϕ (J), and CD31+ endothelial cells (K) (arrowheads).
References
- 1.Lin S.L., Kisseleva T., Brenner D.A., Duffield J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humphreys B.D., Lin S.L., Kobayashi A., Hudson T.E., Nowlin B.T., Bonventre J.V., Valerius M.T., McMahon A.P., Duffield J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:22–24. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao S., Lobov I.B., Vallance J.E., Tsujikawa K., Shiojima I., Akunuru S., Walsh K., Benjamin L.E., Lang R.A. Obligatory participation of macrophages in an angiopoietin 2-mediated cell death switch. Development. 2007;134:4449–4458. doi: 10.1242/dev.012187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl P., Johansson B.R., Leveen P., Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 5.Eardley K.S., Kubal C., Zehnder D., Quinkler M., Lepenies J., Savage C.O., Howie A.J., Kaur K., Cooper M.S., Adu D., Cockwell P. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008;74:495–504. doi: 10.1038/ki.2008.183. [DOI] [PubMed] [Google Scholar]
- 6.Mallamaci F., Benedetto F.A., Tripepi G., Cutrupi S., Pizzini P., Stancanelli B., Seminara G., Bonanno G., Rapisarda F., Fatuzzo P., Malatino L.S., Zoccali C. Vascular endothelial growth factor, left ventricular dysfunction and mortality in hemodialysis patients. J Hypertens. 2008;26:1875–1882. doi: 10.1097/HJH.0b013e328307c3d2. [DOI] [PubMed] [Google Scholar]
- 7.Kim I., Moon S.O., Kim S.H., Kim H.J., Koh Y.S., Koh G.Y. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276:7614–7620. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi R., Shimizu A., Masuda Y., Kitamura H., Ishizaki M., Sugisaki Y., Yamanaka N. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol. 2002;13:1795–1805. doi: 10.1097/01.asn.0000018408.51388.57. [DOI] [PubMed] [Google Scholar]
- 9.Kuhnert F., Tam B.Y., Sennino B., Gray J.T., Yuan J., Jocson A., Nayak N.R., Mulligan R.C., McDonald D.M., Kuo C.J. Soluble receptor-mediated selective inhibition of VEGFR and PDGFRbeta signaling during physiologic and tumor angiogenesis. Proc Natl Acad Sci USA. 2008;105:10185–10190. doi: 10.1073/pnas.0803194105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindblom P., Gerhardt H., Liebner S., Abramsson A., Enge M., Hellstrom M., Backstrom G., Fredriksson S., Landegren U., Nystrom H.C., Bergstrom G., Dejana E., Ostman A., Lindahl P., Betsholtz C. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Cao R., Zhang Y., Jia T., Cao Y., Wahlberg E. Differential roles of PDGFR-alpha and PDGFR-beta in angiogenesis and vessel stability. FASEB J. 2009;23:153–163. doi: 10.1096/fj.08-113860. [DOI] [PubMed] [Google Scholar]
- 12.Eitner F., Bucher E., van Roeyen C., Kunter U., Rong S., Seikrit C., Villa L., Boor P., Fredriksson L., Backstrom G., Eriksson U., Ostman A., Floege J., Ostendorf T. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol. 2008;19:281–289. doi: 10.1681/ASN.2007030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Floege J., Eitner F., Alpers C.E. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23. doi: 10.1681/ASN.2007050532. [DOI] [PubMed] [Google Scholar]
- 14.Rajkumar V.S., Shiwen X., Bostrom M., Leoni P., Muddle J., Ivarsson M., Gerdin B., Denton C.P., Bou-Gharios G., Black C.M., Abraham D.J. Platelet-derived growth factor-beta receptor activation is essential for fibroblast and pericyte recruitment during cutaneous wound healing. Am J Pathol. 2006;169:2254–2265. doi: 10.2353/ajpath.2006.060196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang W.W., Ulich T.R., Lacey D.L., Hill D.C., Qi M., Kaufman S.A., Van G.Y., Tarpley J.E., Yee J.S. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am J Pathol. 1996;148:1169–1180. [PMC free article] [PubMed] [Google Scholar]
- 16.Au P., Tam J., Duda D.G., Lin P.C., Munn L.L., Fukumura D., Jain R.K. Paradoxical effects of PDGF-BB overexpression in endothelial cells on engineered blood vessels in vivo. Am J Pathol. 2009;175:294–302. doi: 10.2353/ajpath.2009.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eremina V., Sood M., Haigh J., Nagy A., Lajoie G., Ferrara N., Gerber H.P., Kikkawa Y., Miner J.H., Quaggin S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darland D.C., Massingham L.J., Smith S.R., Piek E., Saint-Geniez M., D'Amore P.A. Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol. 2003;264:275–288. doi: 10.1016/j.ydbio.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Evensen L., Micklem D.R., Blois A., Berge S.V., Aarsaether N., Littlewood-Evans A., Wood J., Lorens J.B. Mural cell associated VEGF is required for organotypic vessel formation. PLoS One. 2009;4:e5798. doi: 10.1371/journal.pone.0005798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eremina V., Jefferson J.A., Kowalewska J., Hochster H., Haas M., Weisstuch J., Richardson C., Kopp J.B., Kabir M.G., Backx P.H., Gerber H.P., Ferrara N., Barisoni L., Alpers C.E., Quaggin S.E. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Vriese A.S., Stoenoiu M.S., Elger M., Devuyst O., Vanholder R., Kriz W., Lameire N.H. Diabetes-induced microvascular dysfunction in the hydronephrotic kidney: role of nitric oxide. Kidney Int. 2001;60:202–210. doi: 10.1046/j.1523-1755.2001.00787.x. [DOI] [PubMed] [Google Scholar]
- 22.Flyvbjerg A., Dagnaes-Hansen F., De Vriese A.S., Schrijvers B.F., Tilton R.G., Rasch R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51:3090–3094. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- 23.Perrin R.M., Konopatskaya O., Qiu Y., Harper S., Bates D.O., Churchill A.J. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia. 2005;48:2422–2427. doi: 10.1007/s00125-005-1951-8. [DOI] [PubMed] [Google Scholar]
- 24.Simon M., Rockl W., Hornig C., Grone E.F., Theis H., Weich H.A., Fuchs E., Yayon A., Grone H.J. Receptors of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) in fetal and adult human kidney: localization and [125I]VEGF binding sites. J Am Soc Nephrol. 1998;9:1032–1044. doi: 10.1681/ASN.V961032. [DOI] [PubMed] [Google Scholar]
- 25.Grunstein J., Masbad J.J., Hickey R., Giordano F., Johnson R.S. Isoforms of vascular endothelial growth factor act in a coordinate fashion To recruit and expand tumor vasculature. Mol Cell Biol. 2000;20:7282–7291. doi: 10.1128/mcb.20.19.7282-7291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stalmans I., Ng Y.S., Rohan R., Fruttiger M., Bouche A., Yuce A., Fujisawa H., Hermans B., Shani M., Jansen S., Hicklin D., Anderson D.J., Gardiner T., Hammes H.P., Moons L., Dewerchin M., Collen D., Carmeliet P., D'Amore P.A. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–336. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castano A.P., Lin S.L., Surowy T., Nowlin B.T., Turlapati S.A., Patel T., Singh A., Li S., Lupher M.L., Jr., Duffield J.S. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009;1:5ra13. doi: 10.1126/scitranslmed.3000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thurner B., Haendle I., Roder C., Dieckmann D., Keikavoussi P., Jonuleit H., Bender A., Maczek C., Schreiner D., von den Driesch P., Brocker E.B., Steinman R.M., Enk A., Kampgen E., Schuler G. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190:1669–1678. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin S.L., Castano A.P., Nowlin B.T., Lupher M.L., Jr., Duffield J.S. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol. 2009;183:6733–6743. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- 30.Lin S.L., Li B., Rao S., Hudson T.E., Nowlin B.T., Yao H.P., Zheng J.F., Carroll T.J., McMahon A.P., Lang R.A., Duffield J.S. Macrophage Wnt7b is critical in kidney repair and regeneration. Proc Natl Acad Sci U S A. 2010;107:4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X.Y., La Russa V.F., Bao L., Kolls J., Schwarzenberger P., Reiser J. Lentiviral vectors for sustained transgene expression in human bone marrow-derived stromal cells. Mol Ther. 2002;5:555–565. doi: 10.1006/mthe.2002.0585. [DOI] [PubMed] [Google Scholar]
- 32.Ai M., Heeger S., Bartels C.F., Schelling D.K. Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Genet. 2005;77:741–753. doi: 10.1086/497706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhodin J.A. Ultrastructure of mammalian venous capillaries, venules, and small collecting veins. J Ultrastruct Res. 1968;25:452–500. doi: 10.1016/s0022-5320(68)80098-x. [DOI] [PubMed] [Google Scholar]
- 34.Bruns R.R., Palade G.E. Studies on blood capillaries: I General organization of blood capillaries in muscle. J Cell Biol. 1968;37:244–276. doi: 10.1083/jcb.37.2.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rouget C. Memoire sur le developpement, la structure et les proprietes physiologiques des capillaries sanguins et lymphatiques. Arch Physiol Norm Pathol. 1873;5:603–663. [Google Scholar]
- 36.Zimmerman K. Der feinere bau der blutcapillaren. Z Anat Entwicklungsgeschichte. 1923;68:29–36. [Google Scholar]
- 37.Courtoy P.J., Boyles J. Fibronectin in the microvasculature: localization in the pericyte-endothelial interstitium. J Ultrastruct Res. 1983;83:258–273. doi: 10.1016/s0022-5320(83)90133-8. [DOI] [PubMed] [Google Scholar]
- 38.Armulik A., Abramsson A., Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 39.Duffield J.S., Forbes S.J., Constandinou C.M., Clay S., Partolina M., Vuthoori S., Wu S., Lang R., Iredale J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duffield J.S., Tipping P.G., Kipari T., Cailhier J.F., Clay S., Lang R., Bonventre J.V., Hughes J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saunders W.B., Bohnsack B.L., Faske J.B., Anthis N.J., Bayless K.J., Hirschi K.K., Davis G.E. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J Cell Biol. 2006;175:179–191. doi: 10.1083/jcb.200603176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duffield J.S., Humphreys B.D. Origin of new cells in the adult kidney; results from genetic labeling techniques. Kidney Int. 2010 doi: 10.1038/ki.2010.338. in press. [DOI] [PubMed] [Google Scholar]
- 43.Kaissling B., Hegyi I., Loffing J., Le Hir M. Morphology of interstitial cells in the healthy kidney. Anat Embryol (Berl) 1996;193:303–318. doi: 10.1007/BF00186688. [DOI] [PubMed] [Google Scholar]
- 44.Picard N., Baum O., Vogetseder A., Kaissling B., Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol. 2008;130:141–155. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of pericytes and perivascular fibroblasts in normal adult kidney. A: Confocal four-color images of normal kidney cortex showing pericytes (upper and middle panels) and perivascular fibroblasts (lower panel) and their relationship to endothelium (CD31) and basement membranes (laminin). Arrowheads show examples of areas of intimate connection between either pericyte body or pericyte processes and endothelium. In lower panels, g denotes a glomerulus, and the arrows show fibroblasts (green) surrounding an arteriole. B: Electron microscopy images of cortical peritubular capillaries (PTCs). a and b: Low- and high-power images of a PTC showing lumen (L) and endothelial cell body (E). Note clearly visible capillary basement membrane (CBM) and tubule basement membrane and interstitial space. Pericyte processes (Pp; arrows) are shown within the CBM. The pericyte process in b invaginates the endothelium, indicative of a “peg and socket” type junction (arrowhead). c: Image of PTC showing a pericyte body (P) within the CBM. d and e: Low- and high-power images of a PTC showing endothelial cell and three pericyte processes within the CBM. f: PTC lumen showing endothelial cell body and a pericyte process along the capillary. g–i: Low-power (g) and high-power (h and i) images of a PTC showing three pericyte processes on one side of the capillary and a single process (i) on the other side. Note this process overlies a junction between two endothelial cells. j–l: Low-power (j) and high-power (k and l) images of a PTC showing lumen and a long Pp within the CBM. Note that there are numerous points of intimate connection between the endothelial cells and Pp. An invagination of the Pp with a process from the endothelial cell can be seen (arrowhead).
Selective inhibition of PDGFRβ and VEGFR2 signaling by soluble circulating receptors attenuates kidney fibrosis after unilateral ischemia–reperfusion injury (IRI). (A) Time course of circulating adenovirus-mediated sPDGFRβ and sVEGFR2-Fc production by Western blot analysis of 1 μl of plasma following i.v. administration of adenoviruses (1 × 109 plaque-forming units) to adult C57BL/6 mice 3 days before unilateral ischemia–reperfusion injury surgery. B and C: Images of picrosirius red–stained kidney sections for interstitial fibrillar collagens (red) in control (CON) or d15 after IRI (B), and morphometric quantification of fibrillar collagen from whole sagittal kidney sections (C). D and E: Immunohistochemistry of kidney sections CON or d15 after IRI for α-smooth muscle actin (αSMA) (brown) (D), and morphometric quantification of αSMA from whole sagittal kidney sections (E). F: Quantitative PCR (Q-PCR) of transcripts of αSMA, type I collagen (α1) [Col1(α1)], and type 3 collagen (α1) [Col3(α1)] in kidneys of CON or d15 after IRI in mice producing systemic sPDGFRβ, sVEGFR2-Fc, or sFc. G: Q-PCR of transcripts of PDGF-B, TGFβ1, FGF2, and BMP7 in kidneys of CON or d15 after IRI in mice producing systemic sPDGFRβ, sVEGFR2-Fc, or sFc. Scale bar = 25 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 6/group).
Purified sVEGFR2-Fc down-regulates expression of VCAM1 and CX3CL1 in VEGF-stimulated human umbilical venous endothelial cells (HUVEC). HUVECs were treated with VEGF (10 ng/ml) for 6 hours in the presence or absence of affinity-purified sVEGFR2-Fc (5 µg/ml). A: VCAM1, CX3CL1, and GAPDH were analyzed by Western blot analysis. B: Western blot band density normalized to GAPDH. Three independent experiments were performed.
PDGFRβ and VEGFR2 blockade attenuates macrophage infiltration and microvascular rarefaction after ischemia–reperfusion injury (IRI). A and B: Immunofluorescence micrographs showing interstitial F4/80+ macrophages (red) in kidneys CON or d15 after IRI in mice with circulating sFc, sPDGFRβ, or sVEGFR2-Fc (A). Graphs (B) quantifying interstitial F4/80+ macrophages. C: Q-PCR of monocyte chemokine transcripts CCL2 (MCP1) and CX3CL1 (fractalkine) and receptor transcripts CCR2, CX3CR1 in kidneys of CON or d15 after IRI. *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 6/group). D: Immunofluorescence micrographs showing kidney microvasculature (red) in kidneys of CON or d15 after IRI in mice with circulating sFc, sPDGFRβ, or sVEGFR2-Fc. E: Morphometric quantification of peritubular capillary loss (PTC loss). ***P < 0.001 (n = 6/group). F: Quantitative PCR time course for normalized whole-kidney VEGFA transcript expression and the VEGFA splice variants VEGF164, -120, and -188, in days following IRI surgery. *P < 0.05, **P < 0.01, ***P < 0.001 versus normal kidney at day 0 (n = 3/group). G: Time-course Western blot for VEGFA isoforms in kidneys after IRI. Representative blot showed two mice in each time point.
Characterization of VEGFR2 expression in different cellular compartments of the kidney. A: Split-panel immunofluorescence images of control and kidneys 4 days after unilateral ureteral obstruction (UUO) showing VEGFR2 (red) exclusively expressed by CD31+ (green) peritubular capillary endothelial cells. B: Fluorescence-activated cell sorting (FACS) analysis showing cell surface VEGFR2 expression (white) in human umbilical venous endothelial cells (HUVEC) and UUO kidney myofibroblasts compared with isotype control (black). Note no expression of the receptor in myofibroblasts. C: RT-PCR detecting very weak expression of VEGFR2 (310 bp) in UUO kidney myofibroblasts.
Microvascular leak in ureteral obstruction model and VEGFR2-mediated attenuation of the early angiogenic response to unilateral ureteral obstruction (UUO) injury. A: Vascular permeability as detected by retention of Evans Blue (albumin-bound) dye in kidney 1 hour after injection i.v. B: Comparison of Evans Blue dye retention in d4 UUO kidney in mice with circulating receptors. (n = 4/group ***P < 0.001). C: Split-panel immunofluorescence images of kidneys d2 after UUO. Arrows indicate CD31+ endothelial cells that are also positive for pan-cell cycle marker Ki67. Note high blue fluorescent background in epithelial cells. D: Graph showing quantification of CD31+ endothelial cells coexpressing Ki67 in mice treated with different circulating soluble receptors. ***P < 0.001 versus sFc-treated mice.
Selective inhibition of PDGFRβ or VEGFR2 signaling by soluble circulating receptors following the initial angiogenic phase attenuates fibrosis and capillary rarefaction after unilateral ureteral obstruction (UUO). A: Circulating adenovirus-mediated sPDGFRβ and sVEGFR2-Fc production by Western blot analysis of 1 μl of plasma at days 3 and 10 after UUO surgery. Adenoviruses (1 × 109 plaque-forming units) were injected into adult Coll-GFP mice via the tail vein after plasma sampling at day 3 following UUO surgery. Representative images were from three mice in each group. B and C: Images of picrosirius red–stained kidney sections for interstitial fibrillar collagens (red) in control (CON) or d10 after UUO (B), and morphometric quantification of fibrillar collagen from whole sagittal kidney sections (C). Scale bar = 25 μm. ***P < 0.001 (n = 6/group). D: Immunofluorescence micrographs showing kidney microvasculature (red) and pericytes/myofibroblasts (green) in kidneys of d10 after UUO surgery in mice with adenoviruses administration 3 days after UUO surgery. Scale bar = 25 μm. E and F: Morphometric quantification of peritubular capillary loss (PTC loss) (E) and pericytes/fibroblasts (F). ***P < 0.001 (n = 6/group).
Characterization of VEGF and PDGF expression in different cellular compartments of the kidney. A and B: Split-panel immunofluorescence images of VEGF-A in the normal kidney of Coll-GFP mice (A) or wild-type (WT) mice (B). Note VEGF-A was strongly expressed in glomerular podocytes (*) and renal tubules, and weakly detectable in pericytes (arrowhead) and in some resident macrophages (arrow) that lay along the microvasculature. C and D: Split-panel immunofluorescence images of VEGF-A in the kidney d7 after unilateral ureteral obstruction (UUO) of Coll-GFP mice (C) or WT mice (D). In d7 UUO kidneys, there was increased expression of VEGF-A in myofibroblasts (C) and macrophages (D). Note decreased expression in tubular epithelia (indicated by TE). (E, F, and I) Semiquantitative RT-PCR for total VEGF-A (E) or VEGF-A isoforms VEGF120, VEGF 164, and VEGF188 (F), in purified pericytes from normal kidney and purified fibroblasts from d7 UUO kidney. Collagen I (α1) (Coll1a1) and F4/80 transcripts were amplified to assess purity. In purified pericytes, VEGF164 was the predominant isoform, but in cultured myofibroblasts, VEGF 120 and VEGF188 were expressed at higher levels, as assessed by quantitative PCR (Q-PCR) (I). G–I: Semiquantitative RT-PCR for total VEGF-A (G) or VEGF-A isoforms VEGF120, VEGF 164, and VEGF188 (H), in purified peripheral blood monocytes (PBMo) or macrophages from kidney d7 after UUO (UUOMϕ). F4/80, Coll1a1, and Kim1 were amplified to assess purity. Q-PCR for total VEGF-A, VEGF120, VEGF164, and VEGF188 transcripts normalized by GAPDH (I). J and K: Split-panel immunofluorescence images of kidney d7 after UUO, showing expression of PDGF-B in tubules (indicated by TE), F4/80+ Mϕ (J), and CD31+ endothelial cells (K) (arrowheads).