

Abstract
Chronic cholestatic liver injury induced by cholestasis in rodents is associated with hepatic fibrin deposition, and we found evidence of fibrin deposition in livers of patients with cholestasis. Key components of the fibrinolytic pathway modulate cholestatic liver injury by regulating activation of hepatocyte growth factor. However, the exact role of hepatic fibrin deposition in chronic cholestasis is not known. We tested the hypothesis that fibrinogen (Fbg) deficiency worsens liver injury induced by cholestasis. Fbg-deficient mice (Fbgα−/− mice) and heterozygous control mice (Fbgα+/− mice) were fed either the control diet or a diet containing 0.025% α-naphthylisothiocyanate (ANIT), which selectively injures bile duct epithelial cells in the liver, for 2 weeks. Hepatic fibrin and collagen deposits were evident in livers of heterozygous control mice fed the ANIT diet. Complete Fbg deficiency was associated with elevated serum bile acids, periportal necrosis, and increased serum alanine aminotransferase activity in mice fed the ANIT diet. Fbg deficiency was associated with enhanced hepatic expression of the transcription factor early growth response-1 (Egr-1) and enhanced induction of genes encoding the Egr-1–regulated proinflammatory chemokines monocyte chemotactic protein-1, KC growth-regulated protein, and macrophage inflammatory protein-2. Interestingly, peribiliary collagen deposition was not evident near necrotic areas in Fbg-deficient mice. The results suggest that in this model of chronic cholestasis, fibrin constrains the release of bile constituents from injured intrahepatic bile ducts, thereby limiting the progression of hepatic inflammation and hepatocellular injury.
Bile ducts serve as an intrahepatic conduit for the transport of toxic, but physiologically important, molecules in bile to the gallbladder for subsequent distribution to the intestine. Injury to the biliary epithelium disrupts bile flow (ie, cholestasis), and high levels of hydrophobic bile acids spill into the liver sinusoids, where these molecules induce cell injury and promote inflammation.1,2 The initial hepatic response to biliary injury is a compensatory response aimed at preventing the release of toxic bile acids and minimizing exposure of hepatocytes to these cytotoxic mediators. This response includes bile duct proliferation,3 altered expression of bile acid transporters,4,5 and the peribiliary deposition of extracellular matrix (eg, collagens). This last process is exaggerated under chronic conditions, leading to extensive liver fibrosis that compromises liver function.6
Chronic intrahepatic cholestasis can be modeled in mice by feeding a diet containing the xenobiotic α-naphthylisothiocyanate (ANIT), which through selective metabolism and transport achieves toxic levels in the bile, which is required to induce cholestatic liver injury.7,8 This results in a chronic, low-level cytotoxic challenge to the intrahepatic biliary epithelium, causing the release of bile acids and eliciting bile duct epithelial cell proliferation, inflammation, and peribiliary fibrosis.9–12 Of importance, compared with mice given a single, large dose of ANIT,8 hepatocellular injury in the ANIT diet model of cholestasis is mild, most likely as a result of the compensatory changes previously described.
Notable alterations in coagulation and fibrinolysis accompany the progression of chronic cholestatic liver disease.13–15 Similarly, coagulation cascade activation occurs in mice subjected to bile duct ligation (BDL),16 a model of obstructive cholestasis, and in mice give a large dose of ANIT,17 which models massive intrahepatic bile duct epithelial cell injury. Several studies indicate that plasminogen activator inhibitor-1 (PAI-1) expression increases after BDL and contributes to the progression of obstructive cholestasis in mice.16,18 However, PAI-1 deficiency did not affect plasmin activity in livers of mice after BDL,19 suggesting that the role of PAI-1 in this model of liver disease is not mediated by inhibiting fibrinolysis. Indeed, PAI-1 also reduces tissue plasminogen activator-dependent activation of hepatocyte growth factor c-Met signaling, an important hepatoprotective pathway during cholestasis.20 Despite these and other studies evaluating more broadly the role of coagulation and fibrinolysis in cholestasis, the exact role of fibrin in cholestatic liver injury is not known. To this end, we characterized the expression of fibrinogen (Fbg) and hepatic fibrin deposition in the ANIT diet model of intrahepatic cholestasis and used a genetic approach to determine the role of Fbg in the pathogenesis of chronic xenobiotic-induced cholestasis.
Materials and Methods
Mice
All studies were performed with female Fbg-deficient mice (Fbgα+/−) and heterozygous control mice (Fbgα−/−)21 mice between 8 and 16 weeks of age. These mice had been back-crossed six generations onto a C57Bl/6J background and were kindly provided by Dr. Jay Degen (Cincinnati Children's Hospital Medical Center, Cincinnati, OH). Mice were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facility at the University of Kansas Medical Center. Mice were housed at an ambient temperature of 22°C with alternating 12-hour light/dark cycles and were provided water and rodent chow ad libitum (Teklad 8604; Harlan, Indianapolis, IN) before feeding custom diets. All animal procedures were performed according to the guidelines of the American Association for Laboratory Animal Science and were approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee.
Diets and Sample Collection
Diets were prepared by Dyets Inc (Bethlehem, PA). The ANIT diet was an AIN-93M purified diet containing 0.025% ANIT (Sigma-Aldrich, St. Louis, MO). The control diet was the purified AIN-93M diet. Mice were fed the diets for 2 weeks and subsequently anesthetized with isoflurane, and blood was collected from the caudal vena cava into sodium citrate (final, 0.38%) or an empty syringe for the collection of plasma and serum, respectively. The left medial lobe of the liver was affixed to a cork with OCT and frozen for 3 minutes in chilled isopentane. Sections of the left lateral lobe were fixed in neutral-buffered formalin for 48 hours before routine processing. The remaining liver was flash-frozen in liquid nitrogen.
Determination of Serum Alanine Aminotransferase Activity, Bile Acids, and Fbg
Serum alanine aminotransferase (ALT) activity was determined using a commercial reagent (Thermo-Fisher, Waltham, MA). Total bile acids in serum were determined using a colorimetric assay (Bio-Quant, San Diego, CA). Plasma Fbg levels were determined using commercial reagents (Siemens Healthcare Diagnostics, Deerfield, IL) and a ST4 Coagulation Analyzer (Diagnostica-Stago, Parsippany, NJ).
RNA Isolation, cDNA Synthesis, and Real-Time PCR
Total RNA was isolated from 100 mg of snap-frozen liver using TRI Reagent (Molecular Research Center, Cincinnati, OH); 1 μg of total RNA was used for cDNA synthesis, accomplished using a high-capacity cDNA synthesis kit (Applied Biosystems, Foster City, CA). Real-time PCR analysis was performed using a StepOnePlus instrument (Applied Biosystems) and 2X Taqman Master Mix and Taqman gene expression assays (Applied Biosystems). The levels of each gene were adjusted to the levels of 18S RNA and the relative levels of each gene evaluated using the ΔΔCt method in which Fbg+/− mice were the comparator.
Immunofluorescent Staining of Mouse Tissues and Fibrin Western Blotting
Fibrin western blotting was performed using the mouse anti-human fibrin (59D8) monoclonal antibody22 (kindly provided by Dr. Charles Esmon, Oklahoma Medical Research Foundation), as described previously.17 For all immunofluorescent staining, 5-μm frozen sections obtained from livers frozen in isopentane/OCT were used. For Fbg and cytokeratin 19 (CK19) staining, sections were fixed in 4% neutral-buffered formalin containing 2% acetic acid and subsequently blocked with 10% goat serum in PBS for 1 hour at room temperature. The sections were then incubated with primary antibody (rabbit anti-human Fbg; Dako, Carpinteria, CA) diluted 1:5000 in block buffer overnight at 4°C. The sections were then washed with PBS and incubated with the monoclonal rat anti-mouse CK19 antibody (clone, TROMA-3). The TROMA-3 antibody developed by Kemler was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by Department of Biology, The University of Iowa, Iowa City, IA.23 The sections were incubated with Alexa-conjugated secondary antibodies (Invitrogen, Carlsbad, CA), washed with PBS, counterstained with DAPI, and coverslipped. The area of positive CK19 staining was determined in 10 randomly selected ×100 magnification images using Scion Image Software. Neutrophil immunohistochemistry was performed as we have described previously,24 with the substitution of ImmPACT NovaRed HRP substrate (Vector Laboratories, Burlingame, CA). For each tissue, the number of neutrophils per 15 randomly selected 200× fields was determined and the average identified. Early growth response-1 (Egr-1) immunostaining was performed as we have described previously.25 For co-staining sections for macrophage antigens and Egr-1, slides were incubated with primary antibodies, rabbit anti-Egr-1 (Cell Signaling Technology, Danvers, MA), and rat anti-CD68 and rat anti-F4/80 antibodies (BioLegend, La Jolla, CA), each diluted 1:100. Sections were then incubated with secondary antibodies, Alexa 488–conjugated goat anti-rabbit IgG and Alexa 594–conjugated goat anti-rat IgG, each diluted 1:500. For all staining, the slides were then washed with PBS after secondary antibody addition, and fluorescent staining in the livers sections was visualized using an Olympus BX41 microscope (Olympus, Lake Success, NY).
Histopathology and Trichrome Staining
Paraffin-embedded liver sections were cut at 5 μm and stained with H&E for analysis of liver histopathology. Formalin-fixed sections were also stained using a Chromaview Gomori Trichrome staining kit (Thermo-Fisher) as described by the manufacturer's protocol. Liver necrosis and periportal fibrosis in the mouse livers were evaluated by a pathologist with mouse experience (G.H.C.).
Fibrin Staining in Human Liver Samples
Research involving human livers was reviewed by the University of Kansas Medical Center Human Research Protection Program, and the use of deidentified human liver samples was approved. The specimens used in this study were collected by and provided by the KU Liver Center Tissue Bank at The University of Kansas Medical Center. Diseased liver tissue used was from patients with primary biliary cirrhosis (PBC) (2 women, ages 60 and 61 years; 1 man, age 47 years) and patients with primary sclerosing cholangitis (2 men, ages 45 and 55 years; 1 woman, age 70 years). Control liver sections were obtained from liver tissue taken from donor livers before transplantation. The 5-μm frozen sections were fixed for 10 minutes in 4% neutral-buffered formalin, washed with PBS, then blocked for 1 hour in 10% rabbit serum. Sections were then incubated with primary antibodies, goat anti-human CK19 (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:100, and the mouse anti-human fibrin (59D8) monoclonal antibody22 at a concentration of 20 μg/ml in block buffer, overnight at 4°C. Sections were subsequently washed with PBS and then incubated with secondary Alexa 594–conjugated rabbit anti-goat and Alexa 488–conjugated rabbit anti-mouse secondary antibodies, each diluted at 1:500 in block buffer for 1 hour at room temperature. Sections were washed with PBS, counterstained with DAPI, and visualized as described for mouse tissues.
Statistical Analyses
Comparison of two groups was performed using Student's t-test. Comparison of three or more groups was performed using one- or two-way analysis of variance, as appropriate, and the Student-Newman-Keuls post hoc test. The criterion for statistical significance was P < 0.05.
Results
Fbg Expression and Hepatic Fibrin Deposition in Mice Fed the ANIT Diet and Patients with Cholestatic Liver Disease
Compared with Fbg+/− mice fed control diet, the expression of Fbgα, Fbgβ, and Fbgγ mRNAs was not significantly affected by the ANIT diet (Figure 1, A–C). In agreement, plasma Fbg levels were not significantly different in Fbg+/− mice fed the ANIT diet compared with Fbg+/− mice fed the control diet. As anticipated, Fbgα mRNA in liver and Fbg protein in plasma were not detected in Fbg−/− mice (Figure 1, C and D). No fibrin staining was evident in the livers of Fbg−/− mice fed either the control diet or ANIT diet (Figure 2, B and D), confirming specificity of this established protocol.26 Compared with Fbg+/− mice fed the control diet (Figure 2A), marked fibrin deposition occurred in Fbg+/− mice fed the ANIT diet (red, Figure 2C). The fibrin was localized around intrahepatic bile ducts, as indicated by CK19 staining, a selective marker of bile duct epithelial cells in the adult mouse liver (green, Figure 2). Determination of hepatic fibrin levels by Western blotting confirmed significant fibrin deposition in livers from Fbg+/− mice fed the ANIT diet, with minimal fibrin evident in livers from mice fed the control diet (Figure 2E).
Figure 1.
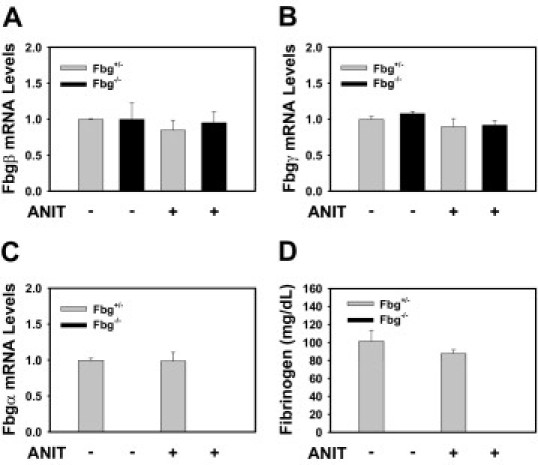
Fbg expression in mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either control diet or identical diet containing 0.025% ANIT for 14 days. Hepatic levels of Fbgβ (A), Fbgγ (B), and Fbgα (C) mRNAs were determined using real-time PCR. D: Plasma Fbg levels were determined as described in Materials and Methods. Data are expressed as mean ± SEM; n = 3 to 7 mice per group.
Figure 2.
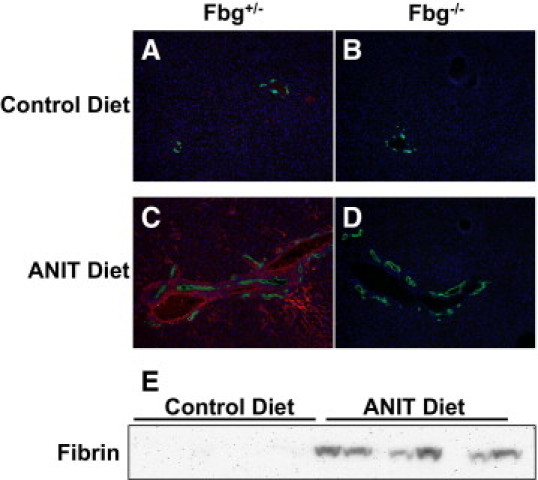
Peribiliary deposition of fibrin in mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either the control diet or an identical diet containing 0.025% ANIT for 14 days. A–D: Representative photomicrographs showing liver sections co-stained for fibrin (red) and CK19 (green), a marker of bile ducts. E: Representative Western blot showing increased fibrin in livers from control mice fed the ANIT diet.
To evaluate whether fibrin deposition increased in livers of patients with cholestatic liver disease, we stained liver sections using the mouse anti-human fibrin antibody 59D8, which specifically detects fibrin generated by thrombin cleavage of Fbg.22 Compared with control livers (see Supplemental Figure S1, A and B, at http://ajp.amjpathol.org), deposition of fibrin was increased in livers from patients with primary sclerosing cholangitis (see Supplemental Figure S1, C and D, at http://ajp.amjpathol.org) and PBC (see Supplemental Figure S1, E and F, at http://ajp.amjpathol.org). Of importance, detection of fibrin using this antibody indicates the presence of local thrombin generation in the liver sufficient to cause fibrin deposition.
Fbg Deficiency Worsens Cholestatic Liver Injury in Mice Fed the ANIT Diet
Periportal necrosis triggered by bile duct injury manifests initially as a consequence of large amounts of hydrophobic bile acids spilling into the parenchyma. Serum bile acid levels increased significantly in Fbg+/− mice fed the ANIT diet, and this increase was larger in Fbg−/− mice fed the ANIT diet (Figure 3A). Serum ALT activity, a biomarker of hepatocyte injury, was not affected by Fbg deficiency in mice fed the control diet (Figure 3B). Serum ALT activity increased to a greater extent in Fbg−/− mice compared with Fbg+/− mice fed the ANIT diet (Figure 3B). Fbg deficiency did not significantly affect liver histology in mice fed the control diet (Figure 3, C and D). In contrast to Fbg+/− mice fed the ANIT diet, periportal hepatocyte necrosis was evident in Fbg−/− mice fed the ANIT diet (Figure 3, E and F). Specifically, of the mice fed the ANIT diet, five of six Fbg−/− mice had grade 2 hepatocyte necrosis (mild, multifocal) and one had no necrosis; in contrast, two of six Fbg+/− mice had grade 1 hepatocyte necrosis (minimal, focal, or at most two foci) and four of six Fbg+/− mice had no necrosis. Although there was evidence of red blood cell accumulation within areas of necrosis, no obvious parenchymal hemorrhage or peliosis was evident in the livers of Fbg−/− mice fed the ANIT diet.
Figure 3.
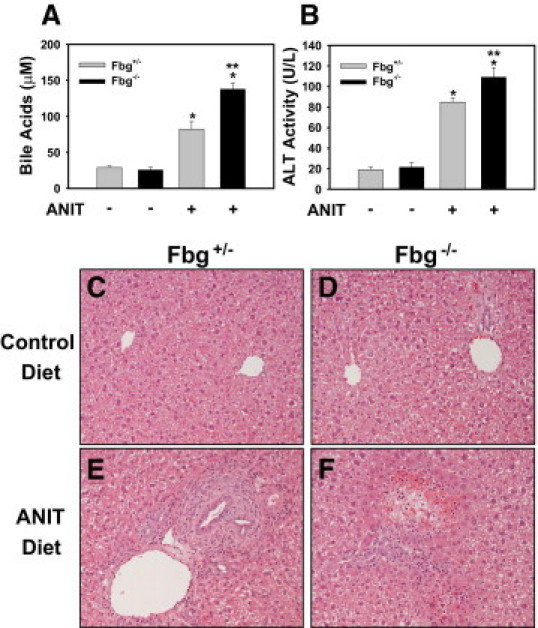
Effect of complete fibrin deficiency on cholestatic liver injury in mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either the control diet or an identical diet containing 0.025% ANIT for 14 days. Serum bile acid levels (A) and ALT activity (B) were determined as described in Materials and Methods. Data are expressed as mean ± SEM; n = 3 to 7 mice per group. *P < 0.05 when compared with the same genotype fed the control diet. **P < 0.05 when compared with Fbg+/− mice fed the ANIT diet. C–F: Representative photomicrographs showing H&E-stained liver sections. C and D: Fbg+/− and Fbg−/− mice, respectively, fed the control diet. E and F: Fbg+/− and Fbg−/− mice, respectively, fed the ANIT diet.
Biliary Hyperplasia in Fbg-Deficient Mice Fed the ANIT Diet
Compensatory biliary hyperplasia occurs in the livers of mice fed the ANIT diet. If there was a failure of bile duct epithelial cell proliferation that could account for increased cholestasis in the Fbg−/− mice. However, the area of CK19-positive staining in the livers of Fbg−/− mice fed the ANIT diet was not significantly reduced compared with Fbg+/− mice fed the ANIT diet (Figure 4, A–E). Similarly, evaluation of liver histopathology revealed a similar extent of biliary hyperplasia in Fbg+/− mice and Fbg−/− mice fed the ANIT diet (Figure 3).
Figure 4.
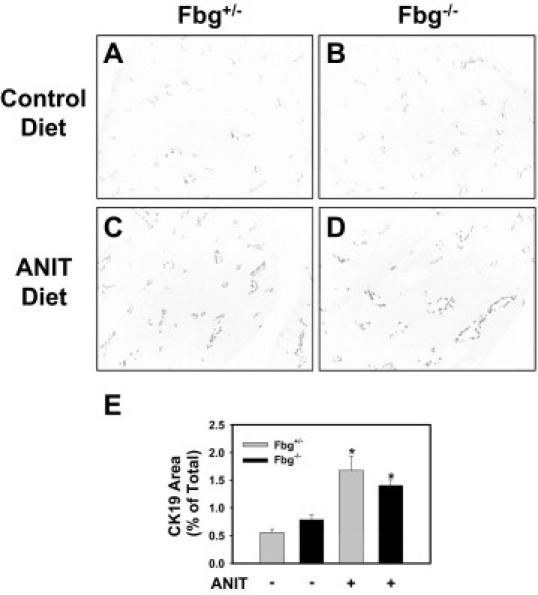
Effect of complete fibrin deficiency on biliary hyperplasia in mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either the control diet or an identical diet containing 0.025% ANIT for 14 days. A–D: Representative photomicrographs showing CK19 staining in liver sections. Images were converted to grayscale and inverted such that CK19 staining is dark. E: Quantification of CK19 staining area as described in Materials and Methods. Data are expressed as mean ± SEM; n = 3 to 7 mice per group. *P < 0.05 when compared with the same genotype fed the control diet.
Fbg Deficiency Enhances the Expression of Egr-1 in Mice Fed the ANIT Diet
Bile acids increase the expression of Egr-1 mRNA and protein in hepatocytes.27 Insofar as we suspected increased release of bile acids into the parenchyma of the livers of Fbg−/− mice, we examined hepatic Egr-1 expression. Egr-1 mRNA levels were not induced in Fbg+/− mice fed the ANIT diet compared with Fbg+/− mice fed the control diet (Figure 5A), although immunofluorescent staining revealed infrequent Egr-1 nuclear staining in periportal regions of the livers of Fbg+/− mice fed the ANIT diet (Figure 5D). Interestingly, Egr-1 mRNA levels increased significantly in livers of Fbg−/− mice fed the ANIT diet compared with Fbg−/− mice fed the control diet (Figure 5A). This was associated with a marked increase in periportal Egr-1 nuclear staining (arrow) and nearby Egr-1 expression in the hepatic parenchyma (asterisk) (Figure 5E). In other models of liver disease, Egr-1 expression by macrophages could contribute to the inflammatory response by inducing genes such as tumor necrosis factor-α.28 Hepatic tumor necrosis factor-α mRNA levels were not increased significantly in mice fed the ANIT diet (data not shown). Co-staining of Egr-1 and macrophage antigens (F4/80 and CD68) in livers of Fbg−/− mice revealed scattered Egr-1–positive macrophages (see Supplemental Figure S2, arrow, at http://ajp.amjpathol.org). Most Egr-1 staining appeared to be in hepatocytes. These results are consistent with the hypothesis that the increased release of bile acids in Fbg−/− mice fed the ANIT diet increases the expression of Egr-1.
Figure 5.
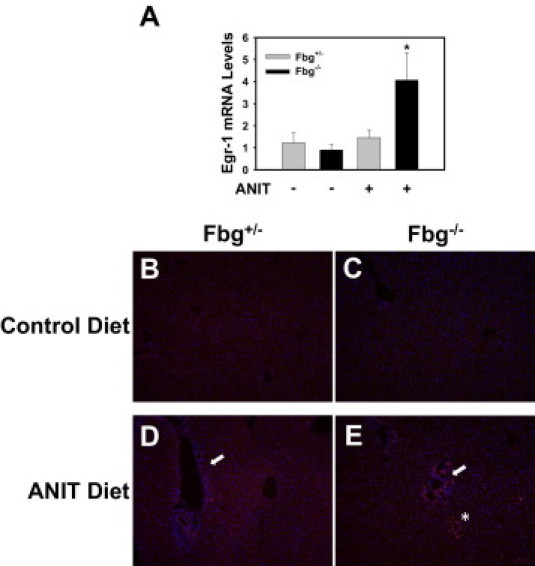
Effect of complete fibrin deficiency on Egr-1 expression in livers of mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either the control diet or an identical diet containing 0.025% ANIT for 14 days. A: Levels of Egr-1 mRNA in liver were determined using real-time PCR. Data are expressed as mean ± SEM; n = 3 to 7 mice per group. *P < 0.05 when compared with the same genotype fed the control diet, as well as when compared to the Fbg+/− mice fed the ANIT diet. B–E: Representative photomicrographs showing liver sections stained for Egr-1 (red) and counterstained with DAPI (blue). Positive Egr-1 staining near bile ducts (arrows). Positive Egr-1 staining in the hepatic parenchyma near the portal area (asterisk).
Fbg Deficiency Enhances Proinflammatory Gene Expression in Livers of Mice Fed the ANIT Diet
Once induced by bile acids in hepatocytes, Egr-1 acts as a transcriptional activator of several inflammatory genes, including those encoding the KC growth-regulated protein-α, monocyte chemoattractant protein-1, macrophage inflammatory protein-2, and intercellular adhesion molecule-1.25 Expression of the mRNAs encoded by each of these genes increased significantly in the livers of Fbg+/− mice fed the ANIT diet (Figure 6, A-D). Hepatic KC growth-regulated protein-α, monocyte chemoattractant protein-1, and macrophage inflammatory protein-2 mRNA levels were significantly higher in Fbg−/− mice fed the ANIT diet compared with the Fbg+/− mice fed the ANIT diet (Figure 6, A-C). Although a similar trend was observed for intercellular adhesion molecule-1, this difference did not achieve statistical significance (Figure 6D).
Figure 6.
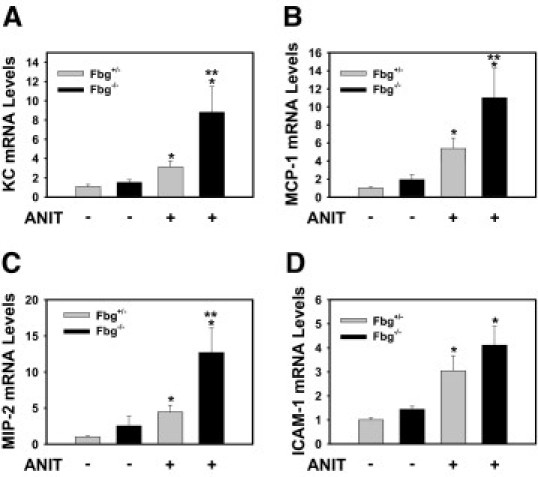
Effect of complete fibrin deficiency on hepatic inflammatory gene induction in mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either the control diet or an identical diet containing 0.025% ANIT for 14 days. Levels of KC (A), monocyte chemoattractant protein-1 (B), macrophage inflammatory protein-2 (C), and intercellular adhesion molecule-1 (D) mRNA in liver were determined using real-time PCR. Data are expressed as mean ± SEM; n = 3 to 7 mice per group. *P < 0.05 when compared with the same genotype fed the control diet. **P < 0.05 when compared with Fbg+/− mice fed the ANIT diet.
Enhanced Hepatic Neutrophil Accumulation in Livers of Fbg−/− Mice Fed the ANIT Diet
A previous study found that deficiency in CXCR2, the primary receptor for macrophage inflammatory protein-2 and KC growth-regulated protein-α in mice, significantly reduced neutrophil accumulation in the livers of mice exposed to ANIT.12 To this end, we determined whether the enhanced expression of these chemokines was associated with enhanced neutrophil accumulation in Fbg−/− mice fed the ANIT diet. Scattered neutrophils were observed in the livers of Fbg+/− and Fbg−/− mice fed the control diet (Figure 7, A and B). Compared with mice fed the control diet, increased neutrophil accumulation was observed in the livers of Fbg+/− mice fed the ANIT diet, primarily adjacent to bile ducts (Figure 7, C and E). The number of neutrophils infiltrating livers of Fbg−/− mice was significantly higher compared with Fbg+/− mice (Figure 7E). Neutrophil accumulation was more evident in periportal areas in Fbg−/− mice fed the ANIT diet (Figure 7D), and focal neutrophil accumulation within regions of hepatic parenchymal cells was also evident in the livers of Fbg−/− mice fed the ANIT diet (Figure 7D).
Figure 7.
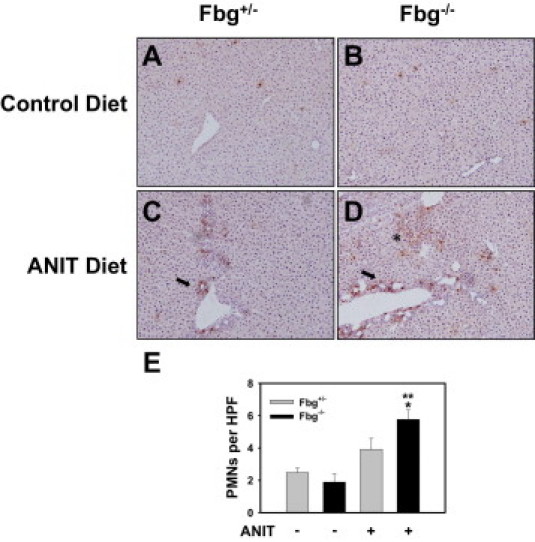
Effect of complete fibrin deficiency on hepatic neutrophil accumulation in mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either the control diet or an identical diet containing 0.025% ANIT for 14 days. Representative photomicrographs showing liver sections stained for neutrophils (A–D) and the quantification of hepatic neutrophil (PMN) accumulation (E) as described in Materials and Methods. Data are expressed as mean ± SEM; n = 3 to 7 mice per group. *P < 0.05 when compared with the same genotype fed the control diet. **P < 0.05 when compared with Fbg+/− mice fed the ANIT diet. Neutrophil accumulation around bile ducts (arrow). Neutrophil accumulation within areas of necrosis (asterisk in D).
Effect of Fbg Deficiency on Collagen Deposition in Mice Fed the ANIT Diet
The levels of Col1α1 and transforming growth factor-β mRNA increased to a similar extent in the livers of Fbg+/− mice and Fbg−/− mice fed the ANIT diet compared with each genotype fed the control diet (Figure 8, A and B). In agreement with this finding, evaluation of type 1 collagen staining in low-magnification photomicrographs by morphometry did not reveal a significant difference in overall hepatic collagen deposition (data not shown). Of interest, examination of Trichrome-stained liver sections revealed an interesting phenomenon. Compared with Fbg+/− mice fed the control diet, peribiliary collagen deposition, but not necrosis, occurred in Fbg+/− mice fed the ANIT diet (Figure 8C). However, in the livers of Fbg−/− mice with moderate necrosis (grade 2), collagen deposition was substantially reduced in areas of biliary hyperplasia proximal to necrotic areas (Figure 8D).
Figure 8.
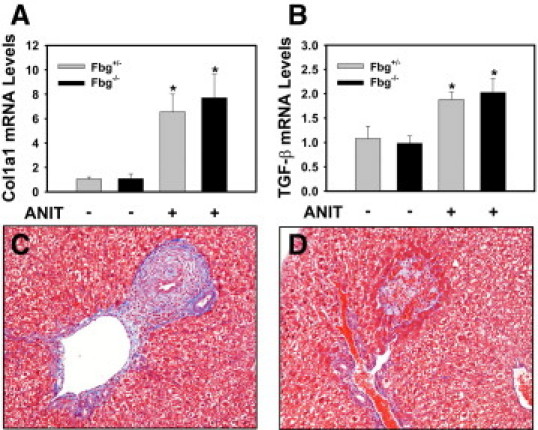
Effect of complete fibrin deficiency on liver fibrosis in mice fed the ANIT diet. Mice heterozygous for Fbgα (Fbg+/−) and mice null for Fbgα (Fbg−/−) were fed either the control diet or an identical diet containing 0.025% ANIT for 14 days. Levels of Col1α1 (A) and transforming growth factor-β (B) mRNA in liver were determined using real-time PCR. Data are expressed as mean ± SEM; n = 3 to 7 mice per group. *P < 0.05 when compared with the same genotype fed the control diet. Representative photomicrographs showing Trichrome-stained liver sections from Fbg+/− mice (C) and Fbg−/− mice (D) fed the ANIT diet.
Discussion
Fibrin deposition in the liver occurs in models of acute and chronic cholestatic liver injury in rodents.17,20 However, few studies have determined the exact role of fibrin in cholestatic liver injury. Inhibition of coagulation cascade activation often reduces both hepatic injury and fibrin deposition in models of liver injury,26,29–31 leaving many to speculate that fibrin deposition is pathological. However, the protection conferred by anticoagulation could relate to inhibition of protease-activated receptor-1 signaling.32–34 For nearly all models of acute and chronic liver injury, it remains unclear whether fibrin is damaging or protective. One study found that plasma Fbg levels in patients with PBC were within the normal range.13 Fibrin deposition was evident in livers of patients with PBC and primary sclerosing cholangitis (see Supplemental Figure S1 at http://ajp.amjpathol.org), a novel finding indicating the presence of active thrombin in the livers of patients with these diseases and consistent with increased thrombin-antithrombin levels in the plasma of patients with cholestatic liver disease.13 We found that fibrin deposits also increased in the livers of mice fed an ANIT diet. Fbg deficiency worsened cholestatic liver injury in this model, suggesting a novel mechanism of hepatoprotection mediated by the coagulation cascade.
The mechanism responsible for worsened injury in Fbg-deficient mice fed the ANIT diet is not known. The pattern of hepatic fibrin deposition in rodent models and humans during cholestasis could yield clues about a mechanism whereby fibrin limits liver injury. Deposition of fibrin near bile ducts in the livers of mice fed the ANIT diet and in patients with cholestatic liver disease is consistent with a hypothesis that fibrin acts to prevent spillage of toxic bile into the liver parenchyma, analogous to the role of fibrin in limiting hemorrhage after vascular injury. Sinusoidal fibrin was also evident in the mouse model and patients with cholestasis, and this fibrin could also function to “seal off” areas of liver containing toxic bile acids. Of importance, the pattern of fibrin alone is insufficient to validate this hypothesis, and additional experiments are warranted. One mechanism whereby fibrin could protect the liver is by forming a preliminary matrix to support protective collagen deposition around the damaged bile ducts.35 Indeed, a lack of collagen deposition around bile ducts in Fbg−/− mice was associated with periportal necrosis. Fibrin could also stabilize damaged bile ducts by interacting with integrins (eg, αVβ3).36 Expression of the β3 integrin in the liver is low,37 although the cellular distribution of this integrin subunit in the liver has not been characterized in detail. Interestingly, a previous study showed that a pharmacological inhibitor of αVβ3 (and αVβ5) worsened fibrosis after BDL, although the inhibitor did not affect serum ALT activity in that model.38 To this end, additional experiments evaluating interactions between β3 integrin and fibrin in the ANIT diet model of cholestasis could be revealing.
The proinflammatory effects of bile acids in models of cholestasis, including chemokine up-regulation and hepatic neutrophil accumulation, are mediated in part by the transcription factor Egr-1.25 Exposure of hepatocytes to bile acids increases Egr-1 mRNA and protein,27 and Egr-1 expression increased in the livers of mice after BDL.25 Unlike in BDL, Egr-1 expression was not induced in the livers of Fbg+/− mice fed the ANIT diet. However, compared with the ANIT diet, serum bile acid levels in mice subjected to BDL achieve much higher levels.39 It is intriguing that Egr-1 induction and Egr-1–dependent proinflammatory events, such as exaggerated neutrophil accumulation, occurred selectively in Fbg-deficient mice fed the ANIT diet. This could relate to excessive release of bile acids into the hepatic parenchyma. Indeed, most Egr-1 staining was observed in hepatic parenchymal cells proximal to bile ducts. Occasional Egr-1–positive macrophages (ie, Kupffer cells) were observed in the livers of Fbg−/− mice. Kupffer cells have been shown to express the bile acid receptor TGR5.40 However, treatment of monocytes and macrophages with bile acids inhibited cytokine production,41 and Kupffer cell function is impaired during cholestasis.42 This suggests that bile acid induction of Egr-1, most likely in hepatocytes but not Kupffer cells, contributes to enhanced inflammatory gene induction in Fbg−/− mice fed the ANIT diet.
The role of fibrin in cholestasis may depend on the degree of insult to the intrahepatic bile ducts. In the long-term ANIT diet model, hepatocellular injury is minimal (ie, bile infarcts are infrequent). To this end, this model provides the opportunity to evaluate endogenous mechanisms that limit modest cholestatic liver injury. Results from the ANIT diet model indicate that Fbg deficiency worsens cholestatic liver injury. However, the role of fibrin may be different in other more severe models of cholestatic liver injury, such as BDL or administration of a large, hepatotoxic dose of ANIT. In the latter two models, substantial periportal necrosis is evident, and these damaged areas are filled with dense fibrin.16,17,20 It is possible that this fibrin is generated as a consequence of the hepatocellular injury. Indeed, necrosis and fibrin deposition were reduced in livers of PAI-1−/− mice after BDL, although activity of the fibrinolytic enzyme plasmin was unaltered.16,19 Of interest, we found that liver necrosis caused by a single hepatotoxic dose of ANIT (60 mg/kg, p.o.) was reduced in Fbg−/− mice,43 suggesting that fibrin may contribute to hepatocyte injury when the cholestatic insult is more severe. In the livers of patients with PBC and primary sclerosing cholangitis, fibrin was evident near bile ducts and in the hepatic parenchyma and deposited within bile infarcts. It is not possible to conclude that this fibrin is protective or damaging in these diseases in humans. However, evidence of fibrin deposition in several rodent models and the demonstration of both protective and damaging effects of fibrin during cholestasis support the need for further studies.
In summary, we found that fibrin deposition occurred in the livers of mice fed the ANIT diet and that Fbg deficiency worsened hepatocellular injury in this model. Expression of the transcription factor Egr-1 and induction of several inflammatory genes were also increased in the Fbg-deficient mice. Taken together, the results suggest that Fbg deficiency worsens chronic ANIT-induced inflammation and cholestatic liver injury in mice. Indicative of a local procoagulant response, fibrin deposition was increased in the livers of patients with cholestatic liver disease. Indeed, abnormalities in coagulation accompanying the progression of liver disease in humans are receiving considerable attention, and ongoing studies have revealed that changes in coagulation and fibrinolysis in liver disease extend beyond the reduced capacity of hepatocytes to produce coagulation factors.44 The novel finding that fibrin could have hepatoprotective effects during cholestasis further supports the need for additional studies evaluating coagulation and fibrinolysis in cholestatic liver disease.
Acknowledgments
We acknowledge Bradley P. Sullivan for critical reading of the manuscript and Mr. Ruipeng Wang for outstanding technical support. We thank Erik Schadde, M.D., Richard Gilroy, M.D., Bashar Abdulkarim, M.D., Ph.D., Jameson Forster, M.D., Mojtaba Olyaee, M.D., and Atta M. Nawabi, M.D., for assistance in collecting human liver specimens used in this study. We thank Natali Navarro-Cazarez, Marsha Danley, and Dr. Ossama Tawfik for help with procuring and preparation of frozen liver sections.
Footnotes
Supported by grants from the National Institutes of Health (R01 ES017537 to J.P.L., DK073566 to B.L.C., and COBRE P20 RR021940).
Supplemental material for this article can be found on http://ajp.amjpathol.org or doi: 10.1016/j.ajpath.2010.11.064.
Supplementary data
Detection of fibrin in the livers of patients with cholestatic liver disease. Representative photomicrographs showing liver sections stained for fibrin (green) and CK19 (red) and counterstained with DAPI from control livers (A-B), livers from patients with PSC (C-D), and livers from patients with PBC (E-F). Bar = 10 μm.
Co-localization of Egr-1 and macrophage antigens in the livers of Fbg−/− mice fed the ANIT diet. Mice null for Fbgα (Fbg−/−) were fed a diet containing 0.025% ANIT for 14 days. Representative photomicrographs from two different liver sections (A-C, section 1; D-F, section 2) stained for Egr-1 (green) and macrophage antigens CD68 and F4/80 (red) and counterstained for DAPI. Bar = 50 μm. Positive Egr-1 staining co-localized with macrophage marker in a few cells (yellow) (arrow). *Positive Egr-1 staining consistent with hepatocytes in many cells (green).
References
- 1.Perez M.J., Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–1689. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia X., Demorrow S., Francis H., Glaser S., Alpini G., Marzioni M., Fava G., Lesage G. Cholangiocyte injury and ductopenic syndromes. Semin Liver Dis. 2007;27:401–412. doi: 10.1055/s-2007-991516. [DOI] [PubMed] [Google Scholar]
- 3.Alvaro D., Gigliozzi A., Attili A.F. Regulation and deregulation of cholangiocyte proliferation. J Hepatol. 2000;33:333–340. doi: 10.1016/s0168-8278(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 4.Pauli-Magnus C., Meier P.J. Hepatobiliary transporters and drug-induced cholestasis. Hepatology. 2006;44:778–787. doi: 10.1002/hep.21359. [DOI] [PubMed] [Google Scholar]
- 5.Lee J., Azzaroli F., Wang L., Soroka C.J., Gigliozzi A., Setchell K.D., Kramer W., Boyer J.L. Adaptive regulation of bile salt transporters in kidney and liver in obstructive cholestasis in the rat. Gastroenterology. 2001;121:1473–1484. doi: 10.1053/gast.2001.29608. [DOI] [PubMed] [Google Scholar]
- 6.Friedman S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietrich C.G., Ottenhoff R., de Waart D.R., Oude Elferink R.P. Role of MRP2 and GSH in intrahepatic cycling of toxins. Toxicology. 2001;167:73–81. doi: 10.1016/s0300-483x(01)00459-0. [DOI] [PubMed] [Google Scholar]
- 8.Becker B.A., Plaa G.L. The nature of alpha-naphthylisothiocyanate-induced cholestasis. Toxicol Appl Pharmacol. 1965;7:680–685. doi: 10.1016/0041-008x(65)90125-0. [DOI] [PubMed] [Google Scholar]
- 9.Moritoki Y., Ueno Y., Kanno N., Yamagiwa Y., Fukushima K., Gershwin M.E., Shimosegawa T. Lack of evidence that bone marrow cells contribute to cholangiocyte repopulation during experimental cholestatic ductal hyperplasia. Liver Int. 2006;26:457–466. doi: 10.1111/j.1478-3231.2006.01250.x. [DOI] [PubMed] [Google Scholar]
- 10.Lesage G., Glaser S., Ueno Y., Alvaro D., Baiocchi L., Kanno N., Phinizy J.L., Francis H., Alpini G. Regression of cholangiocyte proliferation after cessation of ANIT feeding is coupled with increased apoptosis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G182–G190. doi: 10.1152/ajpgi.2001.281.1.G182. [DOI] [PubMed] [Google Scholar]
- 11.Tjandra K., Sharkey K.A., Swain M.G. Progressive development of a Th1-type hepatic cytokine profile in rats with experimental cholangitis. Hepatology. 2000;31:280–290. doi: 10.1002/hep.510310204. [DOI] [PubMed] [Google Scholar]
- 12.Xu J., Lee G., Wang H., Vierling J.M., Maher J.J. Limited role for CXC chemokines in the pathogenesis of alpha-naphthylisothiocyanate-induced liver injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G734–G741. doi: 10.1152/ajpgi.00300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segal H., Cottam S., Potter D., Hunt B.J. Coagulation and fibrinolysis in primary biliary cirrhosis compared with other liver disease and during orthotopic liver transplantation. Hepatology. 1997;25:683–688. doi: 10.1002/hep.510250332. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Ari Z., Panagou M., Patch D., Bates S., Osman E., Pasi J., Burroughs A. Hypercoagulability in patients with primary biliary cirrhosis and primary sclerosing cholangitis evaluated by thrombelastography. J Hepatol. 1997;26:554–559. doi: 10.1016/s0168-8278(97)80420-5. [DOI] [PubMed] [Google Scholar]
- 15.Kloek J.J., Heger M., van der Gaag N.A., Beuers U., van Gulik T.M., Gouma D.J., Levi M. Effect of preoperative biliary drainage on coagulation and fibrinolysis in severe obstructive cholestasis. J Clin Gastroenterol. 2010;44:646–652. doi: 10.1097/MCG.0b013e3181ce5b36. [DOI] [PubMed] [Google Scholar]
- 16.Bergheim I., Guo L., Davis M.A., Duveau I., Arteel G.E. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J Pharmacol Exp Ther. 2006;316:592–600. doi: 10.1124/jpet.105.095042. [DOI] [PubMed] [Google Scholar]
- 17.Luyendyk J.P., Cantor G.H., Kirchhofer D., Mackman N., Copple B.L., Wang R. Tissue factor-dependent coagulation contributes to alpha-naphthylisothiocyanate-induced cholestatic liver injury in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G840–G849. doi: 10.1152/ajpgi.90639.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H., Vohra B.P., Zhang Y., Heuckeroth R.O. Transcriptional profiling after bile duct ligation identifies PAI-1 as a contributor to cholestatic injury in mice. Hepatology. 2005;42:1099–1108. doi: 10.1002/hep.20903. [DOI] [PubMed] [Google Scholar]
- 19.Wang H., Zhang Y., Heuckeroth R.O. PAI-1 deficiency reduces liver fibrosis after bile duct ligation in mice through activation of tPA. FEBS Lett. 2007;581:3098–3104. doi: 10.1016/j.febslet.2007.05.049. [DOI] [PubMed] [Google Scholar]
- 20.Wang H., Zhang Y., Heuckeroth R.O. Tissue-type plasminogen activator deficiency exacerbates cholestatic liver injury in mice. Hepatology. 2007;45:1527–1537. doi: 10.1002/hep.21613. [DOI] [PubMed] [Google Scholar]
- 21.Suh T.T., Holmbäck K., Jensen N.J., Daugherty C.C., Small K., Simon D.I., Potter S.S., Degen J.L. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 22.Weiler-Guettler H., Christie P.D., Beeler D.L., Healy A.M., Hancock W.W., Rayburn H., Edelberg J.M., Rosenberg R.D. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J Clin Invest. 1998;101:1983–1991. doi: 10.1172/JCI2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brulet P., Babinet C., Kemler R., Jacob F. Monoclonal antibodies against trophectoderm-specific markers during mouse blastocyst formation. Proc Natl Acad Sci U S A. 1980;77:4113–4117. doi: 10.1073/pnas.77.7.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luyendyk J.P., Sullivan B.P., Guo G.L., Wang R. Tissue factor deficiency and protease activated receptor-1 deficiency reduce inflammation elicited by diet-induced steatohepatitis in mice. Am J Pathol. 2010;176:177–186. doi: 10.2353/ajpath.2010.090672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim N.D., Moon J.O., Slitt A.L., Copple B.L. Early growth response factor-1 is critical for cholestatic liver injury. Toxicol Sci. 2006;90:586–595. doi: 10.1093/toxsci/kfj111. [DOI] [PubMed] [Google Scholar]
- 26.Copple B.L., Woolley B., Banes A., Ganey P.E., Roth R.A. Anticoagulants prevent monocrotaline-induced hepatic parenchymal cell injury but not endothelial cell injury in the rat. Toxicol Appl Pharmacol. 2002;180:186–196. doi: 10.1006/taap.2002.9394. [DOI] [PubMed] [Google Scholar]
- 27.Allen K., Kim N.D., Moon J.O., Copple B.L. Upregulation of early growth response factor-1 by bile acids requires mitogen-activated protein kinase signaling. Toxicol Appl Pharmacol. 2010;243:63–67. doi: 10.1016/j.taap.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L., Kishore R., McMullen M.R., Nagy L.E. Lipopolysaccharide stimulation of ERK1/2 increases TNF-alpha production via Egr-1. Am J Physiol Cell Physiol. 2002;282:C1205–C1211. doi: 10.1152/ajpcell.00511.2001. [DOI] [PubMed] [Google Scholar]
- 29.Ganey P.E., Luyendyk J.P., Newport S.W., Eagle T.M., Maddox J.F., Mackman N., Roth R.A. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46:1177–1186. doi: 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- 30.Pearson J.M., Schultze A.E., Schwartz K.A., Scott M.A., Davis J.M., Roth R.A. The thrombin inhibitor, hirudin, attenuates lipopolysaccharide-induced liver injury in the rat. J Pharmacol Exp Ther. 1996;278:378–383. [PubMed] [Google Scholar]
- 31.Abdel-Salam O.M., Baiuomy A.R., Ameen A., Hassan N.S. A study of unfractionated and low molecular weight heparins in a model of cholestatic liver injury in the rat. Pharmacol Res. 2005;51:59–67. doi: 10.1016/j.phrs.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Rullier A., Gillibert-Duplantier J., Costet P., Cubel G., Haurie V., Petibois C., Taras D., Dugot-Senant N., Deleris G., Bioulac-Sage P., Rosenbaum J. Protease-activated receptor 1 knockout reduces experimentally induced liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G226–G235. doi: 10.1152/ajpgi.00444.2007. [DOI] [PubMed] [Google Scholar]
- 33.Fiorucci S., Antonelli E., Distrutti E., Severino B., Fiorentina R., Baldoni M., Caliendo G., Santagada V., Morelli A., Cirino G. PAR1 antagonism protects against experimental liver fibrosis: role of proteinase receptors in stellate cell activation. Hepatology. 2004;39:365–375. doi: 10.1002/hep.20054. [DOI] [PubMed] [Google Scholar]
- 34.Copple B.L., Moulin F., Hanumegowda U.M., Ganey P.E., Roth R.A. Thrombin and protease-activated receptor-1 agonists promote lipopolysaccharide-induced hepatocellular injury in perfused livers. J Pharmacol Exp Ther. 2003;305:417–425. doi: 10.1124/jpet.102.046391. [DOI] [PubMed] [Google Scholar]
- 35.Neubauer K., Knittel T., Armbrust T., Ramadori G. Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury. Gastroenterology. 1995;108:1124–1135. doi: 10.1016/0016-5085(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y.P., O'Toole T.E., Leong L., Liu B.Q., az-Gonzalez F., Ginsberg M.H. Beta 3 integrin-mediated fibrin clot retraction by nucleated cells: differing behavior of alpha IIb beta 3 and alpha v beta 3. Blood. 1995;86:2606–2615. [PubMed] [Google Scholar]
- 37.Merono A., Lucena C., Lopez A., Garrido J.J., Perez de L.L., Llanes D. Immunohistochemical analysis of beta3 integrin (CD61): expression in pig tissues and human tumors. Histol Histopathol. 2002;17:347–352. doi: 10.14670/HH-17.347. [DOI] [PubMed] [Google Scholar]
- 38.Patsenker E., Popov Y., Stickel F., Schneider V., Ledermann M., Sagesser H., Niedobitek G., Goodman S.L., Schuppan D. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50:1501–1511. doi: 10.1002/hep.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinugasa T., Uchida K., Kadowaki M., Takase H., Nomura Y., Saito Y. Effect of bile duct ligation on bile acid metabolism in rats. J Lipid Res. 1981;22:201–207. [PubMed] [Google Scholar]
- 40.Keitel V., Donner M., Winandy S., Kubitz R., Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- 41.Calmus Y., Guechot J., Podevin P., Bonnefis M.T., Giboudeau J., Poupon R. Differential effects of chenodeoxycholic and ursodeoxycholic acids on interleukin 1, interleukin 6 and tumor necrosis factor-alpha production by monocytes. Hepatology. 1992;16:719–723. doi: 10.1002/hep.1840160317. [DOI] [PubMed] [Google Scholar]
- 42.Sung J.J., Go M.Y. Reversible Kupffer cell suppression in biliary obstruction is caused by hydrophobic bile acids. J Hepatol. 1999;30:413–418. doi: 10.1016/s0168-8278(99)80099-3. [DOI] [PubMed] [Google Scholar]
- 43.Luyendyk J.P., Mackman N., Sullivan B.P. Role of fibrinogen and protease activated receptors in acute xenobiotic-induced cholestatic liver injury. Toxicol Sci. 2011;119:233–243. doi: 10.1093/toxsci/kfq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lisman T., Caldwell S.H., Burroughs A.K., Northup P.G., Senzolo M., Stravitz R.T., Tripodi A., Trotter J.F., Valla D.C., Porte R.J. Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol. 2010;53:362–371. doi: 10.1016/j.jhep.2010.01.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detection of fibrin in the livers of patients with cholestatic liver disease. Representative photomicrographs showing liver sections stained for fibrin (green) and CK19 (red) and counterstained with DAPI from control livers (A-B), livers from patients with PSC (C-D), and livers from patients with PBC (E-F). Bar = 10 μm.
Co-localization of Egr-1 and macrophage antigens in the livers of Fbg−/− mice fed the ANIT diet. Mice null for Fbgα (Fbg−/−) were fed a diet containing 0.025% ANIT for 14 days. Representative photomicrographs from two different liver sections (A-C, section 1; D-F, section 2) stained for Egr-1 (green) and macrophage antigens CD68 and F4/80 (red) and counterstained for DAPI. Bar = 50 μm. Positive Egr-1 staining co-localized with macrophage marker in a few cells (yellow) (arrow). *Positive Egr-1 staining consistent with hepatocytes in many cells (green).