

Abstract
The identification of somatically acquired tumor mutations is increasingly important in the clinical management of cancer because the sensitivity of targeted drugs is related to the genetic makeup of individual tumors. Thus, mutational profiles of tumors can help prioritize anticancer therapy. We report herein the development and validation of two multiplexed assays designed to detect in DNA from FFPE tissue more than 40 recurrent mutations in nine genes relevant to existing and emerging targeted therapies in lung cancer. The platform involves two methods: a screen (SNaPshot) based on multiplex PCR, primer extension, and capillary electrophoresis that was designed to assess for 38 somatic mutations in eight genes (AKT1, BRAF, EGFR, KRAS, MEK1, NRAS, PIK3CA, and PTEN) and a PCR-based sizing assay that assesses for EGFR exon 19 deletions, EGFR exon 20 insertions, and HER2 exon 20 insertions. Both the SNaPshot and sizing assays can be performed rapidly, with minimal amounts of genetic material. Compared with direct sequencing, in which mutant DNA needs to compose 25% or more of the total DNA to easily detect a mutation, the SNaPshot and sizing assays can detect mutations in samples in which mutant DNA composes 1.56% to 12.5% and 1.56% to 6.25% of the total DNA, respectively. These robust, reliable, and relatively inexpensive assays should help accelerate adoption of a genotype-driven approach in the treatment of lung cancer.
In 2009, nearly 160,000 patients in the United States died of lung cancer, the country's leading cause of cancer-related death.1 Most patients with advanced lung cancer were treated empirically, based on clinical factors and appearance of tumor histological features. Although multiple genetic variants that “drive” lung tumorigenesis have been shown to influence treatment outcomes, mutation analysis of lung tumors has not yet become a part of standard clinical algorithms.
Most genetic alterations involving “driver mutations” have been uncovered in lung adenocarcinoma, a histological subtype of non–small-cell lung cancer (NSCLC). Driver mutations occur in genes that encode signaling proteins critical for cellular proliferation and survival. Approximately 50% of lung adenocarcinomas harbor such recurrent somatic oncogenic mutations in EGFR, HER2, KRAS, PIK3CA, BRAF, MEK1, and ALK. With the exception of PIK3CA mutations, a tumor with an alteration in one of these genes rarely has a mutation in one of the other genes. More importantly, mutations in EGFR, HER2, PIK3CA, BRAF, MEK1, and ALK have already been associated with increased sensitivity to specific kinase inhibitors.2–9 Other mutations, such as those involving AKT1 and PTEN, may influence responses to inhibitors of the EGFR/PI3K/AKT pathway.10,11 The EGFR mutations represent the best example of the benefits of a genotype-driven approach; treatment-naïve patients with metastatic EGFR-mutant tumors experience longer progression-free survival with an EGFR tyrosine kinase inhibitor, whereas patients whose tumors harbor wild-type EGFR have longer progression-free survival with chemotherapy.5
Conventionally, most molecular diagnostic laboratories have tested for mutations in tumor DNA derived from FFPE tissues one gene at a time. A major clinical challenge is prospectively determining the status of multiple clinically relevant genes in FFPE-derived tumor DNA before starting therapy. Further complicating matters, not all types of mutations are readily detected by the same method. For example, missense mutations can be found by one type of assay, but insertions/deletions that add/eliminate nucleotides in specific “hot spots” could be missed. Likewise, protocols to detect insertions/deletions are not optimal for point mutation detection.
We report herein the development and validation of two multiplexed assays designed to detect more than 40 recurrent mutations in nine genes relevant to targeted therapy in lung cancer. The first assay was devised to detect 38 different recurrent point mutations concurrently in eight genes (EGFR, KRAS, BRAF, NRAS, PIK3CA, MEK1, AKT1, and PTEN) and was adapted from a previously implemented genotyping platform designed for targeted mutational analysis of a broader set of tumor types.12 This assay uses technology (SNaPshot; Life Technologies/Applied Biosystem, Foster, CA) that involves multiplexed amplification of DNA targets by PCR with unlabeled oligonucleotide primers, multiplexed single-base primer extension with fluorescently labeled dideoxynucleotide triphosphate (ddNTP), and analysis of labeled primer-extension products by capillary electrophoresis.12 The second assay is a separate PCR-based sizing technique that simultaneously assesses tumors for recurrent insertions in EGFR and HER2 and deletions in EGFR that would not be comprehensively detected by the SNaPshot technique. Compared with direct sequencing, these assays offer higher analytical sensitivity and reduced complexity. They also provide a robust and accessible approach for the rapid identification of important mutations in lung cancer.
Materials and Methods
Cell Lines and Tumor Samples
Genomic DNA was derived from 35 lung cancer cell lines, 73 lung adenocarcinomas, and an additional 34 head and neck cancer cell lines. For cell lines, DNA was isolated using a kit (DNeasy; Qiagen Inc, Valencia, CA). For lung cancers, DNA from 24 specimens with greater than 70% tumor content was obtained from frozen samples using a genomic DNA purification kit (Wizard; Promega Corporation, Madison, WI); and DNA from 49 specimens with 35% to 85% tumor content was extracted from FFPE tissues using a kit (Qiaquick PCR Purification Kit; Qiagen Inc). All lung cancers were analyzed anonymously with informed consent and approval from the local institutional review board. Human male genomic DNA (Promega Corporation) was used as a wild-type control.
SNaPShot Assay
The SNaPShot technique involves multiplexed PCR, multiplexed single-base primer extension, and capillary electrophoresis. The PCR primers are listed in Table 1. An online primer design tool, “primer-BLAST” (National Center for Biotechnology Information), was used to construct primers with minimal chances of cross-reacting with homologous genes or pseudogenes. A search for known polymorphisms was performed for each sequence and accounted for in the design of all primers. Because DNA from FFPE samples is often degraded, PCR amplicons were designed to be approximately 100 bp. Single-base extension primers are listed in Table 2. Extension primers contain approximately 16 to 20 bp, with melting temperatures of higher than 50°C; and ddNTPs were used for primer extension. Random nucleotides (“GACT”) were added to extension primers to adjust their product sizes. Additional details of the rationale and methods used in primer design have been described.12 SNaPshot analysis was performed as previously described.12 Briefly, PCR primers were pooled together to amplify DNA using polymerase (platinum TaqDNA; Invitrogen Corporation, Carlsbad, CA) and the following conditions: 95°C for 8 minutes, followed by 95°C for 20 seconds, 58°C for 30 seconds, and 72°C for 1 minute (40 cycles), with a final extension of 72°C for 3 minutes. Next, corresponding polyacrylamide gel electrophoresis–purified primers were pooled together with a multiplex ready reaction mix (SNaPShot; Applied Biosystems) to perform multiplex extension reactions [96°C for 30 seconds, 96°C for 10 seconds, 50°C for 5 seconds, and 60°C for 30 seconds (for 25 cycles)]. Third, extension products were separated by capillary electrophoresis in an analyzer (ABI 3730) using standard conditions with the following reagents and parameters: dye set, Any5Dye; polymer, POP-7; matrix standard, DS-02; size standard, GeneScan-120 LIZ; injection time, 10 seconds; run time, 1200 seconds; and run voltage, 15 kV. Data were interpreted using computer software (ABI GeneMapper, version 4.0). The ability of the assay to detect all potential mutations was validated with cell lines or spiking primers that contained specific mutations. The latter are listed in Supplemental Table S1 at http://jmd.amjpathol.org.
Table 1.
List of PCR Primers for the SNaPshot Screen
| Name | Sequence |
|---|---|
| AKT1_ex2_a1 | 5′-GAGGGTCTGACGGGTAGAGT-3′ |
| AKT1_ex2_a2 | 5′-TCTTGAGGAGGAAGTAGCGT-3′ |
| BRAF_ex11_a1⁎ | 5′-TCTGTTTGGCTTGACTTGACTT-3′ |
| BRAF_ex11_a2⁎ | 5′-TCACCACATTACATACTTACCATGC-3′ |
| BRAF_ex15_a1⁎ | 5′-TGCTTGCTCTGATAGGAAAATG-3′ |
| BRAF_ex15_a2⁎ | 5′-CTGATGGGACCCACTCCAT-3′ |
| EGFR_ex18_a1⁎ | 5′-CCAACCAAGCTCTCTTGAGG-3′ |
| EGFR_ex18_a2⁎ | 5′-CCTTATACACCGTGCCGAAC-3′ |
| EGFR_ex20_a1⁎ | 5′-TGTTCCCGGACATAGTCCAG-3′ |
| EGFR_ex20_a2⁎ | 5′-ATCTGCCTCACCTCCACCGT-3′ |
| EGFR_ex21_a1⁎ | 5′-CCTCCTTCTGCATGGTATTC-3′ |
| EGFR_ex21_a2⁎ | 5′-GCAGCATGTCAAGATCACAG-3′ |
| KRAS_ex2_a1⁎ | 5′-TCATTATTTTTATTATAAGGCCTGCTG-3′ |
| KRAS_ex2_a2⁎ | 5′-AGAATGGTCCTGCACCAGTAA-3′ |
| KRAS_ex3_a1⁎ | 5′-AATTGATGGAGAAACCTGTCTCTTG-3′ |
| KRAS_ex3_a2⁎ | 5′-TGGTCCCTCATTGCACTGTA-3′ |
| MEK1_ex2_a1 | 5′-AGCGAAAGCGCCTTGAGGCCTT-3′ |
| MEK1_ex2_a2 | 5′-AACACCACACCGCCATTGCCAG-3′ |
| NRAS_ex3_a1 | 5′-ATAGATGGTGAAACCTGTTTGTTGG-3′ |
| NRAS_ex3_a2 | 5′-TGTATTGGTCTCTCATGGCACT-3′ |
| PIK3CA_ex9_a1⁎ | 5′-GACAAAGAACAGCTCAAAGCAA-3′ |
| PIK3CA_ex9_a2⁎ | 5′-TTTAGCACTTACCTGTGACTCCA-3′ |
| PIK3CA_ex20_a1⁎ | 5′-GAGCAAGAGGCTTTGGAGTA-3′ |
| PIK3CA_ex20_a2⁎ | 5′-ATCCAATCCATTTTTGTTGTCC-3′ |
| PTEN_ex7_a1⁎ | 5′-GGTGAAGATATATTCCTCCAATTCA-3′ |
| PTEN_ex7_a2⁎ | 5′-TTCTCCCAATGAAAGTAAAGTACAAA-3′ |
Published previously.12
Table 2.
List of Extension Primers for the SNaPshot Screen⁎
| Name | Sequence† |
|---|---|
| AKT1.49_extR | 5′-ACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTCGCCAGGTCTTGATGTACT-3′ |
| BRAF1397_extF | 5′-TGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACGGGACAAAGAATTGGATCTG-3′ |
| BRAF1406_extF | 5′-AGACTGACTGACTGACTGACTGACTGACTGACTGGAATTGGATCTGGATCATTTG-3′ |
| BRAF1789_extF | 5′-ACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTCAGTAAAAATAGGTGATTTTGGT-3′ |
| BRAF1799_extF‡ | 5′-GACTGACTGACTGACTGACTGACTGTGATTTTGGTCTAGCTACAG-3′ |
| EGFR2155_extF‡ | 5′-GACTGACTGACTGACTGACTGACTGTCAAAAAGATCAAAGTGCTG-3′ |
| EGFR2156_extF | 5′-CTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTCAAAAAGATCAAAGTGCTGG-3′ |
| EGFR2369_extR‡ | 5′-CTGACTGACTGACTGACTGACTGACTGACTAAGGGCATGAGCTGC-3′ |
| EGFR2573_extF‡ | GACTGACTGACTGACTGACTGACTGACAGATCACAGATTTTGGGC-3′ |
| EGFR2582_extR | 5′-TTCTCTTCCGCACCCAGC-3′ |
| KRAS34_extR‡ | 5′-GACTGACTGCTCTTGCCTACGCCAC-3′ |
| KRAS35_extF‡ | 5′-CTGACTCTTGTGGTAGTTGGAGCTG-3′ |
| KRAS37_extF‡ | 5′-TGACTGACTGATGGTAGTTGGAGCTGGT-3′ |
| KRAS38_extF‡ | 5′-GACTGACTGACGGTAGTTGGAGCTGGTG-3′ |
| KRAS181_extF | 5′-CTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTATTCTCGACACAGCAGGT-3′ |
| KRAS182_extF | 5′-GACTGACTGACTGACTGACTATTCTCGACACAGCAGGTC-3′ |
| KRAS183_extR | 5′-ACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACCTCATTGCACTGTACTCCTC-3′ |
| MEK1.167_extF | 5′-GACTGACTGACTGACTCTTGAGGCCTTTCTTACCC-3′ |
| MEK1.171_extR | 5′-CAGTTCTCCCACCTTCTG-3′ |
| MEK1.199_extR | 5′-GACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTCTCACTGATCTTCTCAAAGT-3′ |
| NRAS181_extF‡ | 5′-GACTGACTGACTGACTGACTGACTGACTGACTGACACATACTGGATACAGCTGGA-3′ |
| NRAS182_extF‡ | 5′-CTGACTGACTGACTGACTGACTGACTGACTGACTGCATACTGGATACAGCTGGAC-3′ |
| PIK3CA1624_extR‡ | 5′-TGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTTCTCCTGCTCAGTGATTT-3′ |
| PIK3CA1633_extF‡ | 5′-GACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGACTGATCCTCTCTCTGAAATCACT-3′ |
| PIK3CA3140_extR‡ | 5′-GTCCAGCCACCATGA-3′ |
| PTEN697_extR‡ | 5′-ACTGACTGACTGACTGACTGACTGACTGACTGACTGTGAACTTGTCTTCCCGTC-3′ |
Primers were purified by polyacrylamide gel electrophoresis.
Certain “GACT” nucleotides (italicized) were added to the extension primers to adjust their product sizes.
Published previously.12
Triplex Sizing Assay
An EGFR exon 19 deletion sizing assay, established previously,13 was adapted to allow for further analysis of EGFR and HER2 exon 20 insertions. The EGFR exon 19 deletions range from 9 to 24 nucleotides; EGFR exon 20 insertions, 3 to 12 nucleotides; and HER2 exon 20 insertions, 3 to 12 bp. The following primers were used: for EGFR exon 19 (wild-type amplicon size, 207 bp), there were EGFR_Ex19_FWD1, 5′-GCACCATCTCACAATTGCCAGTTA-3′; and EGFR_Ex19_REV1, 5′-/6FAM/AAAAGGTGGGCCTGAGGTTCA-3′; and for EGFR exon 20 (wild-type amplicon size, 155 bp), there were EGFR_Ex20ins_FWD, 5′-TCTTCACCTGGAAGGGGTCC-3′; and EGFR_Ex20ins_REV, 5′-/HEX/ACGGTGGAGGTGAGGCAGAT-3′. For HER2/ERBB2 exon 20 (wild-type amplicon size, 245 bp), the following were used: ERBB2_Ex20_FWD, 5′-ACCGTGCCCGGCCTAATCTT-3′; and ERBB2_Ex20_REV, 5′-/HEX/TCAGGCAGATGCCCAGAAGG-3′. A 1-ng template DNA was used to perform PCR with a kit (HotStarTaq Master Mix Kit; Qiagen Inc), as follows: 95°C for 15 minutes, followed by 40 cycles of 94°C for 30 seconds, 60°C for 30 seconds, 72°C for 60 seconds, and a final extension of 72°C for 30 minutes. Fluorescently labeled amplicons were separated with a capillary electrophoresis instrument (ABI 3730), and output data were interpreted using software (GeneMapper, version 4.0).
Direct Dideoxynucleotide-Based Sequencing
All mutations detected by SNaPShot and sizing assays were further confirmed with direct sequencing. Exons with positive mutations were amplified using a kit (HotStarTaq Master Mix Kit; Qiagen Inc) and M13-tagged gene-specific primers (see Supplemental Table S2 at http://jmd.amjpathol.org). The following conditions were used: 95°C for 15 minutes, followed by 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 60 seconds, and a final extension of 72°C for 30 minutes. Excess primers and nucleotides were digested using ExoSAP (USB Corporation, Cleveland, OH). ExoSAP is an enzyme mixture of exonuclease I, which removes leftover primers, and Shrimp Alkaline Phosphatase, which removes leftover dNTPs. Sequencing reactions were performed using chemistry (Version 3.1 Big Dye Terminator; Applied Biosystems) and analyzed on a sequencer (model 3730XL; Applied Biosystems). All sequence chromatograms were read in both forward and reverse directions.
Results
Development of a SNaPShot Assay to Assess Multiple Somatic Point Mutations in Lung Cancer
After performing a literature search and reviewing the Catalogue of Somatic Mutations in Cancer database, we decided to include in our SNaPshot screen (v1.0) 38 somatic point mutations occurring at 26 different loci in eight genes potentially relevant to targeted therapy in lung cancer (Table 3). The following criteria were used for selection of mutations: i) they were found in NSCLCs, ii) they occurred at a frequency of 1% or greater, and iii) they could be used as a predictor for targeted therapy. In this study, we included 14 SNaPshot assays derived from a 58-mutation genotyping panel that is being used for clinical testing of FFPE-derived tumor samples12 and designed 12 additional assays (Tables 1 and 2). All mutations were incorporated into five multiplexed panels, each capable of detecting mutations at four (panels 3 and 5) to six (panels 1, 2, and 4) loci. We optimized the concentration of PCR and extension primers in each panel so that all fluorescently labeled fragments displayed roughly the same peak height after capillary electrophoresis (Figure 1A). Each peak was validated with DNA from cell lines containing known mutations or “spiking primers” (ie, oligonucleotides containing mutations of interest; see Supplemental Figure S1 at http://jmd.amjpathol.org). We also developed a “pan-positive” control for the whole screen, using pools of spiking primers (Figure 1B). Spiking primers were mixed with the appropriate PCR products before primer extension reactions. By using normal genomic DNA, we performed the entire SNaPshot screen with all five panels reliably with as little as 2 ng per panel.
Table 3.
SNaPshot Screen Designed to Detect 38 Somatic Point Mutations in Eight Genes Relevant to Targeted Therapy in the NSCLC⁎
| Position | AA mutant | Nucleotide mutant |
|---|---|---|
| EGFR | ||
| G719 | p.G719C† | c.2155G>T |
| p.G719S† | c.2155G>A | |
| p.G719A | c.2156G>C | |
| T790M | p.T790M | c.2369C>T |
| L858 | p.L858R† | c.2573T>G |
| L861 | p.L861Q | c.2582T>A |
| KRAS | ||
| G12 | p.G12C† | c.34G>T |
| p.G12S† | c.34G>A | |
| p.G12R† | c.34G>C | |
| p.G12V† | c.35G>T | |
| p.G12A† | c.35G>C | |
| p.G12D† | c.35G>A | |
| G13 | p.G13C† | c.37G>T |
| p.G13S† | c.37G>A | |
| p.G13R† | c.37G>C | |
| p.G13D† | c.38G>A | |
| p.G13A† | c.38G>C | |
| Q61 | p.Q61K | c.181C>A |
| p.Q61R | c.182A>G | |
| p.Q61L | c.182A>T | |
| p.Q61H | c.183A>T | |
| p.Q61H | c.183A>C | |
| BRAF | ||
| G466 | p.G466V | c.1397G>T |
| G469 | p.G469A | c.1406G>C |
| L597 | p.L597V | c.1789C>G |
| V600 | p.V600E† | c.1799T>A |
| PIK3CA | ||
| H1047 | p.H1047R† | c.3140A>G |
| E542 | p.E542K† | c.1624G>A |
| E545 | p.E545K† | c.1633G>A |
| p.E545Q† | c.1633G>C | |
| NRAS | ||
| Q61 | p.Q61K† | c.181C>A |
| p.Q61L† | c.182A>T | |
| p.Q61R† | c.182A>G | |
| MEK1 (MAP2K1) | ||
| Q56 | p.Q56P | c.167A>C |
| K57 | p.K57N | c.171G>T |
| D67 | p.D67N | c.199G>A |
| AKT1 | ||
| E17 | p.E17K | c.49G>A |
| PTEN | ||
| R233 | p.R233X†‡ | c.697C>T |
The EGFR exon 19 deletions, EGFR exon 20 insertions, and HER2 exon 20 insertions are detected by a separate sizing assay. The PIK3CA H1047L mutation detected in 1 FFPE sample (see text) was not listed because that mutation was not reported in COSMIC (Catalogue of Somatic Mutations in Cancer) in NSCLC when the assay was designed.
Previously published SNaPshot assays.12
Truncation.
Figure 1.
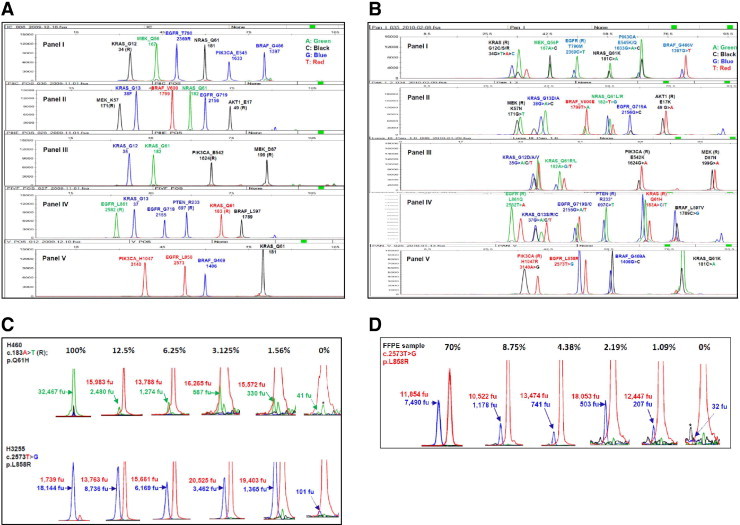
Lung cancer SNaPShot screen (v1.0). A: Human genomic DNA was used as a wild-type control for the multiplex SNaPShot screen, which consists of five panels. Each peak represents a locus where a driver mutation may occur. The name of each gene and the name and position of the amino acid are labeled on the top of each peak. The number under the gene name is the nucleotide position. R designates use of an extension primer encoding the reverse (complementary) strand. B: Pan-positive controls for the SNaPshot screen. Spiking primers were used to display all positive peaks in each locus. C: Sensitivity measurement with cell lines. DNAs from cell lines carrying the known mutations were diluted with human genomic DNA in ratios of 100%, 25% (data not shown), 12.5%, 6.25%, 3.125%, 1.56%, and 0% (wild-type control). Mixtures were then used to perform the SNaPShot screen. Numbers indicate the arbitrary fluorescence units (fu) of wild-type (above) and mutant (underneath) peaks separately. Solid arrows show mutant peaks; and dotted arrows, background peaks. The y axis was adjusted to the appropriate scale to visualize various peaks. Based on previously established criteria,12 the following rules were used to call a mutation: i) A mutation is called confidently if the mutant peak height is 10% or greater of the corresponding wild-type peak [eg, 12.5% dilution of the H460 cell line, as follows: (2480/15,983)×100 = 15.5%]. ii) If the potential mutant peak is less than 10%, the cutoff value [eg, 1.56% dilution of the H460 cell line, as follows: (330/15,572)×100 = 2.1%], a background peak of the same color and size (dotted arrow) in a separate wild-type DNA control (0%) is used as a reference. If the potential mutant peak height is three times or more than the background peak (330/41 = 8.0, >3), a mutation is called positive (see text for further details). *A background peak of the same color but not the same size as a mutant peak. D: Sensitivity measurements with FFPE-derived DNAs. The FFPE-derived DNA from a patient sample containing approximately 70% tumor cells was diluted with FFPE-derived DNA from a patient's normal tissue [ie, 1:1, 1:2, 1:4, 1:8, 1:16, 1:32, and 1:64 corresponded to samples with 70% tumor cells, 35% (data not shown), 17.5% (data not shown), 8.75%, 4.38%, 2.19%, and 1.09%, respectively]; 0% tumor cells (with 100% normal cells) were also analyzed. Numbers indicate the arbitrary fluorescence units (fu) of wild-type (above) and mutant (underneath) peaks separately. Solid arrows show mutant peaks; and dotted arrow, the background peak. The y axis was adjusted to the appropriate scale to visualize various peaks. Asterisks mark background peaks of the same size but not the same color as a mutant peak.
We subsequently validated our SNaPshot screen against a panel of 35 NSCLC cell lines with known mutation status.14–16 All cell lines were shown by us or others previously to harbor specific missense changes. We obtained 100% concordance with published results (Table 4 and Supplemental Table S3 at http://jmd.amjpathol.org). No false-positive or false-negative cases were observed.
Table 4.
Summary of Mutations Detected in Various Lung Cancer Samples
| Genes | Amino acids | Nucleotides | No. (%) of mutations |
|---|---|---|---|
| Cell lines⁎ | |||
| EGFR | T790M | 2368 C>T | 2 (5.7) |
| L858R | 2573 T>G | 3 (8.6) | |
| Exon 19 del (12 bp) | NA | 1 (2.9) | |
| Exon 19 del (15 bp) | NA | 2 (5.7) | |
| KRAS | G12A | 35 G>C | 1 (2.9) |
| G12C | 34 G>T | 3 (8.6) | |
| G12R | 34 G>C | 1 (2.9) | |
| G12S | 34 G>A | 1 (2.9) | |
| G12V | 35 G>T | 1 (2.9) | |
| Q61H | 183 A>T | 1 (2.9) | |
| BRAF | G466V | 1397 T>C | 1 (2.9) |
| NRAS | Q61L | 182 A>T | 1 (2.9) |
| Q61K | 181 C>A | 1 (2.9) | |
| PIK3CA | E545K | 1633 G>A | 1 (2.9) |
| MEK1 | Q56P | 167 A>C | 1 (2.9) |
| HER2 | Exon 20 ins (3 bp) | NA | 1 (2.9) |
| Frozen tissues† | |||
| EGFR | L858R | 2573 T>G | 1 (4.2) |
| Exon 19 del (15 bp) | NA | 1 (4.2) | |
| Exon 20 ins (3 bp) | NA | 1 (4.2) | |
| KRAS | G12A | 35 G>C | 1 (4.2) |
| G12V | 35 G>T | 2 (8.3) | |
| G12C | 34 G>T | 1 (4.2) | |
| G13C | 37 G>T | 1 (4.2) | |
| G13D | 38 G>A | 1 (4.2) | |
| NRAS | Q61K | 181 C>A | 1 (4.2) |
| PIK3CA | E545K | 1633 G>A | 1 (4.2) |
| FFPE samples‡ | |||
| EGFR | G719A | 2156 G>C | 1 (2.0) |
| T790M | 2369 C>T | 1 (2.0) | |
| L858R | 2573 T>G | 2 (4.1) | |
| Exon 19 del (15 bp) | NA | 5 (10.2) | |
| Exon 19 del (18 bp) | NA | 2 (4.1) | |
| Exon 20 ins (6 bp) | NA | 1 (2.0) | |
| KRAS | G12C | 34 G>T | 6 (12.2) |
| G12V | 35 G>T | 3 (6.1) | |
| Q61H | 183 A>C | 1 (2.0) | |
| PIK3CA | H1047L | 3140 A>T | 1 (2.0) |
NA, not available.
Among 35 samples, 23 mutations were found in 20 of them.
Among 24 samples, 11 mutations were found in 10 of them.
Among 49 samples, 23 mutations were found in 21 of them.
Finally, we measured the sensitivity of the SNaPShot assays in serial dilution experiments, using the empirically established method described in detail previously.12 One representative mutation in each of the five panels was studied, using mixtures of DNA from the male human genomic control and positive control cell lines with known mutations. Briefly, for any given locus, a mutation was called confidently if its peak height exceeded 10% of the corresponding heterozygous wild-type peak in the same sample (Figure 1C). If the height of a potential mutation peak was less than 10% of the corresponding wild-type peak, then a mutation was called if the potential mutant peak was three times higher than any background peaks of the same color and size in separate analyses of wild-type DNA controls (Figure 1C). In this way, the calculated sensitivity of our SNaPshot assays ranged from 1.56% to 12.5% (Figure 1C), as previously published.12 Assay sensitivity was further validated on FFPE-derived DNA harboring an EGFR L858R mutation. This sample contained approximately 70% tumor cells and was serially diluted with DNA extracted from FFPE-derived normal tissue. The L858R mutation could be detected in dilutions as low as 1.09% (Figure 1D). Notably, the sensitivity of a specific allele can vary, depending on the quality of the DNA, the level of background noise, and the size (position) of the peak in a panel. Furthermore, because of ploidy differences between cancer cell lines and nonneoplastic cells, sensitivities based on dilutions of DNA should be viewed only as approximations of absolute sensitivity. In both cell lines and FFPE samples, we observed a linear correlation between relative mutant peak intensity and tumor cell content (see Supplemental Figure S2 at http://jmd.amjpathol.org).
Triplex Sizing Assay
Concurrently, we developed a triplex sizing assay to detect recurrent insertions and deletions occurring in EGFR exons 19 and 20 and in HER2 exon 20 (Figure 2)A. This assay is based on length analysis of fluorescently labeled PCR products. By using normal genomic DNA, we performed the assay reliably with as little as 1 ng. In validation studies with 35 NSCLC cell lines with known mutation status, we obtained 100% concordance with published results (Table 4 and Supplemental Table S3 at http://jmd.amjpathol.org). In serial dilution assays using wild-type genomic DNA and positive controls (H1650 for EGFR exon 19 deletion, H1781 for HER2 exon 20 insertion, and a lung tumor sample for EGFR exon 20 insertion), all three mutations could be detected when the starting template was composed of 1.56% to 6.25% mutant DNA (Figure 2B).
Figure 2.
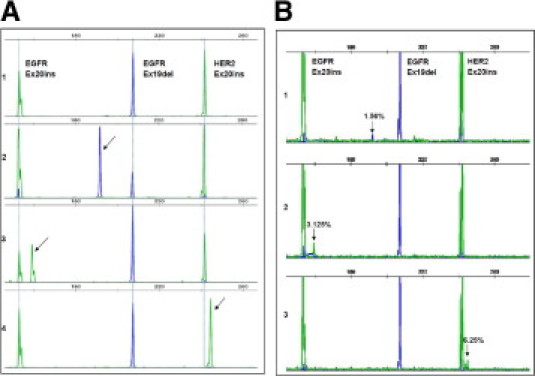
Lung cancer triplex sizing assay. The triplex sizing assay was established to detect simultaneously EGFR exon 19 deletions, EGFR exon 20 insertions, and HER2 20 insertions. A: Examples of results with known positive controls. 1, human genomic DNA was used as a wild-type control (peaks are indicated by dashed lines); 2, H1650 cell line DNA showed a 15-nucleotide deletion in EGFR exon 19 (arrow); 3, DNA from a previously characterized lung adenocarcinoma sample showed a three-nucleotide insertion in EGFR exon 20 (arrow); 4, H1781 cell line DNA showed a homozygous three-nucleotide insertion in HER2 exon 20 (arrow). B: Sensitivity assays. Samples carrying the known mutations were diluted with human genomic DNA in ratios of 100%, 25%, 12.5%, 6.25%, 3.125%, 1.56%, and 0%. Mixtures were then used to perform the sizing assay. The arrows indicate the mutation peaks at the lowest dilution rate.
Application of Multiplex SNaPShot and Sizing Assays
We used both assays to analyze DNA from 24 frozen lung adenocarcinoma samples of previously unknown mutation status. We detected 11 mutations (46%): three (13%) in EGFR, six (25%) in KRAS, one (4.2%) in PIK3CA, and one (4.2%) in NRAS. Consistent with the literature, EGFR and KRAS mutations were mutually exclusive (Table 4 and Supplemental Table S4 at http://jmd.amjpathol.org). The results in 9 of 11 positive cases were confirmed by direct sequencing (Figure 3, remaining data not shown). In the remaining two cases, direct sequencing showed only equivocally positive results, consistent with the notion that SNaPshot assays are more sensitive than direct sequencing (Figure 4).
Figure 3.
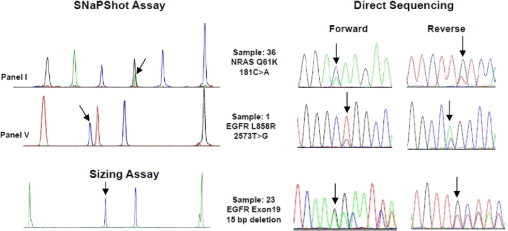
The SNaPshot and sizing assays results confirmed by forward and reverse direct sequencing. Arrows show the positions of mutations. The y axis of the SNaPshot assay involves arbitrary fluorescence units and was adjusted to an appropriate scale for observation of mutant peaks in each panel. Only representative examples are shown; remaining data are not shown.
Figure 4.
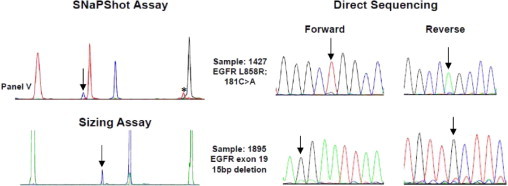
SNaPshot and sizing assays are more sensitive than direct sequencing. Mutations were detected in two samples by SNaPshot and sizing assays, but the calls were only equivocally positive by direct sequencing, consistent with the notion that SNaPshot assays are more sensitive than direct sequencing. An asterisk marks a background peak; no mutant allele exists that will show this position and color (panel V of Figures 1B and Supplemental Figure S1E at http://jmd.amjpathol.org). Arrows indicate mutant peaks.
We next used the SNaPshot and sizing assays to study DNAs from 49 FFPE-derived lung tumor samples, some of which were previously analyzed. Twenty-three changes were detected (Table 4 and Supplemental Table S5 at http://jmd.amjpathol.org). Three KRAS mutations (two KRAS G12C and one KRAS G12V), five EGFR exon 19 deletions, and one EGFR exon 20 insertion matched perfectly with previous results (data not shown). The other 14 mutations detected by SNaPshot and sizing assays (four KRAS G12C, two KRAS G12V, one KRAS Q61H, two EGFR exon 19 deletions, one EGFR G719A, one EGFR T790M, two EGFR L858R, and one PIK3CA H1047L) were further confirmed by direct sequencing (Figure 3, remaining data not shown). Notably, when we designed the assay, the PI3KCA H1047L variant had not yet been reported specifically in NSCLCs; it was detected as part of the screen for the previously reported H1047R mutation.
To evaluate the reproducibility of the lung tests, 19 cell lines and 11 FFPE-derived DNA samples were assayed independently by another operator in the laboratory. We obtained the exact same results (see Supplemental Tables S3 and S5 at http://jmd.amjpathol.org).
Finally, to demonstrate further the specificity of both the SNaPshot and sizing assays, we examined a cohort of 34 head and neck squamous cell cancer cell lines of unknown mutation status. Only three mutations (9% of 34) were found (see Supplemental Table S6 at http://jmd.amjpathol.org), all in PIK3CA. Results were confirmed by direct sequencing (data not shown). The lack of mutations in these samples is consistent with published results17 and demonstrates a low likelihood of detecting false positives using these molecular tests.
Discussion
Historically, cancer treatment decisions have been based on stage and histological classification of tumors, with the choice of chemotherapies guided mostly by empirical data. However, basic and translational research has uncovered molecular abnormalities that drive and sustain cancers. Clinical research18 has shown that patients' tumors respond differently to therapies targeted against these molecular abnormalities. For example, increased sensitivity of tumors harboring EGFR, HER2, PIK3CA, BRAF, MEK1, and ALK alterations to specific kinase inhibitors has already been established (Table 5).2–9 Thus, if physicians can identify genetic abnormalities required for tumor growth before a patient's therapy is chosen, they can begin to administer more appropriate specific agents to destroy tumors in a targeted fashion. Prospective incorporation of molecular tumor testing into everyday practice will prioritize for patients and physicians the treatments with the highest probability of positive outcomes. Unfortunately, clinical implementation of large-scale molecular diagnostics to cancer treatment remains in its infancy. Many hurdles remain, including the need to develop rapid, inexpensive, multiplexed genotyping tests for clinical use.
Table 5.
Frequencies of Mutations Detected by the SNaPshot and Sizing Assays for Lung Cancer⁎
| Gene | Mutation | Frequency | Method used | Reference | Prediction | Reference |
|---|---|---|---|---|---|---|
| EGFR | G719C/S/A | a, 0.5% (AD); b, 0.5% (NSCLC) | a and b, direct sequencing | a, Ding et al19; b, Shigematsu et al20 | Increased sensitivity to EGFR TKIs | Lynch et al29 and Paez et al30 |
| Exon 19 del | a, 13% (AD); b, 10% (NSCLC) | a, sequenom; b, direct sequencing; | a, Chitale et al21; b, Shigematsu et al20 | Increased sensitivity to EGFR TKIs | Lynch et al,29 Paez et al,30 and Pao et al31 | |
| L858R | a, 9% (AD); b, 8% (NSCLC) | a, sequenom; b, direct sequencing | a, Chitale et al21; b, Shigematsu et al20 | Increased sensitivity to EGFR TKIs | Lynch et al,29 Paez et al,30 and Pao et al31 | |
| T790M | a, 0.5% (AD); b, 4% (AD); c, 0.7% (NSCLC) | a, sequenom; b, mutant-enriched PCR; c, NA | a, Chitale et al21; b, Inukai et al22; c, COSMIC | Decreased sensitivity to EGFR TKIs | Pao et al32 and Kobayashi et al33 | |
| Exon 20 ins | a, 3% (AD); b, 2% (NSCLC) | a and b, direct sequencing | a, Ding et al19; b, Shigematsu et al20 | Decreased sensitivity to EGFR TKIs | Wu et al34 | |
| L861Q | a, 0.5% (AD); b, 0.3% (NSCLC) | a, direct sequencing; b, NA | a, Ding et al19; b, COSMIC | Increased sensitivity to EGFR TKIs | Lynch et al29 and Paez et al30 | |
| HER2 | Exon 20 ins | a, 2.6% (AD); b, 2% (NSCLC) | a, PCR-SSCP; b, direct sequencing | a, Sonobe et al23 ; b, Shigematsu et al20 | Increased sensitivity to HER2 TKIs | De Greve et al7 |
| KRAS | G12C/S/R/D/A/V and G13C/S/R/D/A | a, 20% (AD); b, 26% (NSCLC) | a and b, direct sequencing | a, Riely et al24; b, Tsao et al25 | Decreased sensitivity to EGFR TKIs | Pao et al35 |
| Q61H | a, 0.5% (AD); b, 0.2% (NSCLC) | a, direct sequencing; b, NA | a, Ding et al19; b, COSMIC | NA | NA | |
| PIK3CA | E542K, E545K/Q, and H1047R | a, 2% (AD); b, 1.2% (NSCLC) | a, NA; b, direct sequencing | a, COSMIC; b, Yamamoto et al26 | Increased sensitivity to PI3K inhibitors | Shapiro et al8 |
| BRAF | V600E | a, 2% (AD); b, 1% (NSCLC) | a, sequenom; b, sequenom | a, Chitale et al21; b, Pratilas et al27 | Increased sensitivity to BRAF V600E inhibitors | Flaherty et al6 |
| Non-V600E | a, 0.5% (AD); b, 0.7% (NSCLC) | a, sequenom; b, sequenom | a, Chitale et al21; b, Pratilas et al27 | NA | NA | |
| MEK1 | Q56P, K57N, and D67N | 1% (AD) | Direct sequencing | Marks et al3 | Increased sensitivity to MEK inhibitors | Marks et al3 |
| AKT1 | E17K | a, 0.5% (AD); b, 0.5% (NSCLC) | a, direct sequencing; b, NA | a, Ding et al19; b, COSMIC | NA | NA |
| NRAS | Q61L/R | a, 1.6% (AD); b, 0.8% (NSCLC) | a and b, direct sequencing | a, Ding et al19; b, Brose et al28 | NA | NA |
| PTEN | R233† | a, 1% (AD); b, 0.9% (NSCLC) | a and b, NA | a and b, COSMIC | Decreased sensitivity to EGFR TKIs | Sos et al11 |
AD, adenocarcinoma; TKI, tyrosine kinase inhibitor; Sequenom, a method based on Sequenom's MassARRAY system, involving multiplexed PCR, multiplexed single-base primer extension, and analysis of primer extension products using mass spectrometry; NA, not available; COSMIC, Catalogue of Somatic Mutations in Cancer; SSCP, single-strand conformational polymorphism; PI3K, phosphatidylinositol 3-kinase.
ALK fusions predict for sensitivity to ALK inhibitors; however, a different assay (fluorescence in situ hybridization) is used to detect ALK fusions. Version 46 of COSMIC was used as a reference for this table.
Truncation.
For multiplexed analysis of point mutations in DNA from FFPE tissue samples, at least two major platforms are in use. The first relies on Sequenom's MassARRAY system, based on multiplexed PCR, multiplexed single-base primer extension, and analysis of primer extension products using mass spectrometry (ie, matrix-assisted laser desorption/ionization time-of-flight analysis).36 The second is based on a system (SNaPshot; Life Technologies/Applied Biosystems) that depends on the analysis of fluorescently labeled primer-extension products by conventional capillary electrophoresis. In both assays, the identity of the incorporated nucleotide indicates the presence or absence of a mutation. Recently, some of us (D.D.-S., D.R.B., and A.J.I.) developed a fully operational SNaPshot assay that has been used as a clinical test for longer than 1 year to profile FFPE-derived tumor samples; it detects mutations in 58 different loci from 13 cancer genes (APC, BRAF, CTNNB1, EGFR, FLT3, JAK2, KIT, KRAS, NOTCH1, NRAS, PIK3CA, PTEN, and TP53) in eight multiplexed reactions.12 Similarly, others37 have used the SNaPshot technique to simultaneously screen colorectal carcinomas for 22 distinct mutations in four genes (KRAS, NRAS, BRAF, and PIK3CA).
Herein, we extended the SNaPshot technique to develop a multiplexed screen that was designed to assess DNA samples simultaneously for 38 somatic recurrent point mutations in eight genes with relevance to targeted therapy specifically in lung cancer. We chose to adopt the SNaPshot platform for the following reasons: i) familiarity with capillary electrophoresis, ii) ease of data interpretation, and iii) local availability of all necessary equipment. We also developed a triplex sizing assay, based on differential fragment lengths of fluorescently labeled products, to simultaneously assess samples for EGFR exon 19 deletions and EGFR exon 20 and HER2 exon 20 insertions. Such alterations are not amenable to comprehensive detection by the SNaPshot method. Both SNaPshot and sizing assays can be performed rapidly with minimal amounts of starting FFPE-derived DNA material and high sensitivity. Compared with direct sequencing, in which mutant DNA needs to compose 25% or more of the total DNA to easily detect a mutation, the SNaPshot and sizing assays can detect mutations in samples in which mutant DNA composes 1.56% to 12.5% and 1.56% to 6.25% of the total DNA, respectively. More important, the assays are robust, rapid, reliable, and relatively inexpensive. Compared with direct sequencing of all exons involved, it has been estimated that SNaPshot assays cost 80% less.37
The SNaPshot assay does have some limitations. The technique involves multiple primer sets for both PCR amplification and primer extension. The addition of new mutations to existing panels is straightforward but still requires effort (ie, for each additional mutation, the concentrations of PCR and extension primers in a panel may need to be reevaluated because of competition between different primers). Moreover, SNaPshot detects point mutations at specific sites. It is not designed to detect amplifications, insertions, or deletions; it is also not the optimal method if mutations occur at multiple spots across coding exons (eg, in tumor suppressor genes) or for mutations involving fusions (eg, EML4-ALK translocations). For lung cancer specifically, novel platforms are needed that can simultaneously detect gene fusions, point mutations, insertions, deletions, and amplifications.
In its present form, the SNaPshot and sizing assays are designed to detect more than 40 types of recurrent genetic alterations in lung cancer. Together, a mutation in one of these genes can be found in approximately 50% of lung adenocarcinomas. As additional novel mutations are identified through efforts such as The Cancer Genome Atlas,38 we plan to incorporate the clinically relevant ones into our screens. In the meantime, the current screens will be used to assess samples for multiple mutations and, in particular, as part of a 13-center US consortium (http://clinicaltrials.gov/ct2/show/NCT01014286; date of accession, August 18, 2010) among academic cancer centers that seek to genotype 1000 lung cancers nationwide. Mutation results will be used to prioritize therapy for patients, either as part of the standard of care or for clinical trials directed at specific mutation types. Efforts such as these will accelerate the use of genotypic information into standard treatment algorithms for lung cancer.
Acknowledgments
We thank Dr. Jennifer Pietenpol, Dr. David Johnson, Dr. Sam Santoro, Dr. Michael Laposata, Robert Woodhall, Gladys Garrison, Dr. Alfred George, and the Vanderbilt DNA sequencing core facility for support; Ryn Miake-Lye for helpful discussions; and Dr. Margaret Spitz (University of Texas M. D. Anderson Cancer Center, Houston, TX) for sharing DNA samples from tumors collected at her institution on Lung SPORE CA70907.
Footnotes
Supported by the NIH/National Cancer Institute (grants R01 CA121210, P01 CA129243, U54 CA143798, CA102353, and RC2-CA148394-01), the VICC Specialized Program of Research Excellence in Lung Cancer grant (CA90949), the Vanderbilt-Ingram Cancer Center Core grant (CA68485), the V Foundation, the TJ Martell Foundation, the Kleberg Foundation, and an anonymous donor.
D.D.-S. and A.J.I. submitted a patent application for the SNaPshot genotyping methods described herein, which are the subject of licensing discussions; W.P. was a consultant for MolecularMD, and rights to EGFR T790M testing were licensed on behalf of W.P. and others by the Memorial Sloan-Kettering Cancer Center to MolecularMD.
Supplemental material for this article can be found at http://jmd.amjpathol.org and at doi:10.1016/j.jmoldx.2010.11.010.
Current address of C.H.C.: Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, Maryland.
Supplementary data
Validation of individual peaks in each panel of the SNaPshot lung cancer screen. Each of 38 mutation peaks was validated one by one with cell lines carrying known mutations or spiking primers. The wild type peaks are shown on the top of each panel. The arrows indicate the mutations.
Linear correction between relative mutant peak intensity and tumor cell content. DNA from cell lines or an FFPE tumor sample were serially diluted with male human genomic DNA or FFPE-derived normal tissue, respectively. x-axis: relative mutant peak intensity; y-axis: percent tumor cell content. MT: mutant. WT: wildtype. fu: fluorescent units.
References
- 1.Jemal A.J., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Mitsudomi T., Morita S., Yatabe Y., Negoro S., Okamoto I., Tsurutani J., Seto T., Satouchi M., Tada H., Hirashima T., Asami K., Katakami N., Takada M., Yoshioka H., Shibata K., Kudoh S., Shimizu E., Saito H., Toyooka S., Nakagawa K., Fukuoka M. West Japan Oncology Group: Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2009;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 3.Marks J.L., Gong Y.X., Chitale D., Golas B., McLellan M.D., Kasai Y., Ding L., Mardis E.R., Wilson R.K., Solit D., Levine R., Michel K., Thomas R.K., Rusch V.W., Ladanyi M., Pao W. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524–5528. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pratilas C.A., Hanrahan A.J., Halilovic E., Persaud Y., Soh J., Chitale D., Shigematsu H., Yamamoto H., Sawai A., Janakiraman M., Taylor B.S., Pao W., Toyooka S., Ladanyi M., Gazdar A., Rosen N., Solit D.B. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–9383. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mok T.S., Wu Y.L., Thongprasert S., Yang C.H., Chu D.T., Saijo N., Sunpaweravong P., Han B., Margono B., Ichinose Y., Nishiwaki Y., Ohe Y., Yang J.J., Chewaskulyong B., Jiang H., Duffield E.L., Watkins C.L., Armour A.A., Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 6.Flaherty K., Puzanov I., Sosman J., Kim K., Ribas A., McArthur G., Lee R.J., Gripppo J.F., Nolop K., Champman P. Phase I study of PLX4032: proof of concept for V600E BRAF mutation as a therapeutic target in human cancer (abstract) J Clin Oncol. 2009;27:15s. [Google Scholar]
- 7.De Greve J.L., Teugels E., De Mey J., Geers C., Galdermans D., Decoster L., In 't Veld P., Schallier D., Taton M., Shahidi M. Clinical activity of BIBW 2992, an irreversible inhibitor of EGFR and HER2 in adenocarcinoma of the lung with mutations in the kinase domain of HER2neu (abstract) J Thorac Oncol. 2009;4:s307. [Google Scholar]
- 8.Shapiro G., Kwak E., Baselga J., Rodon J., Scheffold C., Laird A.D., Bedell C., Edelman G. Phase I dose-escalation study of XL147, a PI3K inhibitor administered orally to patients with solid tumors (abstract) J Clin Oncol. 2009;27:15s. [Google Scholar]
- 9.Kwak E., Camidge D.R., Clark J., Shapiro G., Maki R.G., Ratain M.J., Solomon B., Bang Y., Ou S., Salgia R. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066 (abstract) J Clin Oncol. 2009;27:15s. [Google Scholar]
- 10.Carpten J.D., Faber A.L., Horn C., Donoho G.P., Briggs S.L., Robbins C.M., Hostetter G., Boguslawski S., Moses T.Y., Savage S., Uhlik M., Lin A.M., Du J., Qian Y.W., Zeckner D.J., Tucker-Kellogg G., Touchman J., Patel K., Mousses S., Bittner M., Schevitz R., Lai M.H., Blanchard K.L., Thomas J.E. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 11.Sos M.L., Koker M., Weir B.A., Heynck S., Rabinovsky R., Zander T., Seeger J.M., Weiss J., Fischer F., Frommolt P., Michel K., Peifer M., Mermel C., Girard L., Peyton M., Gazdar A.F., Minna J.D., Garraway L.A., Kashkar H., Pao W., Meyerson M., Thomas R.K. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi: 10.1158/0008-5472.CAN-08-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dias-Santagata D., Akhavanfard S., David S.S., Vernovsky K., Kuhlmann G., Boisvert S.L., Stubbs H., McDermott U., Settleman J., Kwak E.L., Clark J.W., Isakoff S.J., Sequist L.V., Engelman J.A., Lynch T.J., Haberb D.A., Louis D.N., Ellisen L.W., Borger D.R., Iafrate A.J. Rapid targeted mutational analysis of human tumors: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2:146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Q., Pao W., Ladanyi M. Rapid polymerase chain reaction-based detection of epidermal growth factor receptor gene mutations in lung adenocarcinomas. J Mol Diagn. 2005;7:396–403. doi: 10.1016/S1525-1578(10)60569-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandhi J., Zhang J., Xie Y., Soh J., Shigematsu H., Zhang W., Yamamoto H., Peyton M., Girard L., Lockwood W.W., Lam W.L., Varella-Garcia M., Minna J.D., Gazdar A.F. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4:e4576. doi: 10.1371/journal.pone.0004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sos M.L., Michel K., Zander T., Weiss J., Frommolt P., Peifer M., Li D., Ullrich R., Koker M., Fischer F., Shimamura T., Rauh D., Mermel C., Fischer S., Stückrath I., Heynck S., Beroukhim R., Lin W., Winckler W., Shah K., LaFramboise T., Moriarty W.F., Hanna M., Tolosi L., Rahnenführer J., Verhaak R., Chiang D., Getz G., Hellmich M., Wolf J., Girard L., Peyton M., Weir B.A., Chen T.H., Greulich H., Barretina J., Shapiro G.I., Garraway L.A., Gazdar A.F., Minna J.D., Meyerson M., Wong K.K., Thomas R.K. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forbes S.A., Tang G., Bindal N., Bamford S., Dawson E., Cole C., Kok C.Y., Jia M., Ewing R., Menzies A., Teague J.W., Stratton M.R., Futreal P.A. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38(Database issue):D652–D657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu W., Schönleben F., Li X., Ho D.J., Close L.G., Manolidis S., Bennett B.P., Su G.H. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:1441–1446. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDermott U., Settleman J. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol. 2009;27:5650–5659. doi: 10.1200/JCO.2009.22.9054. [DOI] [PubMed] [Google Scholar]
- 19.Ding L., Getz G., Wheeler D.A., Mardis E.R., McLellan M.D., Cibulskis K., Sougnez C., Greulich H., Muzny D.M., Morgan M.B., Fulton L., Fulton R.S., Zhang Q., Wendl M.C., Lawrence M.S., Larson D.E., Chen K., Dooling D.J., Sabo A., Hawes A.C., Shen H., Jhangiani S.N., Lewis L.R., Hall O., Zhu Y., Mathew T., Ren Y., Yao J., Scherer S.E., Clerc K., Metcalf G.A., Ng B., Milosavljevic A., Gonzalez-Garay M.L., Osborne J.R., Meyer R., Shi X., Tang Y., Koboldt D.C., Lin L., Abbott R., Miner T.L., Pohl C., Fewell G., Haipek C., Schmidt H., Dunford-Shore B.H., Kraja A., Crosby S.D., Sawyer C.S., Vickery T., Sander S., Robinson J., Winckler W., Baldwin J., Chirieac L.R., Dutt A., Fennell T., Hanna M., Johnson B.E., Onofrio R.C., Thomas R.K., Tonon G., Weir B.A., Zhao X., Ziaugra L., Zody M.C., Giordano T., Orringer M.B., Roth J.A., Spitz M.R., Wistuba I.I., Ozenberger B., Good P.J., Chang A.C., Beer D.G., Watson M.A., Ladanyi M., Broderick S., Yoshizawa A., Travis W.D., Pao W., Province M.A., Weinstock G.M., Varmus H.E., Gabriel S.B., Lander E.S., Gibbs R.A., Meyerson M., Wilson R.K. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shigematsu H., Lin L., Takahashi T., Nomura M., Suzuki M., Wistuba I.I., Fong K.M., Lee H., Toyooka S., Shimizu N., Fujisawa T., Feng Z.D., Roth J.A., Herz J., Minna J.D., Gazdar A.F. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 21.Chitale D., Gong Y., Taylor B.S., Broderick S., Brennan C., Somwar R., Golas B., Wang L., Motoi N., Szoke J., Reinersman J.M., Major J., Sander C., Seshan V.E., Zakowski M.F., Rusch V., Pao W., Gerald W., Ladanyi M. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28:2773–2783. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inukai M., Toyooka S., Ito S., Asano H., Ichihara S., Soh J., Suehisa H., Ouchida M., Aoe K., Aoe M., Kiura K., Shimizu N., Date H. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66:7854–7858. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 23.Sonobe M., Manabe T., Wada H., Tanaka F. Lung adenocarcinoma harboring mutations in the ERBB2 kinase domain. J Mol Diagn. 2006;8:351–356. doi: 10.2353/jmoldx.2006.050132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riely G.J., Kris M.G., Rosenbaum D., Marks J., Li A., Chitale D.A., Nafa K., Riedel E.R., Su M.H., Pao W., Miller V.A., Ladanyi M. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14:5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsao M.S., Aviel-Ronen S., Ding K., Lau D., Liu N., Sakurada A., Whitehead M., Zhu C.Q., Livingston R., Johnson D.H., Rigas J., Seymour L., Winton T., Shepherd F.A. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non-small-cell lung cancer. J Clin Oncol. 2007;25:5240–5247. doi: 10.1200/JCO.2007.12.6953. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto H., Shigematsu H., Nomura M., Lockwood W.W., Sato M., Okumura N., Soh J., Suzuki M., Wistuba I.I., Fong K.M., Lee H., Toyooka S., Date H., Lam W.L., Minna J.D., Gazdar A.F. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68:6913–6921. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pratilas C.A., Hanrahan A.J., Halilovic E., Persaud Y., Soh J., Chitale D., Shigematsu H., Yamamoto H., Sawai A., Janakiraman M., Taylor B.S., Pao W., Toyooka S., Ladanyi M., Gazdar A., Rosen N., Solit D.B. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–9383. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brose M.S., Volpe P., Feldman M., Kumar M., Rishi I., Gerrero R., Einhorn E., Herlyn M., Minna J., Nicholson A., Roth J.A., Albelda S.M., Davies H., Cox C., Brignell G., Stephens P., Futreal P.A., Wooster R., Stratton M.R., Weber B.L. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 29.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G., Louis D.N., Christiani D.C., Settleman J., Haber D.A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 30.Paez J.G., Janne P.A., Lee J.C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F.J., Lindeman N., Boggon T.J., Naoki K., Sasaki H., Fujii Y., Eck M.J., Sellers W.R., Johnson B.E., Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 31.Pao W., Miller V., Zakowski M., Doherty J., Politi K., Sarkaria I., Singh B., Heelan R., Rusch V., Fulton L., Mardis E., Kupfer D., Wilson R., Kris M., Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pao W., Miller V.A., Politi K.A., Riely G.J., Somwar R., Zakowski M.F., Kris M.G., Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi S., Boggon T.J., Dayaram T., Janne P.A., Kocher O., Meyerson M., Johnson B.E., Eck M.J., Tenen D.G., Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 34.Wu J.Y., Wu S.G., Yang C.H., Gow C.H., Chang Y.L., Yu C.J., Shih J.Y., Yang P.C. Cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res. 2008;14:4877–4882. doi: 10.1158/1078-0432.CCR-07-5123. [DOI] [PubMed] [Google Scholar]
- 35.Pao W., Wang T.Y., Riely G.J., Miller V.A., Pan Q., Ladanyi M., Zakowski M.F., Heelan R.T., Kris M.G., Varmus H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:57–61. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas R.K., Baker A.C., Debiasi R.M., Winckler W., Laframboise T., Lin W.M., Wang M., Feng W., Zander T., MacConaill L., Lee J.C., Nicoletti R., Hatton C., Goyette M., Girard L., Majmudar K., Ziaugra L., Wong K.K., Gabriel S., Beroukhim R., Peyton M., Barretina J., Dutt A., Emery C., Greulich H., Shah K., Sasaki H., Gazdar A., Minna J., Armstrong S.A., Mellinghoff I.K., Hodi F.S., Dranoff G., Mischel P.S., Cloughesy T.F., Nelson S.F., Liau L.M., Mertz K., Rubin M.A., Moch H., Loda M., Catalona W., Fletcher J., Signoretti S., Kaye F., Anderson K.C., Demetri G.D., Dummer R., Wagner S., Herlyn M., Sellers W.R., Meyerson M., Garraway L.A. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 37.Lurkin I., Stoehr R., Hurst C.D., van Tilborg A.A., Knowles M.A., Hartmann A., Zwarthoff E.C. Two multiplex assays that simultaneously identify 22 possible mutation sites in the KRAS, BRAF, NRAS and PIK3CA genes. PLoS One. 2010;5:e8802. doi: 10.1371/journal.pone.0008802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., Alexe G., Lawrence M., O'Kelly M., Tamayo P., Weir B.A., Gabriel S., Winckler W., Gupta S., Jakkula L., Feiler H.S., Hodgson J.G., James C.D., Sarkaria J.N., Brennan C., Kahn A., Spellman P.T., Wilson R.K., Speed T.P., Gray J.W., Meyerson M., Getz G., Perou C.M., Hayes D.N. Cancer Genome Atlas Research Network: integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of individual peaks in each panel of the SNaPshot lung cancer screen. Each of 38 mutation peaks was validated one by one with cell lines carrying known mutations or spiking primers. The wild type peaks are shown on the top of each panel. The arrows indicate the mutations.
Linear correction between relative mutant peak intensity and tumor cell content. DNA from cell lines or an FFPE tumor sample were serially diluted with male human genomic DNA or FFPE-derived normal tissue, respectively. x-axis: relative mutant peak intensity; y-axis: percent tumor cell content. MT: mutant. WT: wildtype. fu: fluorescent units.