

Abstract
Kawasaki disease (KD) is an acute inflammatory illness marked by coronary arteritis. However, the factors increasing susceptibility to coronary artery lesions are unknown. Because transforming growth factor (TGF) β increases elastin synthesis and suppresses proteolysis, we hypothesized that, in contrast to the benefit observed in aneurysms forming in those with Marfan syndrome, inhibition of TGF-β would worsen inflammatory-induced coronary artery lesions. By using a murine model of KD in which injection of Lactobacillus casei wall extract (LCWE) induces coronary arteritis, we show that LCWE increased TGF-β signaling in the coronary smooth muscle cells beginning at 2 days and continuing through 14 days, the point of peak coronary inflammation. By 42 days, LCWE caused fragmentation of the internal and external elastic lamina. Blocking TGF-β by administration of a neutralizing antibody accentuated the LCWE-mediated fragmentation of elastin and induced an overall loss of medial elastin without increasing the inflammatory response. We attributed these increased pathological characteristics to a reduction in the proteolytic inhibitor, plasminogen activator inhibitor-1, and an associated threefold increase in matrix metalloproteinase 9 activity compared with LCWE alone. Therefore, our data demonstrate that in the coronary arteritis associated with KD, TGF-β suppresses elastin degradation by inhibiting plasmin-mediated matrix metalloproteinase 9 activation. Thus, strategies to block TGF-β, used in those with Marfan syndrome, are unlikely to be beneficial and could be detrimental.
Kawasaki disease (KD) is an acute inflammatory disease marked by vasculitis of small and medium arteries, most commonly affecting the coronary arteries. Kawasaki disease remains a leading cause of acquired heart disease in children,1,2 with approximately 25% of untreated and 5% of treated patients developing coronary artery lesions (CALs), including aneurysms. Although episodic outbreaks suggest an infectious cause, a specific pathogen has not been identified.3 Genetic factors appear to influence disease susceptibility and severity, with an increased incidence in patients of Japanese heritage and in children whose relatives have a history of4; in addition, there is a greater risk of giant coronary artery aneurysms in patients with recurrent KD.5 Therefore, it is likely that an inflammatory stimulus in combination with specific host factors is required for CAL development.
In Marfan syndrome (MS), aortic aneurysms form as a result of a genetic defect in fibrillin-1, a microfibrillar protein important in elastin assembly. Fibrillin-1 binds the cytokine transforming growth factor (TGF) β in an inactive form. Deficiency of fibrillin-1 leads to inappropriate activation of TGF-β and appears to be critical to the development of aneurysms observed in MS. However, the mechanisms responsible for this pathological effect remain unknown. In the murine model of MS, TGF-β blockade with either a neutralizing antibody (nAb) or the angiotensin-II receptor antagonist, losartan, effectively prevented aortic dilation and induced regression of established disease.6 Furthermore, in a clinical study7 in pediatric patients with MS, losartan therapy was effective in decreasing the rate of aortic root dilation. As a result of these studies, there has been speculation that increased TGF-β activation is related to non-MS aneurysm formation, leading to interest in extrapolating the use of TGF-β blockade to treat conditions such as abdominal aortic aneurysm and KD.8
However, this therapeutic strategy may be detrimental in aneurysms that form in response to inflammatory stimuli, such as KD, considering the anti-inflammatory properties of TGF-β (ie, inhibition of T-cell proliferation and inflammatory cytokine expression).9,10 Moreover, TGF-β induces genes important in elastin fiber assembly (eg, tropoelastin)11–13 and limits elastin degradation by increasing tissue inhibitors of matrix metalloproteinases (TIMPs) and plasminogen activator inhibitor (PAI) 1.14,15 In fact, in a xenograft model of aortic aneurysm marked by inflammation and proteolysis, transfection of TGF-β limited aortic dilation by preserving medial elastin and decreasing matrix metalloproteinase (MMP) 2 and MMP-9 activity.16 Therefore, better defining the role of TGF-β in diseases marked by inflammation and aneurysm formation will have major therapeutic implications. The role of TGF-β in the pathogenesis of aneurysms occurring in young patients is particularly unclear, given that our knowledge is almost exclusively derived from animal models of aneurysm formed by caustic chemical infusions17,18 or that develop in association with atherosclerosis.19
To address these issues, we used a murine model of KD, in which the injection of Lactobacillus casei wall extract (LCWE) induces coronary arteritis.20–22 Because KD is almost exclusively a pediatric illness, we treated mice in the first few weeks of life with LCWE and determined the effect of blocking TGF-β during this process. We hypothesized that, in contrast to the effect in MS, the induction of TGF-β in response to LCWE would be protective, stimulating new tropoelastin synthesis and decreasing proteolytic activity. Consistent with this hypothesis, we show that TGF-β blockade worsens elastin degradation and decreases levels of medial elastin, without affecting the expression of tropoelastin or increasing the degree of coronary arteritis. We attribute this protective effect to the ability of TGF-β to preserve the levels of the plasmin inhibitor, PAI-1, and thereby to suppress activation of the elastolytic protease, MMP-9. Interestingly, LCWE induces similar pathological characteristics in adult mice that are also aggravated by TGF-β inhibition, suggesting that, in addition to KD, these results may be generalizable to forms of inflammatory aneurysms affecting older patients.
Materials and Methods
Murine Model of KD
Coronary arteritis was induced in neonatal (7-day) C57/BL6/129S mice by injection of 20 mg/kg (rhamnose equivalent) of LCWE i.p. or an equal volume of vehicle (PBS), as used previously in an accepted animal model of KD.21,22 Mice were sacrificed at 2 and 14 days, the hearts were perfused with PBS, and then either prepared for histological analysis or snap frozen for RNA and protein extraction. At 42 days, mice were anesthetized and sacrificed by exsanguination and the coronary circulation was perfused under physiological perfusion pressure by injecting barium gelatin through the left ventricle after ligating the ascending aorta. The heart and aortic root were then fixed en bloc in formalin for assessment of coronary artery morphological features and elastin integrity. All surgical and animal care procedures and experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Stanford University, Stanford, California.
Elastin Staining and Determination of Elastin Content
The integrity of the coronary elastin was visualized by light microscopy after Hart's elastin staining.23 Cross sections of the large coronary arteries under ×40 magnification were evaluated in a blinded fashion, and the number of visible breaks was measured per 1000-μm length of vessel wall. The amount of medial elastin was quantified by determining the total amount of stained elastin versus total medial area using a commercially available system (Bioquant True Color Windows Image Analysis system; R & M Biometrics, Nashville, TN) in cross sections of the large and medium coronary branches in a blinded fashion.
Masson Trichrome Staining
To assess collagen and the nonelastin matrix, formalin-fixed sections of the large coronary arteries were soaked in Bouin's solution overnight and then stained using a kit (Accustain Trichrome Kit; Sigma, St Louis, MO). Representative images of the large coronary arteries were obtained under ×40 magnification. The total percentage matrix area versus the total cross-sectional area of the vessel was determined in a blinded fashion using Metamorph Image Analysis software (Leeds Precision Instruments, Minneapolis, MN).
Immunostaining for Phosphorylated Smad2/3
Immunohistochemical staining was performed on FFPE sections after antigen retrieval by heating sections in citrate buffer at 95°C for 10 minutes. Sections were then incubated with anti–phosphorylated mothers against decapentaplegic homolog 2/3 (pSmad2/3) antibody (1:200; Cell Signaling, Danvers, MA) overnight at 4°C, followed by detection with a commercially available system (Vectastain ABC system; Vector Laboratories, Burlingame, CA), and counterstained with hematoxylin (Sigma Aldrich, Mountain View, CA). Images were quantified by determining the percentage of brown- versus total-stained nuclei in a blinded fashion with computer software (MetaMorph Imaging).
Inhibition of TGF-β Signaling
Additional neonatal mice were used in experiments to determine the effect of blocking TGF-β activity on LCWE-mediated coronary pathological features. Mice were injected with either 10 mg/kg i.p. pan-sensitive TGF-β nAb or rabbit IgG (R&D Systems, Minneapolis), daily (three doses), beginning 24 hours before LCWE treatment, and then weekly thereafter for 2 weeks, a strategy previously described as effective in blocking the TGF-β activity observed in the murine model of MS.6 Additional groups of animals were treated with either the TGF-β nAb or rabbit IgG on an identical schedule but without LCWE. The mice were sacrificed at 2, 14, and 42 days after treatment for the analyses previously described. A similar experimental design was used to study the impact of LCWE with or without TGF-β nAb in adult mice (aged 5 weeks).
Immunoblotting
Protein was extracted from frozen heart tissue using a kit (NE-PER kit; Pierce, Rockford, IL) containing protease inhibitors, and 60 μg was resolved on 4% to 12% gels (NuPage Bis-Tris gels; Invitrogen, Carlsbad, CA) and electrotransferred to nitrocellulose membranes.24 Membranes were incubated overnight at 4°C with primary antibodies for PAI-1 (1:1000; BD Biosciences, San Jose, CA), incubated with anti–mouse horseradish peroxidase (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA) and Enhanced Chemiluminescence (ECL) substrate (GE Health Care, Piscataway, NJ), and the signal was then developed by exposure to Hyperfilm. Membranes were stripped and reprobed for α-tubulin (1:10,000; Sigma) as a housekeeping gene.
Quantitative PCR
Total RNA was extracted from the upper heart (including the aortic root), and 2 μg of RNA was reverse transcribed to cDNA using (Superscript III; Invitrogen) per the manufacturer's protocol. Quantitative PCR was performed using primers (TaqMan; Applied Biosystems, Foster City, CA) for tumor necrosis factor (TNF) α (Mm00443258_m1), MMP-9 (Mm00442991_m1), collagen1-α1 (Mm011302043_g1), collagen1-α2 (Mm00483937_m1), collagen6-α1 (Mm00487160_m1), fibrillin-1 (Mm00514908_m1), laminin-α5 (Mm01222029_m1), and laminin-β2 (Mm00493080_m1), using the relative standard curve method of analysis with expression of the target genes normalized to 18S and a real-time PCR instrument (model 7900HT; Applied Biosystems).
Gelatin Zymography
Total heart tissue obtained from the four groups (IgG, TGF-β nAb, LCWE plus IgG, and LCWE plus TGF-β nAb) was homogenized in ice-cold PBS. A total of 30 μg of protein was then resolved on 10% SDS gels containing 0.1% gelatin (Invitrogen).25 The gels were stained with Coomassie blue, and the bands of gelatinolytic activity were quantified using imaging software (Quantity One; Bio-Rad, Hercules, CA).
Statistical Analysis
All data are presented as the mean ± SEM. The number of animals used in each determination is given in the legends for the figures. Statistical differences between the two groups were determined by the Student's t-test. Statistical differences involving comparison of more than two groups were determined by one-way analysis of variance, followed by Bonferroni's multiple comparison post hoc analysis. P ≤ 0.05 was considered statistically significant.
Results
LCWE Increases TGF-β Signaling in the Coronary Wall and Induces Coronary Arteritis
To study to role of TGF-β in inflammatory aneurysm formation, we injected neonatal mice with LCWE, an agent that produces coronary arteritis and elastin degradation similar to that observed in KD.21,22,26,27 We first examined the activation of TGF-β in the coronary arteries by performing immunohistochemistry to detect pSmad2/3, the intracellular molecule that translocates into the nucleus in response to TGF-β signaling. A moderate amount of nuclear pSmad2/3 was observed in the neonatal coronary artery endothelium, adventitia, and cardiac myocytes in vehicle-treated controls 2 days after the start of the experiment (Figure 1A). The LCWE induced marked nuclear pSmad2/3 staining in the coronary smooth muscle cells and augmented basal signaling in the endothelium, adventitial cells, and cardiac myocytes (P < 0.0001). By 14 days, the nuclear pSmad2/3 had decreased in the LCWE-treated mice, with a similar degree of pSmad2/3 expression observed in the coronary endothelium and smooth muscle cells in LCWE-treated and control animals (Figure 1B). However, persistent prominent immunoreactivity remained in the perivascular cells of the LCWE mice. At this same point, examination of H&E-stained sections revealed that LCWE treatment caused a modest degree of perivascular inflammation in the coronary arteries (Figure 1C).
Figure 1.
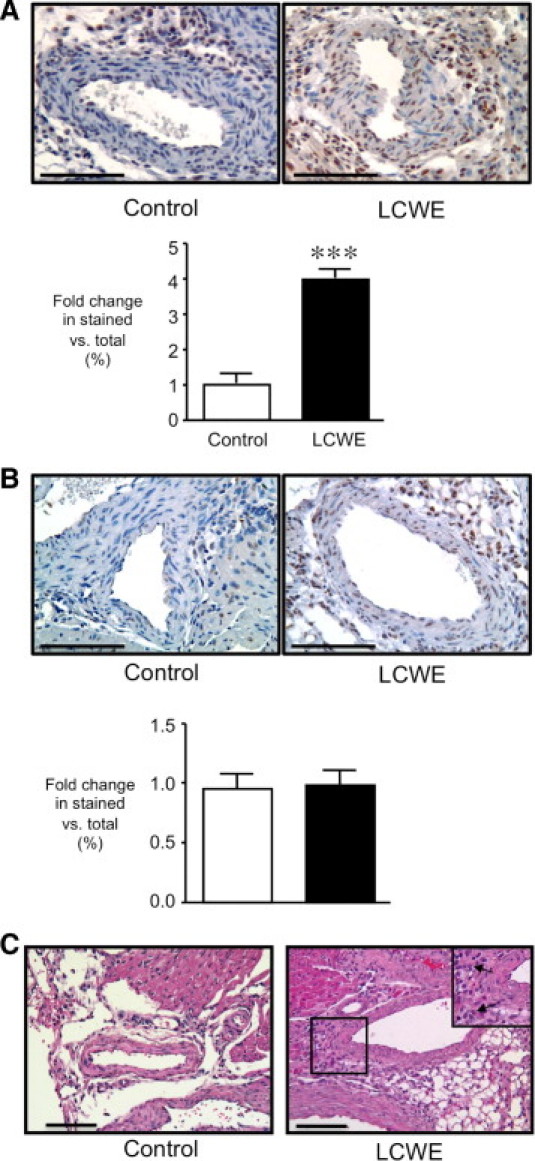
The LCWE induces TGF-β signaling in the coronary wall and coronary arteritis in neonatal mice. A: Representative sections of the coronary arteries after immunostaining to detect pSmad2/3 after either PBS or LCWE injection at 2 days (A) and 14 days (B). Total area of brown-stained versus total nuclei quantified within the coronary wall and expressed as fold change over control. ***P < 0.0001 versus control. Bars represent mean ± SEM (n = 4 to 11 sections counted per group). C: Representative H&E sections of the coronary arteries 14 days after PBS or LCWE demonstrate perivascular inflammatory cell infiltration (arrows) in the LCWE-treated animals. Scale bars = 100 μm.
LCWE Induces Fragmentation of the Coronary Artery Elastin
Next, we evaluated the integrity of the coronary elastin by Hart's staining 42 days after LCWE, the point when previous studies21,22 have demonstrated that LCWE causes elastin breakdown, an early feature of CAL. Compared with vehicle controls, the coronary arteries of the LCWE-treated mice demonstrated thinning and flattening of the internal elastic lamina and fraying of the external elastic lamina, with multiple areas of fragmentation (Figure 2A). When we quantified the number of visible breaks in the elastic lamina and the total amount of elastin within the coronary media, we found that while LCWE caused a significant disruption of the integrity of the external elastic lamina (Figure 2B), the total amount of medial elastin was not significantly lower than that found in control mice (Figure 2C).
Figure 2.
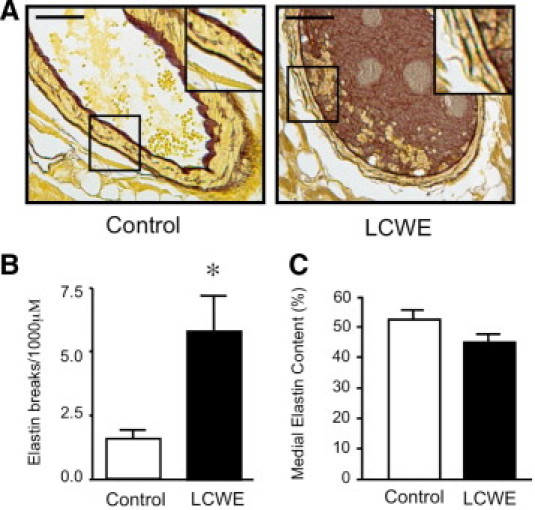
The LCWE induces fragmentation of coronary elastin. A: Representative sections of the coronary arteries after Hart's stain to stain the elastin brown, 42 days after PBS or LCWE. Insets demonstrate visible breaks in the external elastic lamina in LCWE-treated mice. Scale bars = 50 μm. B: Quantification of the number of visible breaks in the elastic lamina under ×40 magnification per 1000 μm. Bars represent mean ± SEM (n = 5 to 8 sections counted per group). *P > 0.05 versus control. C: Quantification of the percentage of elastin-stained media versus the entire medial area using imaging software (Bioquant) in sections of the coronary arteries in PBS- and LCWE-injected mice at 42 days. Bars represent mean ± SEM (n = 4 to 6 per group).
TGF-β Blockade Increases LCWE-Mediated Elastin Degradation Without Increasing Arteritis or Suppressing Tropoelastin Expression
To determine whether the LCWE-mediated induction of TGF-β contributed to the pathological features observed (ie, inflammation and elastin fragmentation) or served a protective role, we blocked TGF-β by administering a pan-sensitive TGF-β nAb to the neonatal mice beginning 1 day before LCWE treatment and continuing through the induction of arteritis at 14 days. Additional groups of mice were treated with isotype control IgG in addition to LCWE or with either IgG or TGF-β nAb in the absence of LCWE. Immunostaining for nuclear pSmad2/3 demonstrated that TGF-β nAb administration reduced the LCWE-mediated induction of TGF-β signaling in the coronary wall at 2 days (P = 0.013) (Figure 3).
Figure 3.
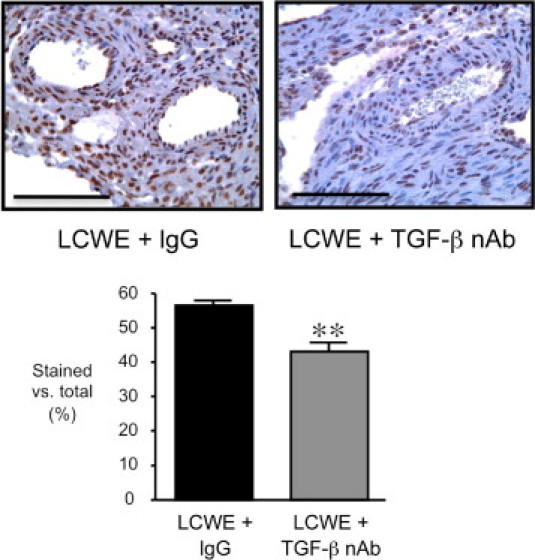
Administration of TGF-β nAb suppresses the LCWE-mediated induction of TGF-β in the coronary arteries. Representative sections of the coronary arteries are shown after immunostaining to detect pSmad2/3, 2 days after LCWE plus IgG or LCWE plus TGF-β nAb. Total area of brown-stained nuclei versus total nuclei was quantified within the coronary wall. **P = 0.0013 versus LCWE plus IgG. Bars represent mean ± SEM (n = 6 to 7 sections counted per group). Scale bars = 100 µm.
Transforming growth factor β is an important regulator of inflammation; therefore, we examined whether blocking TGF-β altered the extent of coronary arteritis induced by LCWE at 14 days. Evaluation of H&E-stained sections of the coronary arteries taken from LCWE plus IgG and LCWE plus TGF-β nAb-treated mice at 14 days demonstrated similar degrees of perivascular inflammation in both groups (Figure 4A). As an additional measure of the inflammatory response, we determined whether the expression of TNF-α, a proinflammatory cytokine increased in the heart after LCWE and necessary for the induction of LCWE-mediated coronary arteritis,20 was modulated by neutralizing TGF-β. Increased TNF-α mRNA levels were found in the hearts of the LCWE-treated mice as early as 7 days, and persisted to 14 days, coinciding with the development of coronary arteritis (Figure 4B). Inhibition of TGF-β did not further increase TNF-α mRNA levels but rather decreased levels slightly so that they were no longer significantly elevated at either 7 or 14 days after LCWE administration, although a strong trend persisted.
Figure 4.
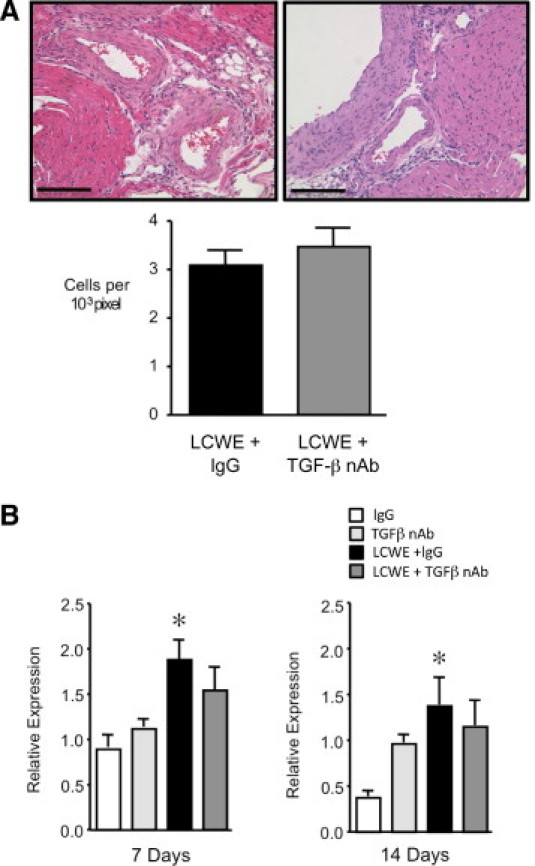
Inhibition of TGF-β does not increase LCWE-mediated coronary arteritis. A: Representative H&E sections of the coronary arteries 14 days after LCWE plus IgG or LCWE plus TGF-β nAb demonstrate a similar degree of perivascular inflammatory cell infiltration. Scale bars = 100 μm. B: Quantitative PCR to detect the expression of TNF-α in the heart 7 and 14 days after LCWE. Bars represent mean ± SEM (n = 4 to 6 per group). *P < 0.05 versus IgG.
Then, we evaluated the effect of inhibiting TGF-β on the degree of elastin degradation induced by LCWE at 42 days. Interestingly, blocking the basal TGF-β activity in the controls resulted in marked dilation of the coronary vessels (P < 0.001) and thinning of the coronary arterial media in these developing mice (Figure 5, A and B). As observed in the previous experiments, treatment with IgG in addition to LCWE resulted in thinning, fraying, and fragmentation of the external elastic lamina. In accordance with our hypothesis, inhibition of TGF-β accentuated the LCWE-mediated pathological characteristics in these animals, with areas of complete loss of the internal elastic lamina and marked disruption and dissolution of the external lamina. Interestingly, in contrast to the dilated coronary arteries of the mice treated with the TGF-β nAb alone, the coronary arteries of the mice receiving LCWE in addition to TGF-β appeared relatively constricted, despite the marked disruption of the elastic lamina evident in this group.
Figure 5.
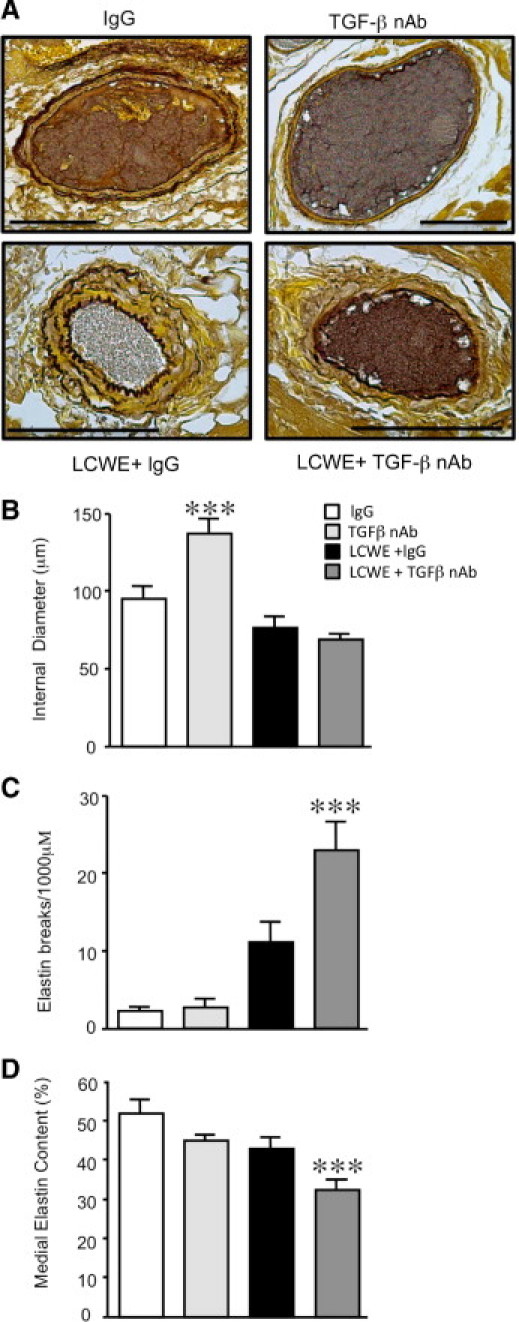
Inhibition of TGF-β increases LCWE-mediated elastin degradation. A: Representative sections of the coronary arteries after Hart's elastin staining 42 days after IgG, TGF-β nAb, LCWE plus IgG, or LCWE plus TGF-β nAb. B: Quantification of the internal diameter of the cross sections of the coronary arteries in each group. ***P < 0.001 versus IgG, LCWE plus IgG, and LCWE plus TGF-β nAb. C: Quantification of the number of visible breaks in the elastic lamina under ×40 magnification per 1000 μm. Bars represent mean ± SEM (n = 5 to 11 sections counted per group). ***P < 0.001 versus IgG. D: Quantification of the percentage of media staining for elastin versus the entire medial area using imaging software (Bioquant) in sections of the large coronary arteries in each group at 42 days. Bars represent mean ± SEM (n = 4 to 6 per group). ***P < 0.001 versus IgG. Scale bars = 100 μm.
Quantification of elastin integrity and overall elastin content morphometrically demonstrated that although administration of the TGF-β nAb alone resulted in thinning and dilation of the coronary wall, it did not result in fragmented elastin nor did it significantly decrease the level of medial elastin (Figure 5, C and D). Similarly, although LCWE plus IgG induced fragmentation of the coronary elastin in the mice, the overall elastin content was not changed. However, blocking TGF-β activity in combination with LCWE resulted in significantly greater fragmentation of the elastin compared with mice treated with LCWE plus IgG (Figure 5C) and significantly reduced the medial elastin content (Figure 5D) (P < 0.001). Because TGF-β can regulate elastin fiber assembly by stimulating the expression of tropoelastin,13,28 we determined whether blocking TGF-β induced loss of the coronary elastin by suppressing tropoelastin protein expression. However, inhibition of TGF-β did not affect tropoelastin protein levels in the LCWE-treated mice (data not shown).
Treatment of adult mice with LCWE with or without TGF-β nAb induced similar vascular pathological features to those observed in the neonates, except that TGF-β nAb alone did not cause dilation of the arteries, suggesting that this effect was developmentally restricted. However, increased fragmentation of elastin was evident in the adult mice treated with LCWE plus TGF-β nAb. In addition, the trend toward reduced medial elastin observed with either LCWE or TGF-β nAb in the neonates became significant in the adult; like the infants, the effect was greatest when the two agents were combined (see Supplemental Figure S1 at http://ajp.amjpathol.org).
To further explore the mechanism by which TGF-β inhibition caused dilation of the developing coronary arteries, we evaluated the expression of matrix proteins that are essential to the strength and integrity of the vessel wall and regulate vascular tone and diameter.29 There were no significant changes in the gene expression of the various matrix components that we examined after either TGF-β nAb or LCWE treatment at 7 days (see Supplemental Figure S2 at http://ajp.amjpathol.org). By 14 days, TGF-β nAb resulted in a marked reduction in laminin-α5 (P < 0.05), an important regulator of vascular tone,29 but did not alter levels of collagen1-α1 and collagen-α2 or laminin β2 (Figure 6). Although LCWE plus IgG induced an increase in collagen1-α1 and collagen1-α2, only the increase in collagen1-α2 was evident with LCWE plus TGF-β nAb. There were no significant changes in collagen6-α1 or fibrillin-1 in any of the four treatment groups nor did LCWE with or without TGF-β nAb alter levels of laminin.
Figure 6.
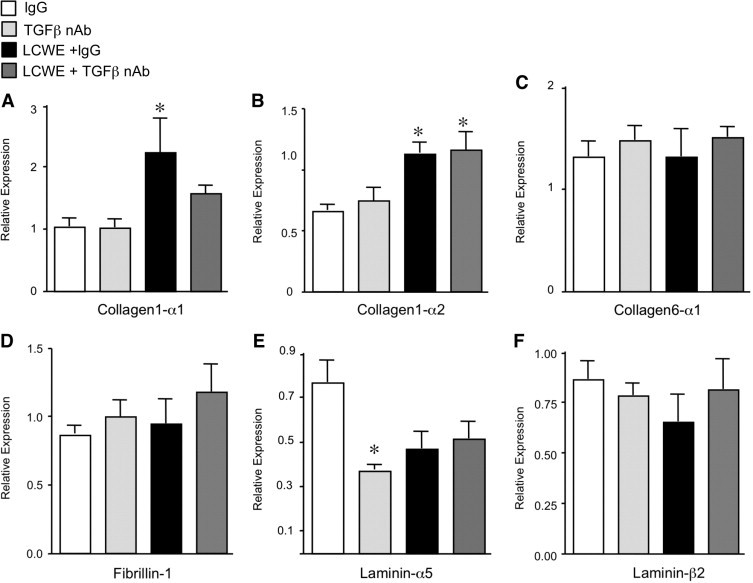
The LCWE increases collagen-1 expression. Quantitative PCR to detect the expression of matrix components at 14 days: collagen1-α1 (A), collagen1-α2 (B), collagen6-α1 (C), fibrillin-1 (D), laminin-α5 (E), and laminin-β2 (F). Bars represent mean ± SEM (n = 4 to 6 per group). *P < 0.05 versus IgG.
Then, we evaluated the deposition of extracellular matrix components in the coronary adventitia by Masson's trichrome staining, 42 days after LCWE, the point when TGF-β nAb caused a significant loss of medial elastin. Treatment with the TGF-β nAb in the control animals resulted in a significant reduction of adventitial matrix versus IgG-treated controls (P < 0.001) (Figure 7B). Despite the LCWE-mediated increase in collagen1-α1 and collagen1-α2 mRNA expression at 14 days, extracellular matrix deposition was not significantly increased in the LCWE plus IgG-treated animals. In contrast, LCWE plus TGF-β nAb increased matrix deposition in the coronary adventitia (P < 0.05), perhaps serving to prevent the vascular dilation despite the severe fragmentation and loss of the coronary elastin (Figure 7B).
Figure 7.
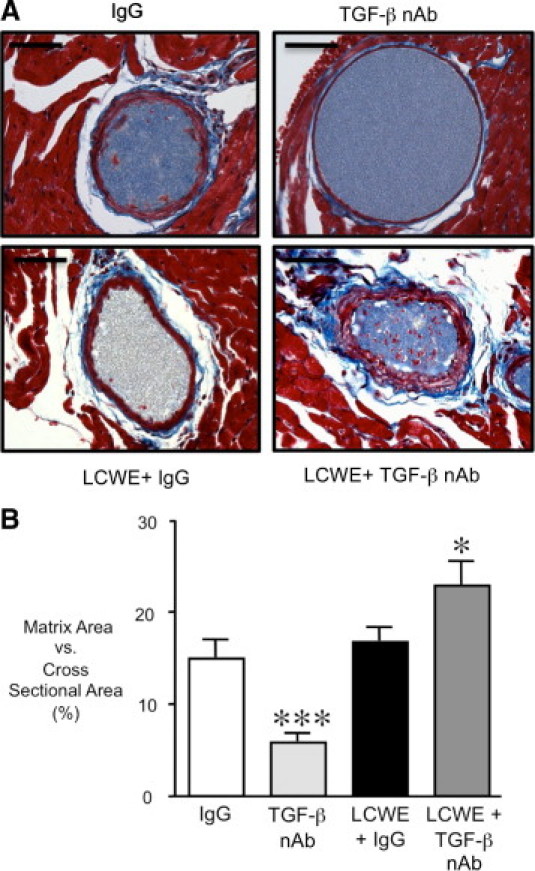
Increased matrix deposition in the coronary adventitia of LCWE plus TGF-β nAb–treated mice. A: Representative sections of the coronary arteries taken at ×40 magnification after Masson's trichrome staining 42 days after IgG, TGF-β nAb, LCWE plus IgG, or LCWE plus TGF-β nAb. B: Quantification of the percentage matrix versus total cross-sectional area of the coronary arteries for each group. Bars represent mean ± SEM (n = 4 to 6 per group). *P < 0.05 and ***P < 0.001 versus IgG. Scale bars = 50 μm.
TGF-β Blockade Increases MMP-9 Activity and Reduces PAI-1 Expression
Blocking TGF-β in combination with LCWE caused a significant loss of medial elastin without affecting tropoelastin levels, thereby suggesting that the mechanism was the result of enhanced degradation of insoluble elastin rather than impaired synthesis. Matrix metalloproteinases 2 and 9 have direct elastolytic activity, and their levels are increased in animal models of aneurysm and in patients with an abdominal aortic aneurysm.30,31 Furthermore, TNF-α–mediated induction of MMP-9 activity is necessary for the elastin breakdown observed in the LCWE murine model of KD.22 We evaluated the expression of MMP-9 by quantitative RT-PCR in the heart at 14 days and found no significant difference in MMP-9 expression in any of the four treatment groups (Figure 8A).
Figure 8.
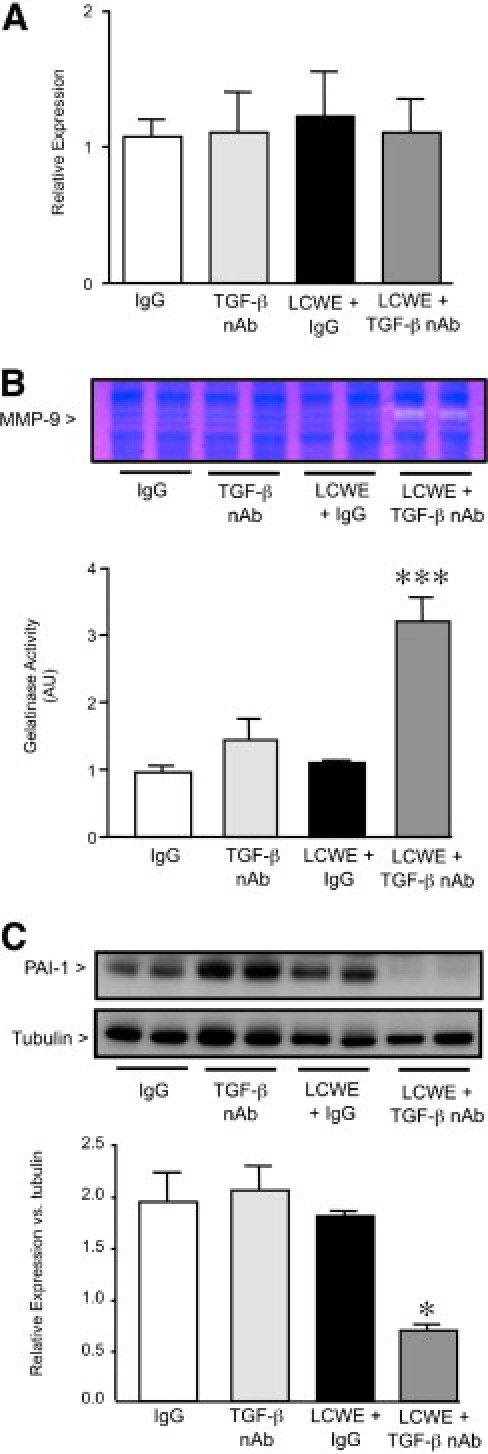
Inhibition of TGF-β increases MMP-9 activity and reduces plasminogen activator inhibitor-1 in LCWE-treated mice. A: Quantitative PCR to detect MMP-9 expression in the heart at 14 days. B: Gelatin zymography was performed using total heart homogenates obtained 14 days after IgG, TGF-β nAb, LCWE plus IgG, or LCWE plus TGF-β nAb injection to detect active MMP-9. Bars represent mean ± SEM (n = 4 to 6 per group). ***P < 0.001 versus IgG controls. AU indicates arbitrary unit. C: Western immunoblot analysis was performed to detect PAI-1 protein expression at 14 days after IgG, TGF-β nAb, LCWE plus IgG, or LCWE plus TGF-β nAb. Bars represent mean ± SEM (n = 4 to 6 per group). *P < 0.05 versus IgG.
Transforming growth factor β can inhibit proteolytic degradation of the extracellular matrix in two ways: i) by inducing the expression of TIMPs; and ii) by increasing inhibitors of plasmin, such as PAI-1, thereby decreasing plasmin-mediated MMP activation.14,15,32,33 Both are mechanisms that affect MMP activity, independent of mRNA levels. Therefore, to determine whether TGF-β inhibition increased LCWE-mediated loss of elastin by augmenting either MMP-2 or MMP-9 activity, we performed gelatin zymography on heart homogenates 14 days after LCWE. We found no difference in either pro- or active MMP-2 in any of the groups (data not shown). Furthermore, neither TGF-β nAb nor LCWE alone significantly increased active MMP-9 levels in the mice at 14 days. However, blocking TGF-β in addition to LCWE resulted in a greater than threefold increase in MMP-9 activity (P < 0.001) (Figure 8B).
The mechanism accounting for this increase in MMP-9 activity could not be attributed to changes in the protein expression of the MMP-9 inhibitors, TIMP-1 and TIMP-2, as determined by Western immunoblot (data not shown). However, TGF-β blockade markedly reduced levels of PAI-1 in the LCWE-treated mice (P < 0.05) (Figure 8C), suggesting that plasmin-mediated activation of MMP-9 likely contributes to the loss of medial elastin in this group.
Discussion
In this study, we demonstrate that TGF-β signaling protects the coronary arteries against the destruction of elastin induced by coronary arteritis. This is in contrast to the pathological role of TGF-β in the aortic aneurysms that form in MS,6 as assessed by the efficacy of TGF-β blockade in both preventing aneurysms in the murine model of MS and slowing disease progression in patients with MS.7 In marked contrast to the MS model, our data suggest that with an inflammatory stimulus, loss of elastin is prevented by induction of TGF-β and PAI-1–mediated repression of MMP-9 activation by plasmin. An unexpected observation is that TGF-β signaling is essential during this postnatal window of coronary artery development because TGF-β inhibition caused marked coronary wall thinning and dilation. We attribute this dilation to suppression of adventitial matrix deposition, including reduced expression of laminin-α5 (not to accentuated MMP-9 activity).
In this model of KD, LCWE caused an activation of TGF-β beginning at 2 days and continuing through 14 days, concurrent with the induction of inflammatory cytokines and the development of coronary arteritis previously demonstrated.21 Tissue samples from the coronary arteries of patients with KD are limited and were obtained only from patients with fatal complications, making identification of the molecular mediators leading to CALs challenging. Evidence suggests that dysregulation of TGF-β signaling occurs in patients with KD. Increased TGF-β1 protein expression is found in the coronary smooth muscle cells in patients with KD.34,35 However, although one study36 found that serum levels of TGF-β1 were increased in patients with acute KD compared with febrile controls, another study37 documented that levels were decreased. Therefore, the role that TGF-β plays in the pathogenesis of inflammatory arteritis, such as KD, remains unclear.
We observed that the LCWE-mediated induction of TGF-β was followed by the fragmentation of coronary elastin and that the inhibition of TGF-β markedly increased the loss of elastin in association with accentuation of MMP-9 activity. Degradation of elastin is a key feature of aneurysm development, and extensive clinical and experimental evidence demonstrates that an imbalance between the proteinases and their endogenous inhibitors influences the development and progression of arterial aneurysms.38 Matrix metalloproteinase 9 appears to be a key factor affecting this balance, with increased activity found in aneurysmal tissue taken from patients with an abdominal aortic aneurysm and MS,39 in the coronary arteries of children with fatal KD,30 and in animal models of abdominal aortic aneurysm.39–41 Heightened MMP-9 activity is also found in the coronary arteries of LCWE-treated animals, localizing to the vascular smooth muscle cells.22 Interestingly, we observed an increase in active MMP-9 in the animals treated with LCWE and TGF-β nAb by 14 days, before the point when MMP-9 activity is increased in mice treated with LCWE alone.22 This suggests that TGF-β blockade both enhances and accelerates the proteolytic process.
In addition to suppressing proteolysis, TGF-β can modulate inflammation by repressing the transcription10 and mRNA stability42 of inflammatory cytokines. The critical role of TGF-β in immune regulation is evident in the phenotype of TGF-β1–null mice, demonstrating widespread tissue inflammation and persistent T-cell activation.43 Dysregulation of T-cell activation, resulting from a polymorphism within the inositol 1,4,5-triphosphate 3-kinase C gene, has recently been identified as a key factor determining KD susceptibility and risk for coronary artery aneurysms.44 Patients with the C allele have reduced levels of inositol 1,4,5-triphosphate 3-kinase C, a protein that suppresses T-cell activation by decreasing inositol triphosphate-mediated calcium influx and nuclear factor of activated T cells signaling. Transforming growth factor β can decrease inositol triphosphate receptor expression and calcium influx in vascular smooth muscle.45 Therefore, although the TGF-β blockade did not increase the number of infiltrating inflammatory cells or the expression of TNF-α, future studies could be important to determine whether TGF-β blockade in this model increases T-cell activation by derepressing inositol triphosphate receptor expression and enhancing nuclear factor of activated T cells signaling.
Interestingly, in the absence of LCWE, TGF-β blockade in the young mice caused dilation of the coronary arteries. Active TGF-β signaling in the coronary arteries was found beginning at the age of 1 week and continuing through the age of 3 weeks. Taken together, these data suggest that local activation of TGF-β during the early postnatal period is a requirement of normal coronary arterial development. Transforming growth factor β regulates matrix components, such as collagen and laminin,46–49 that serve to limit blood vessel distention in response to pulsatile flow. Although we found no difference in collagen expression in this group, blocking TGF-β signaling in the developing coronary artery was associated with a significant reduction of laminin-α5 and a decrease in adventitial matrix deposition. The laminins have a specific role in the regulation of blood vessel diameter because deletion of either laminin-α4 or laminin-α5 results in vessels with larger vascular lumens.46,50 Laminin-α5 expression in the vascular basement membrane begins in the mouse at E13.5 and increases postnatally, supporting its role in stabilizing the developing vasculature.
The administration of LCWE in combination with TGF-β blockade reduced PAI-1, increased MMP-9 activity, and caused severe fragmentation of the internal elastic lamina and almost complete dissolution of the external elastic lamina; however, coronary dilation did not occur. The increase in matrix deposition demonstrated by trichrome staining likely limited dilation. However, collagen synthesis, as judged by mRNA expression, was not greater in the group treated with LCWE plus TGF-β compared with those treated with LCWE plus IgG, suggesting that other matrix components (eg, fibronectin or osteopontin) predominated in this constrictive remodeling response, similar to that seen in the adventitial fibrosis that occurs after other types of vascular injury.51 Furthermore, although loss of elastin is an early feature of aneurysm formation, recent evidence suggests that other factors are likely required for the vessel dilation that occurs in aneurysm formation, including apoptosis of vascular smooth muscle cells52 and alterations in smooth muscle cell contractile proteins.53
It is also possible that a transient phase of coronary dilation preceded the constrictive remodeling observed in the LCWE-treated mice, similar to that seen in the clinical setting. Up to 10% of patients with a history of KD and CAL subsequently develop coronary stenosis, suggesting that persistent remodeling occurs even after the initial inflammation resolves. It is this later constrictive remodeling that accounts for most of the morbidity and mortality from KD, with affected patients at risk for myocardial ischemia and sudden death and often requiring interventions such as balloon angioplasty, coronary bypass, or cardiac transplantation.54 Interestingly, even patients with KD without a history of CAL have evidence of increased stiffness of the coronary arteries compared with controls55,56; and increased stiffness is observed in association with reduced elastin.57 In addition, patients with KD have high levels of endothelin-1 and impaired coronary artery vasodilation, suggesting that an imbalance in vasoconstrictive and vasodilatory factors contributes to the disease.58 The coronary circulation of the mice was not injected with vasodilators before injection of barium; therefore, it is possible that heightened vasoconstriction in the LCWE-treated animals was an additional factor limiting coronary distention in these groups.
In summary, we demonstrate that TGF-β inhibition in a murine model of KD worsens disease severity, increasing elastin fragmentation and loss by suppressing PAI-1 and markedly increasing MMP-9 activity. We also identified a novel role for TGF-β in the stabilization of the growing coronary arteries because inhibition during this developmental window caused marked thinning and dilation. This study has important implications, particularly in light of the recent identification of TGF-β as a critical pathological mediator in the aneurysms forming in MS. First, it demonstrates that the molecular mechanisms leading to aneurysms in genetic and inflammatory diseases are distinct. Second, it suggests that therapeutic strategies to block TGF-β are unlikely to be of benefit in KD and could be detrimental, particularly during early development.
Acknowledgments
We thank Robert P. Mecham, Ph.D., for providing the tropoelastin antibody.
Footnotes
Supported by grants from the American Heart Association Fellow to Faculty Award (C.M.A.); the Canadian Institutes for Health ResearchFRN 53245 (R.S.M.Y.); Arthritis Society of Canada Investigator Award (R.S.M.Y.); the H.A. and Edna Benning Foundation (D.Y.L.); the Burroughs Wellcome Foundation (D.Y.L.); the Juvenile Diabetes Research Foundation (D.Y.L.); the American Heart Association (D.Y.L.); the National Heart, Lung, and Blood Institute (D.Y.L.); the National Institutes of Health (grant RO1 HL074186 to M.R.); and the Dunlevie Chair in Pediatric Cardiology (M.R.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.11.054.
Supplementary data
Morphometric analysis of elastin-stained coronary arteries of adult mice 42 days after IgG, TGF-β Nab, LCWE plus IgG, or LCWE plus TGF-β Nab treatment. A: Quantification of the internal diameter of the cross sections of the coronary arteries in each group. B: Quantification of the number of visible breaks in the elastic lamina under ×40 magnification per 1000 μmol/L. Bars represent SEM (n = 6-13 sections counted per group). *P < 0.05 versus IgG. C: Quantification of the percentage of media staining for elastin versus the entire medial area using imaging software (Bioquant) in sections of the large coronary arteries in each group at 42 days. Bars represent SEM (n = 4-5 per group). *P < 0.05 versus IgG.
Quantitative PCR to detect the expression of matrix components at 7 days: collagen1-α1 (A), collagen1-α2 (B), collagen6-α1 (C), fibrillin-1 (D), laminin-α5 (E), and laminin-β2 (F). Bars represent SEM (n = 4-6 per group).
References
- 1.Falcini F. Kawasaki disease. Curr Opin Rheumatol. 2006;18:33–38. doi: 10.1097/01.bor.0000197998.50450.f6. [DOI] [PubMed] [Google Scholar]
- 2.Newburger J.W., Takahashi M., Gerber M.A., Gewitz M.H., Tani L.Y., Burns J.C., Shulman S.T., Bolger A.F., Ferrieri P., Baltimore R.S., Wilson W.R., Baddour L.M., Levison M.E., Pallasch T.J., Falace D.A., Taubert K.A., Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease. Council on Cardiovascular Disease in the Young. American Heart Association Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114:1708–1733. doi: 10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 3.Sakalihasan N., Limet R., Defawe O.D. Abdominal aortic aneurysm. Lancet. 2005;365:1577–1589. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu K., Mitchell R.N., Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987–994. doi: 10.1161/01.ATV.0000214999.12921.4f. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura Y., Yanagawa H., Ojima T., Kawasaki T., Kato H. Cardiac sequelae of Kawasaki disease among recurrent cases. Arch Dis Child. 1998;78:163–165. doi: 10.1136/adc.78.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Habashi J.P., Judge D.P., Holm T.M., Cohn R.D., Loeys B.L., Cooper T.K., Myers L., Klein E.C., Liu G., Calvi C., Podowski M., Neptune E.R., Halushka M.K., Bedja D., Gabrielson K., Rifkin D.B., Carta L., Ramirez F., Huso D.L., Dietz H.C. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooke B.S., Habashi J.P., Judge D.P., Patel N., Loeys B., Dietz H.C., 3rd Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nichols L., Lagana S., Parwani A. Coronary artery aneurysm: a review and hypothesis regarding etiology. Arch Pathol Lab Med. 2008;132:823–828. doi: 10.5858/2008-132-823-CAAARA. [DOI] [PubMed] [Google Scholar]
- 9.DiChiara M.R., Kiely J.M., Gimbrone M.A., Jr, Lee M.E., Perrella M.A., Topper J.N. Inhibition of E-selectin gene expression by transforming growth factor beta in endothelial cells involves coactivator integration of Smad and nuclear factor kappaB-mediated signals. J Exp Med. 2000;192:695–704. doi: 10.1084/jem.192.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinberg M.W., Shimizu K., Lebedeva M., Haspel R., Takayama K., Chen Z., Frederick J.P., Wang X.F., Simon D.I., Libby P., Mitchell R.N., Jain M.K. Essential role for Smad3 in regulating MCP-1 expression and vascular inflammation. Circ Res. 2004;94:601–608. doi: 10.1161/01.RES.0000119170.70818.4F. [DOI] [PubMed] [Google Scholar]
- 11.Boak A.M., Roy R., Berk J., Taylor L., Polgar P., Goldstein R.H., Kagan H.M. Regulation of lysyl oxidase expression in lung fibroblasts by transforming growth factor-beta 1 and prostaglandin E2. Am J Respir Cell Mol Biol. 1994;11:751–755. doi: 10.1165/ajrcmb.11.6.7946403. [DOI] [PubMed] [Google Scholar]
- 12.Kuang P.P., Joyce-Brady M., Zhang X.H., Jean J.C., Goldstein R.H. Fibulin-5 gene expression in human lung fibroblasts is regulated by TGF-beta and phosphatidylinositol 3-kinase activity. Am J Physiol Cell Physiol. 2006;291:C1412–C1421. doi: 10.1152/ajpcell.00087.2006. [DOI] [PubMed] [Google Scholar]
- 13.McGowan S.E., Jackson S.K., Olson P.J., Parekh T., Gold L.I. Exogenous and endogenous transforming growth factors-beta influence elastin gene expression in cultured lung fibroblasts. Am J Respir Cell Mol Biol. 1997;17:25–35. doi: 10.1165/ajrcmb.17.1.2686. [DOI] [PubMed] [Google Scholar]
- 14.Akool el S., Doller A., Muller R., Gutwein P., Xin C., Huwiler A., Pfeilschifter J., Eberhardt W. Nitric oxide induces TIMP-1 expression by activating the transforming growth factor beta-Smad signaling pathway. J Biol Chem. 2005;280:39403–39416. doi: 10.1074/jbc.M504140200. [DOI] [PubMed] [Google Scholar]
- 15.Kutz S.M., Higgins C.E., Samarakoon R., Higgins S.P., Allen R.R., Qi L., Higgins P.J. TGF-beta 1-induced PAI-1 expression is E box/USF-dependent and requires EGFR signaling. Exp Cell Res. 2006;312:1093–1105. doi: 10.1016/j.yexcr.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 16.Dai J., Losy F., Guinault A.M., Pages C., Anegon I., Desgranges P., Becquemin J.P., Allaire E. Overexpression of transforming growth factor-beta1 stabilizes already-formed aortic aneurysms: a first approach to induction of functional healing by endovascular gene therapy. Circulation. 2005;112:1008–1015. doi: 10.1161/CIRCULATIONAHA.104.523357. [DOI] [PubMed] [Google Scholar]
- 17.Thompson R.W., Curci J.A., Ennis T.L., Mao D., Pagano M.B., Pham C.T. Pathophysiology of abdominal aortic aneurysms: insights from the elastase-induced model in mice with different genetic backgrounds. Ann N Y Acad Sci. 2006;1085:59–73. doi: 10.1196/annals.1383.029. [DOI] [PubMed] [Google Scholar]
- 18.Yoshimura K., Aoki H., Ikeda Y., Fujii K., Akiyama N., Furutani A., Hoshii Y., Tanaka N., Ricci R., Ishihara T., Esato K., Hamano K., Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 19.Lemaitre V., Soloway P.D., D'Armiento J. Increased medial degradation with pseudo-aneurysm formation in apolipoprotein E-knockout mice deficient in tissue inhibitor of metalloproteinases-1. Circulation. 2003;107:333–338. doi: 10.1161/01.cir.0000044915.37074.5c. [DOI] [PubMed] [Google Scholar]
- 20.Hui-Yuen J.S., Duong T.T., Yeung R.S. TNF-alpha is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J Immunol. 2006;176:6294–6301. doi: 10.4049/jimmunol.176.10.6294. [DOI] [PubMed] [Google Scholar]
- 21.Duong T.T., Silverman E.D., Bissessar M.V., Yeung R.S. Superantigenic activity is responsible for induction of coronary arteritis in mice: an animal model of Kawasaki disease. Int Immunol. 2003;15:79–89. doi: 10.1093/intimm/dxg007. [DOI] [PubMed] [Google Scholar]
- 22.Lau A.C., Duong T.T., Ito S., Yeung R.S. Matrix metalloproteinase 9 activity leads to elastin breakdown in an animal model of Kawasaki disease. Arthritis Rheum. 2008;58:854–863. doi: 10.1002/art.23225. [DOI] [PubMed] [Google Scholar]
- 23.Bland R.D., Ertsey R., Mokres L.M., Xu L., Jacobson B.E., Jiang S., Alvira C.M., Rabinovitch M., Shinwell E.S., Dixit A. Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice: prelude to defective alveolar septation during lung development? Am J Physiol Lung Cell Mol Physiol. 2008;294:L3–L14. doi: 10.1152/ajplung.00362.2007. [DOI] [PubMed] [Google Scholar]
- 24.Alvira C.M., Abate A., Yang G., Dennery P.A., Rabinovitch M. Nuclear factor-kappaB activation in neonatal mouse lung protects against lipopolysaccharide-induced inflammation. Am J Respir Crit Care Med. 2007;175:805–815. doi: 10.1164/rccm.200608-1162OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones P.L., Crack J., Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the alpha v beta 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol. 1997;139:279–293. doi: 10.1083/jcb.139.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenkranz M.E., Schulte D.J., Agle L.M., Wong M.H., Zhang W., Ivashkiv L., Doherty T.M., Fishbein M.C., Lehman T.J., Michelsen K.S., Arditi M. TLR2 and MyD88 contribute to Lactobacillus casei extract-induced focal coronary arteritis in a mouse model of Kawasaki disease. Circulation. 2005;112:2966–2973. doi: 10.1161/CIRCULATIONAHA.105.537530. [DOI] [PubMed] [Google Scholar]
- 27.Schulte D.J., Yilmaz A., Shimada K., Fishbein M.C., Lowe E.L., Chen S., Wong M., Doherty T.M., Lehman T., Crother T.R., Sorrentino R., Arditi M. Involvement of innate and adaptive immunity in a murine model of coronary arteritis mimicking Kawasaki disease. J Immunol. 2009;183:5311–5318. doi: 10.4049/jimmunol.0901395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kucich U., Rosenbloom J.C., Abrams W.R., Rosenbloom J. Transforming growth factor-beta stabilizes elastin mRNA by a pathway requiring active Smads, protein kinase C-delta, and p38. Am J Respir Cell Mol Biol. 2002;26:183–188. doi: 10.1165/ajrcmb.26.2.4666. [DOI] [PubMed] [Google Scholar]
- 29.Jakobsson L., Domogatskaya A., Tryggvason K., Edgar D., Claesson-Welsh L. Laminin deposition is dispensable for vasculogenesis but regulates blood vessel diameter independent of flow. FASEB J. 2008;22:1530–1539. doi: 10.1096/fj.07-9617com. [DOI] [PubMed] [Google Scholar]
- 30.Gavin P.J., Crawford S.E., Shulman S.T., Garcia F.L., Rowley A.H. Systemic arterial expression of matrix metalloproteinases 2 and 9 in acute Kawasaki disease. Arterioscler Thromb Vasc Biol. 2003;23:576–581. doi: 10.1161/01.ATV.0000065385.47152.FD. [DOI] [PubMed] [Google Scholar]
- 31.Longo G.M., Xiong W., Greiner T.C., Zhao Y., Fiotti N., Baxter B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamanaka M., Shegogue D., Pei H., Bu S., Bielawska A., Bielawski J., Pettus B., Hannun Y.A., Obeid L., Trojanowska M. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J Biol Chem. 2004;279:53994–54001. doi: 10.1074/jbc.M410144200. [DOI] [PubMed] [Google Scholar]
- 33.Allaire E., Hasenstab D., Kenagy R.D., Starcher B., Clowes M.M., Clowes A.W. Prevention of aneurysm development and rupture by local overexpression of plasminogen activator inhibitor-1. Circulation. 1998;98:249–255. doi: 10.1161/01.cir.98.3.249. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki A., Miyagawa-Tomita S., Komatsu K., Nakazawa M., Fukaya T., Baba K., Yutani C. Immunohistochemical study of apparently intact coronary artery in a child after Kawasaki disease. Pediatr Int. 2004;46:590–596. doi: 10.1111/j.1442-200x.2004.01943.x. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki A., Miyagawa-Tomita S., Komatsu K., Nishikawa T., Sakomura Y., Horie T., Nakazawa M. Active remodeling of the coronary arterial lesions in the late phase of Kawasaki disease: immunohistochemical study. Circulation. 2000;101:2935–2941. doi: 10.1161/01.cir.101.25.2935. [DOI] [PubMed] [Google Scholar]
- 36.Terai M., Yasukawa K., Narumoto S., Tateno S., Oana S., Kohno Y. Vascular endothelial growth factor in acute Kawasaki disease. Am J Cardiol. 1999;83:337–339. doi: 10.1016/s0002-9149(98)00864-9. [DOI] [PubMed] [Google Scholar]
- 37.Matsubara T., Umezawa Y., Tsuru S., Motohashi T., Yabuta K., Furukawa S. Decrease in the concentrations of transforming growth factor-beta 1 in the sera of patients with Kawasaki disease. Scand J Rheumatol. 1997;26:314–317. doi: 10.3109/03009749709105322. [DOI] [PubMed] [Google Scholar]
- 38.Jones J.A., Beck C., Barbour J.R., Zavadzkas J.A., Mukherjee R., Spinale F.G., Ikonomidis J.S. Alterations in aortic cellular constituents during thoracic aortic aneurysm development: myofibroblast-mediated vascular remodeling. Am J Pathol. 2009;175:1746–1756. doi: 10.2353/ajpath.2009.081141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung A.W., Au Yeung K., Sandor G.G., Judge D.P., Dietz H.C., van Breemen C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101:512–522. doi: 10.1161/CIRCRESAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- 40.Ikonomidis J.S., Gibson W.C., Butler J.E., McClister D.M., Sweterlitsch S.E., Thompson R.P., Mukherjee R., Spinale F.G. Effects of deletion of the tissue inhibitor of matrix metalloproteinases-1 gene on the progression of murine thoracic aortic aneurysms. Circulation. 2004;110:II268–II273. doi: 10.1161/01.CIR.0000138384.68947.20. [DOI] [PubMed] [Google Scholar]
- 41.Kaito K., Urayama H., Watanabe G. Doxycycline treatment in a model of early abdominal aortic aneurysm. Surg Today. 2003;33:426–433. doi: 10.1007/s10595-002-2513-0. [DOI] [PubMed] [Google Scholar]
- 42.Dai Y., Datta S., Novotny M., Hamilton T.A. TGFbeta inhibits LPS-induced chemokine mRNA stabilization. Blood. 2003;102:1178–1185. doi: 10.1182/blood-2002-12-3771. [DOI] [PubMed] [Google Scholar]
- 43.Kulkarni A.B., Huh C.G., Becker D., Geiser A., Lyght M., Flanders K.C., Roberts A.B., Sporn M.B., Ward J.M., Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onouchi Y., Gunji T., Burns J.C., Shimizu C., Newburger J.W., Yashiro M., Nakamura Y., Yanagawa H., Wakui K., Fukushima Y., Kishi F., Hamamoto K., Terai M., Sato Y., Ouchi K., Saji T., Nariai A., Kaburagi Y., Yoshikawa T., Suzuki K., Tanaka T., Nagai T., Cho H., Fujino A., Sekine A., Nakamichi R., Tsunoda T., Kawasaki T., Nakamura Y., Hata A. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. 2008;40:35–42. doi: 10.1038/ng.2007.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma K., McGowan T.A., Wang L., Madesh M., Kaspar V., Szalai G., Thomas A.P., Hajnoczky G. Inhibition of type I and III IP(3)Rs by TGF-beta is associated with impaired calcium release in mesangial cells. Am J Physiol Renal Physiol. 2000;278:F1022–F1029. doi: 10.1152/ajprenal.2000.278.6.F1022. [DOI] [PubMed] [Google Scholar]
- 46.Thyboll J., Kortesmaa J., Cao R., Soininen R., Wang L., Iivanainen A., Sorokin L., Risling M., Cao Y., Tryggvason K. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol. 2002;22:1194–1202. doi: 10.1128/MCB.22.4.1194-1202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poschl E., Schlotzer-Schrehardt U., Brachvogel B., Saito K., Ninomiya Y., Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 48.Liu X., Wu H., Byrne M., Krane S., Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rahkonen O., Su M., Hakovirta H., Koskivirta I., Hormuzdi S.G., Vuorio E., Bornstein P., Penttinen R. Mice with a deletion in the first intron of the Col1a1 gene develop age-dependent aortic dissection and rupture. Circ Res. 2004;94:83–90. doi: 10.1161/01.RES.0000108263.74520.15. [DOI] [PubMed] [Google Scholar]
- 50.Miner J.H., Cunningham J., Sanes J.R. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michel J.B., Thaunat O., Houard X., Meilhac O., Caligiuri G., Nicoletti A. Topological determinants and consequences of adventitial responses to arterial wall injury. Arterioscler Thromb Vasc Biol. 2007;27:1259–1268. doi: 10.1161/ATVBAHA.106.137851. [DOI] [PubMed] [Google Scholar]
- 52.Henderson E.L., Geng Y.J., Sukhova G.K., Whittemore A.D., Knox J., Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99:96–104. doi: 10.1161/01.cir.99.1.96. [DOI] [PubMed] [Google Scholar]
- 53.Guo D.C., Pannu H., Tran-Fadulu V., Papke C.L., Yu R.K., Avidan N., Bourgeois S., Estrera A.L., Safi H.J., Sparks E., Amor D., Ades L., McConnell V., Willoughby C.E., Abuelo D., Willing M., Lewis R.A., Kim D.H., Scherer S., Tung P.P., Ahn C., Buja L.M., Raman C.S., Shete S.S., Milewicz D.M. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 54.Kato H., Sugimura T., Akagi T., Sato N., Hashino K., Maeno Y., Kazue T., Eto G., Yamakawa R. Long-term consequences of Kawasaki disease: a 10- to 21-year follow-up study of 594 patients. Circulation. 1996;94:1379–1385. doi: 10.1161/01.cir.94.6.1379. [DOI] [PubMed] [Google Scholar]
- 55.Cheung Y.F., O K., Woo C.W., Armstrong S., Siow Y.L., Chow P.C., Cheung E.W. Oxidative stress in children late after Kawasaki disease: relationship with carotid atherosclerosis and stiffness. BMC Pediatr. 2008;8:20. doi: 10.1186/1471-2431-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheung Y.F., Wong S.J., Ho M.H. Relationship between carotid intima-media thickness and arterial stiffness in children after Kawasaki disease. Arch Dis Child. 2007;92:43–47. doi: 10.1136/adc.2006.096628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shifren A., Durmowicz A.G., Knutsen R.H., Faury G., Mecham R.P. Elastin insufficiency predisposes to elevated pulmonary circulatory pressures through changes in elastic artery structure. J Appl Physiol. 2008;105:1610–1619. doi: 10.1152/japplphysiol.90563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morise T., Takeuchi Y., Takeda R., Karayalcin U., Yachie A., Miyawaki T. Increased plasma endothelin levels in Kawasaki disease: a possible marker for Kawasaki disease. Angiology. 1993;44:719–723. doi: 10.1177/000331979304400908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Morphometric analysis of elastin-stained coronary arteries of adult mice 42 days after IgG, TGF-β Nab, LCWE plus IgG, or LCWE plus TGF-β Nab treatment. A: Quantification of the internal diameter of the cross sections of the coronary arteries in each group. B: Quantification of the number of visible breaks in the elastic lamina under ×40 magnification per 1000 μmol/L. Bars represent SEM (n = 6-13 sections counted per group). *P < 0.05 versus IgG. C: Quantification of the percentage of media staining for elastin versus the entire medial area using imaging software (Bioquant) in sections of the large coronary arteries in each group at 42 days. Bars represent SEM (n = 4-5 per group). *P < 0.05 versus IgG.
Quantitative PCR to detect the expression of matrix components at 7 days: collagen1-α1 (A), collagen1-α2 (B), collagen6-α1 (C), fibrillin-1 (D), laminin-α5 (E), and laminin-β2 (F). Bars represent SEM (n = 4-6 per group).