

Abstract
Cancer cachexia is a severe wasting syndrome characterized by the progressive loss of lean body mass and systemic inflammation. It occurs in approximately 80% of patients with advanced malignancy and is the cause of 20% to 30% of all cancer-related deaths. The mechanism by which striated muscle loss occurs is the tumor release of pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α. These cytokines interact with their cognate receptors on muscle cells to enhance NF-κB signaling, which then mediates muscle loss and significant cardiac dysfunction. Genetic inhibition of NF-κB signaling has demonstrated its predominant role in skeletal muscle loss. Therefore, we tested two novel drugs designed to specifically inhibit NF-κB by targeting the IκB kinase (IKK) complex: Compound A and NEMO binding domain (NBD) peptide. Using an established mouse model of cancer cachexia (C26 adenocarcinoma), we determined how these drugs affected the development of tumor-induced cardiac atrophy and function. Echocardiographic and histological analysis revealed that both Compound A and NBD inhibit cardiac NF-κB activity and prevent the development of tumor-induced systolic dysfunction and atrophy. This protection was independent of any effects of the tumor itself (Compound A) or tumor-secreted cytokines (NBD). This study identifies for the first time, to our knowledge, that drugs targeting the IKK complex are cardioprotective against cancer cachexia–induced cardiac atrophy and systolic dysfunction, suggesting therapies that may help reduce cardiac-associated morbidities found in patients with advanced malignancies.
Cancer cachexia is a severe wasting syndrome characterized by the progressive loss of lean body mass and fat. It occurs in as many as 80% of patients with advanced malignancy, and accounts for an estimated 20% to 30% of all cancer-related deaths.1–3 There are essentially no therapies currently available that can be used broadly to prevent the high morbidity associated with cancer cachexia. In the present study, we investigate two novel compounds that selectively inhibit NF-κB to determine whether they can prevent the cardiac sequelae of cancer cachexia using an established mouse model of cancer cachexia.
The C26 adenocarcinoma model of cancer cachexia was established in 1975 to create a model system that could be used to test biological and chemotherapies in vivo.4 Over the past three decades, it has been widely used to delineate the natural history of carcinoma-induced cachexia and antitumor therapies in vivo, as recently reviewed by Aulino et al, 2010.5 This model has been used to carefully characterize mechanisms of cancer cachexia, which has been found to closely parallel human disease. Specifically, the C26 adenocarcinoma cells can be placed in mice, where they form tumors that secrete proinflammatory cytokines. Initial studies found a prominent role of IL-6, but later studies identified that, in addition to IL-6, TNF-α and IL-1 can be identified circulating in vivo.6,7 These proinflammatory cytokines interact with their cognate receptors on muscle to activate the transcription factor NF-κB.8 Although these cytokines interact with their own unique receptors, these receptors have in common this activation of NF-κB. Recent studies have identified that NF-κB activation up-regulates the ubiquitin-dependent degradation of the sarcomere, which is central to the pathophysiology of striated muscle atrophy. For example, the use of dominant-negative IKKα/β-GFP, which inhibits NF-κB, markedly reduces the atrophy induced by disuse (up to 70%) in vivo.9 Pharmacological and genetic inhibition of NF-κB similarly inhibits atrophy, in part by inhibiting the expression of ubiquitin ligases, including MuRF1 and Atrogin-1, that mediate muscle atrophy.10 Proinflammatory cytokines such as IL-1, IL-6, and TNF-α interact with their cognate receptors on muscle to initiate NF-κB, which regulates the degradation of the sarcomere, resulting in muscle atrophy.11,12 These mechanisms parallel findings in the C26 adenocarcinoma model used in the current studies. The C26 model has been used to identify specific alterations in myofibrillar proteins in skeletal muscle during atrophy,13 the selective degradation of myosin,14 the down-regulation of dystrophin,15 and the participation of Peg3/PW1 and p53 in cancer-induced muscle atrophy.16 Anti-muscle wasting therapies have also been tested in the C26 model, including indomethacin, ibuprofen, appetite stimulants,17,18 IL-27 treatment,6,19 myostatin,20 erythropoietin,21 exercise,22 and high-protein diets.23
Recent studies have described therapeutic interventions that specifically target only acute increases in NF-κB, without affecting basal NF-κB activities. They have been shown to be useful experimentally in cardiac ischemia reperfusion injury and in reversing cardiac hypertrophy in vivo. In the present study, we test the hypothesis that Compound A and NEMO binding domain (NBD) peptides, two novel specific NF-κB inhibitors targeting the IκB kinase (IKK) complex, can prevent C26 adenocarcinoma–induced cardiac atrophy and dysfunction.
Materials and Methods
Cell Lines
The transplantable C26 colon adenocarcinoma cells13,24 were maintained and implanted as previously described in our laboratory.25
Experimental Protocol
Seventy-five male BALB/c mice were obtained from the Charles River Laboratories (Wilmington, MA). In our first set of studies (Group A), 36 mice aged 43 to 63 days (weight 22 to 24 g) were evenly and randomly divided into groups: healthy control mice, tumor-bearing mice (dimethyl sulfoxide [DMSO] sham or mutant NBD peptide [500 μg] sham), tumor-bearing plus NBD peptide (80 μg, 200 μg, or 500 μg), and tumor-bearing plus Compound A (2 mg/kg, 5 mg/kg, or 10 mg/kg). Mice were housed five animals per cage in a temperature-controlled room on a 12-hour light:dark cycle. All animals had access to unlimited food and water. Mice were allowed to acclimate to their new environment for approximately 3 days before beginning the study.
On day 0, the mice in tumor-bearing groups were injected subcutaneously in the right flank with 100 μl (approximately 500,000 cells) of C26 adenocarcinoma cells. Body weight, tumor volume, and food consumption were measured every other day from inoculation to completion of the study. One animal in the tumor-bearing group was sacrificed early due to tumor volume in excess of Institutional Animal Care and Use Committee (IUCAC) guidelines. On day 6, tumor-bearing mice began receiving single intraperitoneal injections of NBD peptide, Compound A, mutant NBD peptide, or sterile DMSO, based on their respective intervention, dosage, and dosing schedule (Figure 1A). On day 17, tumor-bearing mice had a significant tumor burden with clinical signs of cachexia and underwent cardiac echocardiography. In our second group of studies (Group B), 27 age-matched (to Group A) mice were used in echocardiographic and cardiac perfusion studies described below (n = 3 to 6 per group as outlined in Supplemental Table S1 at http://ajp.amjpathol.org). In a third set of studies (Group C), 12 mice were used in echocardiographic evaluations of drug therapies in the absence of tumor (Supplemental Table S2 at http://ajp.amjpathol.org). In our last group of studies (Group D), 15 mice (three mice per group) were assayed for circulating cytokines.
Figure 1.
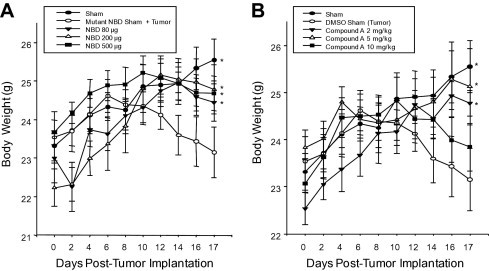
Inhibition of NF-κB with the NEMO-binding domain (NBD) peptide and Compound A inhibits C26 adenocarcinoma tumor-induced cachexia. The C26 adenocarcinoma cell line was injected at day 0, allowed to grow in vivo for 6 days before the development of cachexia, at which point daily treatment was begun. Both NBD and Compound A treatment inhibited the loss of body mass as determined by carcass weight (A and B, respectively). A one-way analysis of variance was performed to determine significance, followed by a multiple comparison procedure (Holm-Sidak method) to determine significance between groups. *P < 0.05 versus tumor on day 17. (n = 4/group, 36 total, Group A outlined in the Materials and Methods section).
At the completion of echocardiography or perfusion studies, all animals were euthanized. A final total body weight measurement was obtained. Tumors were resected, measured, and weighed, and a total carcass weight (total body weight minus the tumor weight) was calculated. Hind limbs were weighed separately. Heart muscle was excised from each animal immediately after sacrifice, snap frozen in liquid nitrogen, and stored at −80°C. All animal protocols were reviewed and in compliance with the IACUC.
Echocardiography
Echocardiography was performed on conscious mice using a VisualSonics Vevo 770 ultrasound biomicroscopy system (VisualSonics, Toronto, ON, Canada) as previously described.26,27
Real-Time PCR Determination of mRNA Expression
Total RNA was isolated from cardiac ventricular tissue as previously described.26 Briefly, mRNA expression was determined using a two-step reaction. cDNA was made using a High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). PCR products were amplified on an ABI Prism 7900HT Sequence Detection System using cDNA and the TaqMan probe set in TaqMan Universal PCR Master Mix (Applied Biosystems). The TaqMan probes used in these studies included ANF (Mm01255747_g1), βMHC (Mm00600555_m1), BNP (Mm00435304_g1), smooth muscle α-actin (Mm00808218_g1), MuRF1 (Mm01188690_m1), Atrogin-1/MAFbx/Fbxo32 (Mm00499518_m1), Nfkbia (Mm00477798_m1), and 18S (Hs99999901_s1) (Applied Biosystems).
Histology and Lectin Staining
Hearts were perfused with 4% paraformaldehyde in PBS and processed for histology and stained with H&E, Masson's Trichrome, or Triticum vulgaris lectin TRITC conjugate as previously described.26–29 Myocyte cross-sectional area was determined on lectin-stained sections using NIH Image J (Version 1.38X) based on parallel photomicrographs of a standard graticule ruler. Each cross-sectional area was determined from 300 measurements per group from at least 10 sections from three mice per group.
NF-κB ELISA Assay
Nuclear extracts were isolated from cardiac ventricles immediately after harvest using a commercially available nuclear extraction kit (Cayman Chemical, Ann Arbor, MI). Samples were homogenized using a glass tissue homogenizer (Kimble-Kontes, 885450-0022; Fisher Scientific, Waltham, MA), and nuclear extracts were then isolated according to the manufacturer's protocols and stored at −80°C. Fifteen micrograms of ventricular nuclear extracts were then assayed for NF-κB (p65) activity. NF-κB activity was determined using an ELISA assay based on the manufacturer's protocol (Cayman Chemical). Briefly, this assay utilizes a specific double-stranded DNA sequence containing a NF-κB response element immobilized to a 96-well plate. Isolated nuclei from each animal were added to the wells in triplicate, allowing nuclear transcription factors to bind to NF-κB response elements. After capture, an antibody specific for the p65 NF-κB subunit is used to identify the presence of p65 in the nucleus, indicative of activity. The activity of the samples was calculated relative to experimental control using the following formula: (sample average − average of non-specific binding wells, OD450)/(control average, OD450 − NSB average). Four animals per group were used, and all assays were performed in triplicate.
Multiplex Cytokine Analysis
At the completion of the experiment, before euthanasia, approximately 200 μl of blood was collected via a submandibular bleed from each mouse. The blood was allowed to clot at room temperature for 1 hour, serum was separated by centrifugation, and then stored at −80°C until analysis. Serum cytokine and chemokine concentrations were determined using a custom Bioplex Protein Array system (BioRad, Hercules, CA). For each cytokine, the minimum detectable concentration was 10 pg/ml.
Statistical Methods
Results are presented throughout as group mean values plus or minus SE. The significance of observed differences in group mean values was determined using analysis of variance with Holm-Sidak pairwise post hoc analysis. All analyses were performed using Sigma Stat 3.5 (Systat Software, Inc., San Jose, CA) and basic statistics on Microsoft Excel 2007 (Microsoft, Seattle, WA). Results are expressed as averages + SE, with statistical significance defined as P < 0.05.
Results
C26 Adenocarcinoma–Induced Cachexia in BALB/c Mice Results in Cardiac Atrophy
To determine whether NF-κB inhibition might be cardioprotective during cancer-induced cardiac atrophy, we used the C26 adenocarcinoma murine model of cancer-induced cachexia, which has been extensively characterized over the past 30 years.5 Nine groups of mice were used in these studies, including three control groups (an untreated control, a DMSO control sham, and a mutant NBD peptide in DMSO sham), three groups of increasing NBD peptide doses, and three groups of increasing Compound A doses. The C26 adenocarcinoma cell line was injected into the back flank at day 0 and allowed to implant and grow for 5 days; the respective treatments were begun on day 6. Daily treatment with either NBD peptide or Compound A protected against total body weight loss (Figure 1, A and B). In tumor-burdened animals, the measured whole left ventricular (LV) mass/tibia-length ratio decreased 24.3% compared with control hearts (3.1 mg/mm versus 4.1 mg/mm) after 17 days, indicating significant cardiac atrophy (Supplemental Table S1 at http://ajp.amjpathol.org).
Compound A and NBD Peptide Protect against Tumor-Induced Loss of Cardiac Mass and Decreased Systolic Dysfunction
Seventeen days after the implantation of the C26 tumors, echocardiography and histological analysis were performed on all nine groups of animals. Daily treatment with 10 mg/kg of Compound A prevented the loss of cardiac mass after 17 days (4.1 mg/mm versus 3.1 mg/mm in sham mice and 4.1 mg/mm in control) (Supplemental Table S1 at http://ajp.amjpathol.org). Tumor-burdened animals receiving daily mutant NBD peptide sham lost 26.8% of total heart weight (3.0 mg/mm versus 4.1 mg/mm in control mice), which contrasted with mice treated with 500 μg of NBD peptide, who had hearts that maintained their baseline heart weight, actually exceeding the weight of sham controls by 9.7%. Structural assessment of the left ventricular mass using echocardiography identified similar trends of protection by the NBD peptide and Compound A. By LV mass determination, sham-treated groups underwent a significant loss in mass, which was prevented with daily treatment with Compound A or NBD peptide (Figure 2A). This was reflected in preserved anterior and posterior wall thickness (Figure 2, B–D). Compound A was protective in a dose-dependent manner, whereas all doses of NBD tested exerted approximately the same protection (Figure 2).
Figure 2.
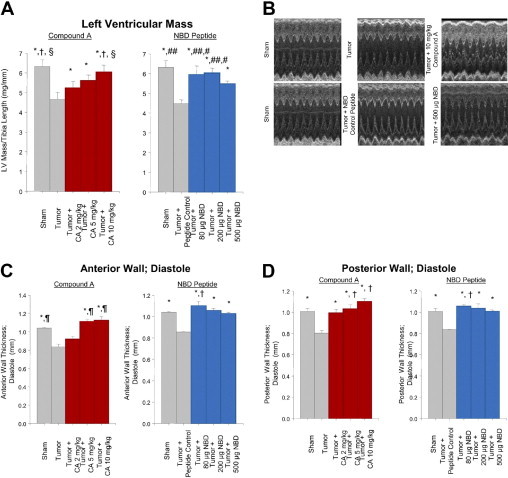
Tumor-induced cardiac atrophy is inhibited using therapeutics inhibiting NF-κB signaling by targeting IκB. A: Conscious echocardiographic analysis of left ventricular mass normalized to tibia length. LV mass (index) = [1.055 * ((ExLVD;d)3 − (LVEDD;d)3); where ExLVD = external left ventricle dimension and LVEDD = left ventricle end-diastolic dimension. *P < 0.01 versus Tumor or Tumor + NBD Control; †P < 0.02 versus Tumor + Compound A 2 mg/kg; §P < 0.05 versus Tumor + Compound A 5 mg/kg; #P < 0.05 versus Sham; and ##P < 0.02 versus Tumor NBD Peptide 500 μg. B: Representative M-mode echocardiographic tracings. C: Anterior wall thickness. *P < 0.01 versus Tumor or Tumor + Mutant NBD Control; ¶P < 0.05 versus Tumor + 2 mg/kg Compound A; and †P < 0.05 versus all other groups. D: Posterior wall thickness of the heart in vivo. *P < 0.01 versus Tumor or Tumor + Mutant NBD Control; and †P < 0.05 versus all other groups.
To demonstrate that these changes in wall thickness reflected changes in cardiomyocyte size, we determined the cardiomyocyte cross-sectional area of control, tumor, NBD peptide–treated, and Compound A–treated hearts (Figure 3, A and B). We found that the tumor-induced loss of cardiomyocyte area was prevented with either NBD or Compound A treatment. Interestingly, treatment with the highest level of Compound A (10 mg/kg) for 11 days in tumor-challenged mice promoted a significant increase in cardiomyocyte size compared to sham control hearts (Figure 3B). Tumor-induced atrophic hearts displayed a significant decrease in cardiac function (Figure 3, C and D), consistent with previous data demonstrating that many of the proinflammatory cytokines released during cancer cachexia, such as IL-1, IL-6, and TNF-α, directly induce cardiac dysfunction.30 Treatment with both Compound A and the NBD peptide protected against this cardiac dysfunction, as evidenced by improvement in both fractional shortening (Figure 3C) and ejection fraction in treated hearts (Figure 3D). These findings demonstrate that NF-κB inhibition, using either NBD peptide or Compound A treatment, can prevent tumor-induced cardiac dysfunction in addition to preventing the cardiac atrophy normally associated with cancer cachexia.
Figure 3.
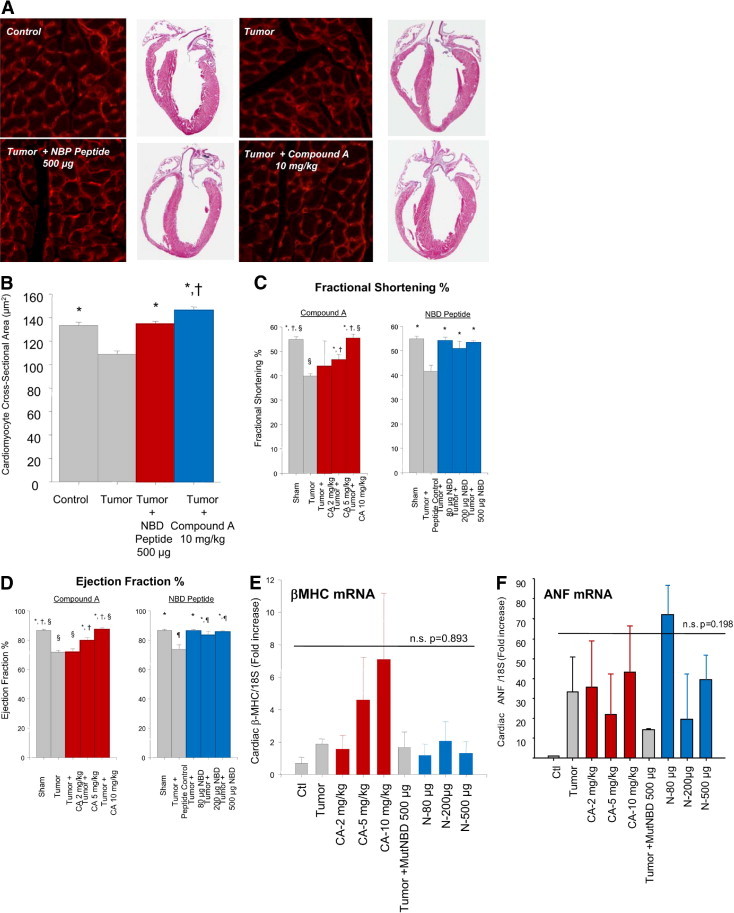
A: Histological analysis of cardiomyocytes cross-sectional area and gross histological representation of control, tumor, and tumors treated with either 500 μg of NBD peptide or 10 mg/kg of Compound A. B: Calculated cross-sectional areas of control, tumor, and tumors treated with either 500 μg of NBD peptide or 10 mg/kg of Compound A. Each cross-sectional area was determined from 300 measurements per group from at least 10 sections from 2 to 4 mice/group. *P < 0.01 versus Tumor; and †P < 0.05 versus all other groups. C: Echocardiography determination of fractional shortening percentage. *P < 0.01 versus Tumor or Tumor + NBD Control; †P < 0.05 versus Tumor + 2 mg/kg; and §P < 0.05 versus Tumor + 5 mg/kg. D: Echocardiographic determination of ejection fraction percentage. *P < 0.01 versus Tumor or Tumor + NBD Control; †P < 0.02 versus Tumor + 2 mg/kg; §P < 0.05 versus Tumor + 5 mg/kg; and ¶P < 0.05 versus Sham. E: Quantitative real-time PCR analysis of βMHC mRNA and (F) ANF mRNA. A one-way analysis of variance was performed to determine significance, followed by a multiple comparison procedures (Holm-Sidak method) to determine significance between groups.
Compound A and NBD Peptide Protect against Tumor Cachexia–Induced Loss in Cardiomyocyte Size
In hearts treated with the highest dose of Compound A (10 mg/kg), increases in both anterior and posterior wall thickness (Figure 2) and cardiomyocyte area (Figure 3, A and B) were greater than that seen in non-tumor controls, suggesting that the cardioprotective mechanism of compound A may involve more than just prevention of tumor-induced cardiac atrophy. These increases in cardiomyocyte size and LV anterior and posterior wall thickness, however, did not correlate with an increase in cardiac mass, as shown by LV mass/tibia length (Figure 2A) or heart weight/body weight (see Supplemental Table S1 at http://ajp.amjpathol.org). Without an increase in LV and total heart mass, the apparent increase in cardiomyocyte cross-sectional area and increased LV wall thickness in response to Compound A in mice challenged with tumor could not be interpreted as cardiac hypertrophy. Our interpretation of these findings are that Compound A may have had an effect on cardiomyocyte relaxation, allowing the cardiomyocytes and heart wall to appear larger, while no change in heart mass was seen.
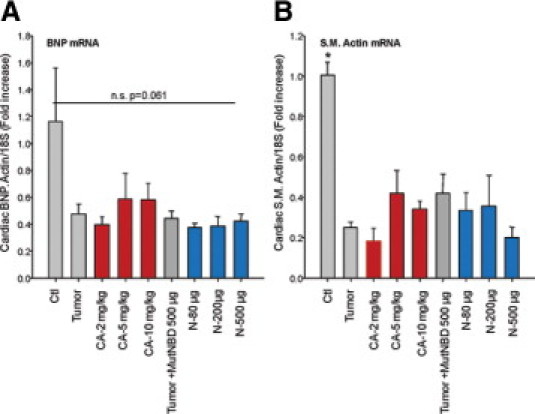
To further rule out that there was not an activation of a pathological cardiac hypertrophy program, we investigated how both Compound A and NBD affected mRNA levels of fetal genes commonly up-regulated in pathological cardiac hypertrophy. Complicating the interpretation of these findings is the fact that these same “fetal gene” pathways have previously been described to increase in models of cardiac atrophy.31 At day 17 after tumor implantation, cardiac β-myosin heavy chain (MHC) and atrial natriuretic factor (ANF) mRNA were not significantly increased in groups receiving a tumor (Figure 3, E and F). Cardiac BNP and smooth muscle actin, however, actually decreased in the presence of tumor in our model (see Supplemental Figure S1 at http://ajp.amjpathol.org). Despite the lack of statistical significance, some individual animals had increases in β-MHC beyond the tumor-related increases that were identified in the highest doses of Compound A (10 mg/kg). And both β-MHC and ANF mRNA levels were increased in some animals with tumor, especially at the 80-μg dose of NBD, but NBD peptide appeared to enhance ANF mRNA levels beyond the tumor alone (Figure 3, E and F). These findings reveal that Compound A and NBD peptides may differentially regulate the expression of specific genes associated with pathological cardiac hypertrophy and atrophy.31,32 However, these changes are not statistically significant and do not consistently show increases consistent with hypertrophy. When either Compound A or NBD peptide treatment is given in the absence of tumor for the 11 days duration used in these studies, no apparent increase in LV wall thickness is induced in additional studies (see Supplemental Table S2 at http://ajp.amjpathol.org). The Compound A–related increases in cardiomyocyte cross-sectional area and LV wall thickness occur only in the context of C26 adenocarcinoma–induced cachexia. These findings are consistent with Compound A's ability to affect cardiomyocyte relaxation in the face of tumor challenge, and not hypertrophy.
Both Compound A and NBD Peptide Inhibit Tumor-Induced Increases in NF-κB Activity in Vivo
Both Compound A and the NBD peptide were given systemically, allowing NF-κB to be inhibited at the level of the heart, skeletal muscle, or other organs. We therefore determined how each of these therapies affected the tumor-induced increase in NF-κB activity, using an assay to quantitatively determine the amount of nuclear p65 that specifically recognized the p65 binding site. Nuclear extracts were prepared from freshly harvested hearts and subsequently assayed for the amount of p65 recognizing its response element (see Materials and Methods for details). We identified that tumor challenge increased NF-κB activity over threefold (Figure 4A). Tumor challenge in the presence of the highest levels of Compound A and NBD (the only doses tested) completely inhibited this activity (Figure 4A). In additional experiments, we identified the expression of the gene Nfkbia, a gene regulated by NF-κB as another surrogate of cardiac NF-κB activity.33 Consistent with our NF-κB activity assay, we identified that the tumor-induced increases in Nfkbia were inhibited by the higher two doses of Compound A and by all three doses of NBD peptide tested (Figure 4B). These findings illustrate how Compound A and NBD peptides inhibit cardiac NF-κB activity, which may be one mechanism by which these therapies are cardioprotective.
Figure 4.
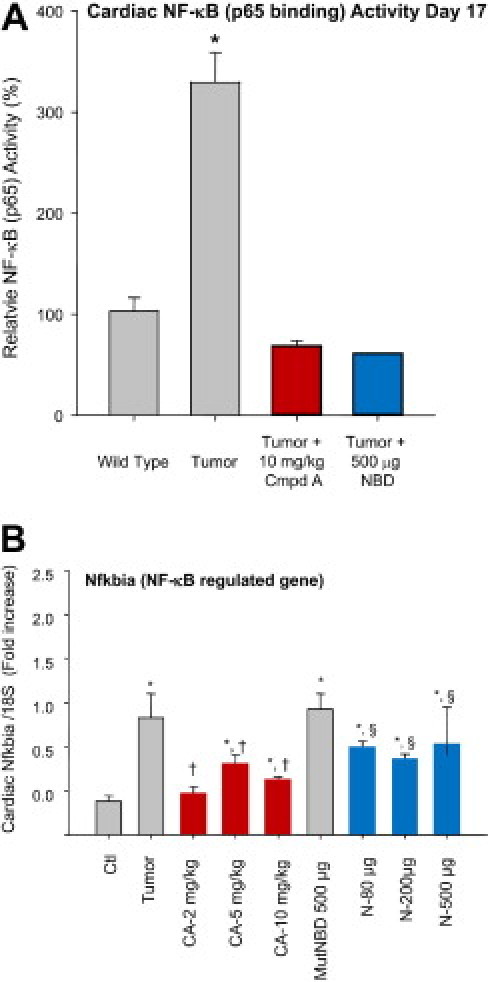
Compound A and NBD peptide inhibit cardiac NF-κB activity. A: NF-κB activity was determined by assaying cardiac nuclear extracts for the ability to bind NF-κB response elements using an ELISA-based assay, and identifying p65 binding (see Materials and Methods for details). At day 17, tumor-induced increases in NF-κB are inhibited significantly by Compound A (10 mg/kg) and the NBD peptide (500 μg). *P < 0.02 versus all other groups. n = 4/group. B: Compound A (CA) and NBD peptide treatment reduced cardiac NF-κB mRNA after 11 days of treatment (day 17). *P < 0.05 versus Ctrl; †P < 0.05 versus Tumor; and §P < 0.05 versus Mut NBD Peptide. n = 3/group. A one-way analysis of variance was performed to determine significance, followed by a multiple comparison procedure (Holm-Sidak method) to determine significance between groups.
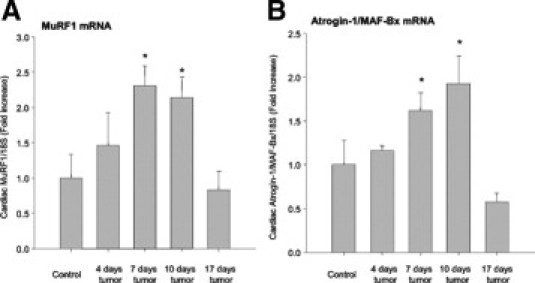
Cardiac MuRF1 and Atrogin-1 Expression Peak at Day 10 after Tumor Implantation
Activation of the NF-κB transcription pathway by factors inducing cachexia, such as TNF-α, has been shown to be sufficient to induce skeletal muscle atrophy, in part by up-regulation of the ubiquitin ligases MuRF1 and Atrogin-1.34 We recently reported that mice lacking MuRF1 (MuRF1−/−) were unable to undergo cardiac atrophy induced by dexamethasone, indicating MuRF1's significant role in cardiac atrophy.27 We therefore assayed levels of MuRF1 and its associated ubiquitin ligase Atrogin-1 by real-time PCR after C26 adenocarcinoma tumor challenge. (see Supplemental Figure S2, A and B, at http://ajp.amjpathol.org). We identified that MuRF1 and Atrogin-1 increased maximally by approximately day 10, returning to baseline levels by 17 days after tumor implantation. These findings describe MuRF1 and Atrogin-1 expression in cardiac atrophy for the first time, paralleling studies in skeletal muscle,35 indicating that MuRF1 and Atrogin-1 are significantly increased during the initiation of atrophy in heart as well as skeletal muscle.
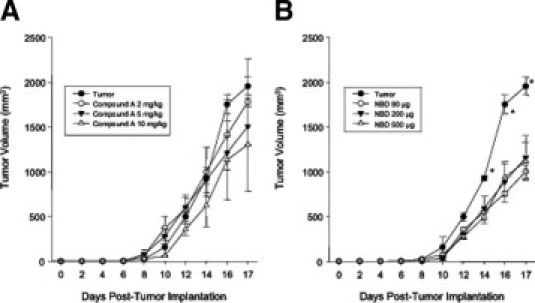
NBD, But Not Compound A, Inhibits Tumor Size But Not the Release of Tumor-Derived Cytokines Mediating Cardiac Atrophy
The volume of the C26 adenocarcinoma tumors were measured throughout the study in mice with and without increasing doses of Compound A and NBD peptides (see Supplemental Figure S3 at http://ajp.amjpathol.org). No dose of Compound A affected the C26 tumor size (see Supplemental Figure S3A at http://ajp.amjpathol.org). Conversely, we identified that all three doses of NBD peptides (80 to 500 μg) resulted in a decrease in tumor volume (see Supplemental Figure S3B at http://ajp.amjpathol.org). We therefore tested the circulating cytokine levels in both control and tumor-challenged mice with and without NBD treatment (Table 1). Although we identified tumor-induced increases in circulating TNF-α, IL-6, and IL-10, we did not find that NBD inhibited these circulating cytokines. In fact, we found significant increases in IL-6 and MIP-2 in tumor-treated mice given 500 μg of NBD peptide compared to tumors alone (Table 1). Despite NBD-mediated decreases in tumor size, the cytokines mediating cardiac atrophy were not decreased. This suggests that both Compound A, and likely NBD, exerted most of its protective effects by inhibiting atrophy at the level of the heart.
Table 1.
NBD Peptide Treatment Does Not Inhibit Circulating Chemokine and Cytokine Levels (Reported as pg/ml) at Day 17
| DMSO sham |
NBD peptide treatment |
||||
|---|---|---|---|---|---|
| Control | Tumor | 80 μg | 200 μg | 500 μg | |
| TNF-α | 6.4 ± 0.7 | 7.7 ± 1.6⁎ | 7.0 ± 1.5 | 7.4 ± 1.4 | 7.3 ± 1.3 |
| IL-1β | 25.1 ± 0.0 | 22.3 ± 5.7 | 24.5 ± 4.5 | 20.3 ± 5.0 | 20.3 ± 3.8 |
| IL-2 | <10 | <10 | <10 | <10 | <10 |
| IL-4 | 185.4 ± 16.0 | 153.1 ± 66.6 | 125.4 ± 12.8 | 126.6 ± 7.2 | 124 ± 14.6 |
| IL-5 | 13.8 ± 7.1 | 8.65 ± 3.5 | 10.1 ± 2.7 | 8.4 ± 2.9 | 9.1 ± 3.9 |
| IL-6 | 5.1 ± 0.0 | 141.5 ± 245⁎ | 991.6 ± 822 | 361.1 ± 228 | 378.2 ± 309† |
| IL-10 | 6.8 ± 1.6 | 9.34 ± 3.4⁎ | 14.6 ± 6.6 | 12.1 ± 4.5 | 12.7 ± 5.5 |
| IL-12p70 | 74.0 ± 20.3 | 88.6 ± 68.4 | 238.0 ± 152 | 440.2 ± 829 | 92.9 ± 50.1 |
| IL-13 | <10 | <10 | <10 | <10 | <10 |
| IL-17 | <10 | <10 | <10 | <10 | <10 |
| IFN-γ | 39.6 ± 6.7 | 37.1 ± 4.1 | 35.6 ± 5.9 | 33.4 ± 3.2 | 34.3 ± 3.5 |
| GM-CSF | 5.5 ± 1.2 | 4.2 ± 1.3 | 7.0 ± 6.0 | 18.4 ± 32.6 | 3.5 ± 1.5 |
| JE/MCP-1 | 81.0 ± 53.1 | 43.0 ± 2.7 | 48.6 ± 50.0 | 56.5 ± 74.9 | 81.0 ± 53.1 |
| MIP-2 | 10.7 ± 2.8 | 8.53 ± 2.7 | 10.3 ± 1.8 | 10.0 ± 1.3 | 10.3 ± 1.2† |
| VEGF | 164.4 ± 137 | 127.1 ± 29.4 | 738 ± 1173 | 2601 ± 5257 | 381.1 ± 587 |
Serum was collected from tumor, or tumor-treated age-matched mice 17 days after tumor implantation and treated with NBD or control peptide (±SE). A one-way ANOVA was performed to determine significance, followed by a Holm-Sidak pairwise comparison to determine significance between groups. (n = 3/group, Group D, outlined in Materials and Methods section). TNF-α, tumor necrosis factor alpha; GM-CSF, granulocyte macrophage colony stimulating factor; G-CSF, granulocyte-colony stimulating factor; JE/MCP-1, monocyte chemotactant protein-1; MIP-2, macrophage inflammatory protein-2; VEGF, vascular endothelial growth factor.
Significantly increased versus control animals (P < 0.05);
Significantly increased versus tumor-bearing animals (P < 0.05).
Discussion
NF-κB exists as a heterodimer of two subunits (p65 and p50), which are sequestered in an inactive form in the cytoplasm by the repressor molecule inhibitory κBα (IκBα). When activated by cytokines such as IL-1, IL-6, TNFα, or other tumor-related factors, the IKK complex, composed of IKKα, IKKβ, and IKKγ (NEMO) subunits, phosphorylates IκBα. Once phosphorylated, IκBα is targeted for ubiquitination and degradation by the 26S proteasome, allowing translocation of NF-κB to the nucleus, and subsequent transcription of a number of pro-inflammatory cytokines. Traditionally, broad inhibition of the proteasome has been one way in which NF-κB activity has been inhibited. However, due to the vital nature of the proteasome in maintaining key cellular processes and protein quality control in the heart,36,37 proteasome inhibition is not an efficient way to protect the heart against atrophy, and has been associated with lethal cardiac side effects.38 This led to our study of the efficacy of novel NF-κB–specific compounds in cancer-induced cardiac atrophy.
The molecular mechanisms by which Compound A and NBD peptides act to specifically inhibit NF-κB activation have been determined at the molecular level. Compound A is a small ATP-competitive inhibitor that selectively targets IKKβ to inhibit its phosphorylation and degradation in cells.39 Moreover, Compound A's inhibition is focused on stress-induced NF-κB activation, but not basal levels of NF-κB activation.39 Similarly, the NBD peptides used in this study have been shown to block the association of NEMO (IKKγ) and the IKK complex, which prevents the phosphorylation and degradation of the IKK complex.40 Similar to Compound A, the NBD peptide potently inhibits stress-induced NF-κB activation without affecting basal NF-κB activity.40 The strategies these molecules employ are distinctly different from proteasome inhibitors, which also inhibit NF-κB activity, among many other cellular processes. Proteasome inhibitors used to treat myeloma, for example, block basal NF-κB activity41 and prevent a host of vital ubiquitin proteasome functions from occurring,42–45 and can result in significant cardiac side effects and lethality in humans.38
In the present study, we identified that Compound A inhibited cardiac atrophy without affecting tumor growth. We also found that NBD inhibited cardiac atrophy without affecting circulating levels of tumor-derived cytokines (Table 1), although it had an effect on tumor size (see Supplemental Figure S3 at http://ajp.amjpathol.org). Since many studies have established these tumor-derived circulating cytokines, such as IL-6, as the main mechanism by which C26 adenocarcinoma induces atrophy in mice and human disease, the mouse models used in these studies were ideal to test the efficacy of novel NF-κB inhibitors in protecting against cancer-induced cardiac atrophy. We identified that these inhibitors were able to inhibit cardiac NF-κB activation in the heart, to potently prevent cancer-induced dysfunction and atrophy.
Several recent studies from our laboratory and others have demonstrated the therapeutic potential of inhibiting cardiac NF-κB; the use of the IKKβ inhibitor Compound A used in the present study has been reported to be cardioprotective in inhibiting ischemia reperfusion injury and in promoting the regression of pressure overload–induced cardiac hypertrophy.46,47 The use of cell-permeable NBD peptides has shown efficacy in inhibiting disease in models of pancreatitis, synovitis, and inflammatory colitis.48–52 In the present study, both Compound A and the NBD peptides demonstrated cardioprotection against tumor-induced cardiac atrophy and cardiac dysfunction in vivo. Although the activity of these therapeutics is quite specific, we did confirm that both drugs inhibited cardiac NF-κB activity in the presence of tumor (Figure 4, A and B).
Proinflammatory cytokines, including TNFα, are potent inducers of the ubiquitin ligases MuRF1 and Atrogin-1.35 Moreover, recent studies have implicated NF-κB, which is activated by proinflammatory cytokines, including TNFα, in the regulation of these ubiquitin ligases.10,34 Therefore, it is plausible that NF-κB inhibition prevented the up-regulation of MuRF1 and Atrogin-1. However, these ubiquitin ligases are regulated by other transcription factors such as FOXO53–55 and have not been good indicators of muscle proteolysis/atrophy.56 Therefore, future studies will need to focus on genetically altered mice, such as MuRF1−/− and Atrogin-1−/− mice, to delineate their role in mediating Compound A and NBD cardioprotective effects in vivo.
Although both Compound A and NBD specifically inhibit IKK, it is important to point out that non–NF-κB targets of IKK have recently been described, including FOXO3a,57 mTOR,58 A20,59 and p53.60 Constitutive expression of IKKβ in primary tumor cells has been reported to increase cell proliferation and the development of tumors, which can be attenuated by expression of FOXO3a, implicating IKKβ's regulation of FOXO3a.57 In prostate cancer cells, IKKα has been shown to interact with mTOR, as part of the TORC1 complex, to efficiently induce mTOR activity when constitutive Akt activation is present.58 Recent studies have also described that IKKβ phosphorylates A20 in vitro and in vivo at serine 381, to enhance A20's ability to inhibit NF-κB signaling, representing a feedback loop that attenuates NF-κB signaling following activation.59 IKKβ has recently been shown to phosphorylate p53 at serines 362 and 366, which leads to its being targeted by β-TrCP1 for ubiquitination.60 These studies identified blocking IKKβ as a way to stabilize p53 and modulate its biological activity, vital to tumorigenesis. Although the identification of these non–NF-κB targets of the IKK complex have been made in cancer, FOXO3a, p53, and mTOR are signaling pathways that have been implicated in muscle atrophy,16,61–65 so it is important to leave the possibility that the beneficial effects identified in the present study may be due to the drug's effects on one of these other signaling pathways, in addition to NF-κB.
Our findings suggest that NF-κB inhibition targeting the IκB complex is able to protect against cancer-associated cardiac atrophy and dysfunction by inhibiting the cytokine-induced NF-κB activity in the heart. Our findings highlight the therapeutic potential of these drugs in treating the associated cardiac morbidity related to cancer-associated cachexia beyond their protective affects against the loss of lean body mass and fat mass.
Acknowledgment
We acknowledge Janice Weaver (Animal Histopathology Laboratory, University of North Carolina) for assistance in preparing histological specimens.
Footnotes
Supported by the Howard Hughes Medical Institute Research Training Fellowship (A.W.). This work was supported by University of North Carolina Program in Translational Science grant (M.C.), the American Heart Association Scientist Development grant (M.W.), and the National Heart, Lung, and Blood Institute grant R01HL104129 (M.W.).
None of the authors disclosed any relevant financial relationships.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2010.12.009.
Supplementary data
Supplemental Figure S1.
MuRF1 and Atrogin-1/MAFBx expression significantly increases at day 7 to 10 after implantation of the C26 tumor. MuRF1 (A) and MAFBx/Atrogin-1 (B) expression peaks at approximately day 10 after tumor implantation, returning to baseline levels by day 17. *P < 0.05 versus Control and 17 days tumor. A one-way analysis of variance was performed to determine significance, followed by a multiple comparison procedures (Holm-Sidak method) to determine significance between groups.
Supplemental Figure S2.
NF-κB inhibition has negligible effects on C26 tumor volumes during the course of the study. Tumor volume after treatment with (A) Compound A; or (B) NBD peptide. Tumors were implanted on day 0, with treatment starting on day 6 through day 17. A one-way analysis of variance was performed to determine significance, followed by a multiple comparison procedures (Holm-Sidak method) to determine significance between groups. *P < 0.01 versus all other groups.
Supplemental Figure S3.
Tumor inhibits BNP and smooth muscle actin. Quantitative real-time PCR determination of (A) cardiac BNP and (B) smooth muscle actin mRNA. A one-way analysis of variance was performed to determine significance, followed by a multiple comparison procedures (Holm-Sidak method) to determine significance between groups. *P < 0.01 versus all other groups.
References
- 1.Tisdale M.J. Biology of cachexia. J Natl Cancer Inst. 1997;89:1763–1773. doi: 10.1093/jnci/89.23.1763. [DOI] [PubMed] [Google Scholar]
- 2.Inagaki J., Rodriguez V., Bodey G.P. Proceedings: causes of death in cancer patients. Cancer. 1974;33:568–573. doi: 10.1002/1097-0142(197402)33:2<568::aid-cncr2820330236>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Tisdale M.J. Cachexia in cancer patients. Nat Rev Cancer. 2002;2:862–871. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 4.Corbett T.H., Griswold D.P., Jr., Roberts B.J., Peckham J.C., Schabel F.M., Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35:2434–2439. [PubMed] [Google Scholar]
- 5.Aulino P., Berardi E., Cardillo V.M., Rizzuto E., Perniconi B., Ramina C., Padula F., Spugnini E.P., Baldi A., Faiola F., Adamo S., Coletti D. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer. 2010;10:363. doi: 10.1186/1471-2407-10-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujita J., Tsujinaka T., Yano M., Ebisui C., Saito H., Katsume A., Akamatsu K., Ohsugi Y., Shiozaki H., Monden M. Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. Int J Cancer. 1996;68:637–643. doi: 10.1002/(SICI)1097-0215(19961127)68:5<637::AID-IJC14>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 7.Yasumoto K., Nukaida N., Harada A., Kuno K., Akiyama M., Nakashima E., Fujioka N., Mai M., Kasahara T., Fujimoto-Ouchi K., Mori K., Tanaka Y., Matsushima K. Molecular analysis of the cytokine network involved in cachexia in colon 26 adenocarcinoma-bearing mice. Cancer Res. 1995;55:921–927. [PubMed] [Google Scholar]
- 8.Vallabhapurapu S., Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 9.Van Gammeren D., Damrauer J.S., Jackman R.W., Kandarian S.C. The IkappaB kinases IKKalpha and IKKbeta are necessary and sufficient for skeletal muscle atrophy. FASEB J. 2009;23:362–370. doi: 10.1096/fj.08-114249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai D., Frantz J.D., Tawa N.E., Jr., Melendez P.A., Oh B.C., Lidov H.G., Hasselgren P.O., Frontera W.R., Lee J., Glass D.J., Shoelson S.E. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 11.Tisdale M.J. Catabolic mediators of cancer cachexia. Curr Opin Support Palliat Care. 2008;2:256–2561. doi: 10.1097/spc.0b013e328319d7fa. [DOI] [PubMed] [Google Scholar]
- 12.Tisdale M.J. Mechanisms of cancer cachexia. Physiol Rev. 2009;89:381–410. doi: 10.1152/physrev.00016.2008. [DOI] [PubMed] [Google Scholar]
- 13.Acharyya S., Ladner K.J., Nelsen L.L., Damrauer J., Reiser P.J., Swoap S., Guttridge D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest. 2004;114:370–378. doi: 10.1172/JCI20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diffee G.M., Kalfas K., Al-Majid S., McCarthy D.O. Altered expression of skeletal muscle myosin isoforms in cancer cachexia. Am J Physiol Cell Physiol. 2002;283:C1376–C1382. doi: 10.1152/ajpcell.00154.2002. [DOI] [PubMed] [Google Scholar]
- 15.Acharyya S., Butchbach M.E., Sahenk Z., Wang H., Saji M., Carathers M., Ringel M.D., Skipworth R.J., Fearon K.C., Hollingsworth M.A., Muscarella P., Burghes A.H., Rafael-Fortney J.A., Guttridge D.C. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 2005;8:421–432. doi: 10.1016/j.ccr.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Schwarzkopf M., Coletti D., Sassoon D., Marazzi G. Muscle cachexia is regulated by a p53-PW1/Peg3-dependent pathway. Genes Dev. 2006;20:3440–3452. doi: 10.1101/gad.412606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weyermann P., Dallmann R., Magyar J., Anklin C., Hufschmid M., Dubach-Powell J., Courdier-Fruh I., Hennebohle M., Nordhoff S., Mondadori C. Orally available selective melanocortin-4 receptor antagonists stimulate food intake and reduce cancer-induced cachexia in mice. PLoS ONE. 2009;4:e4774. doi: 10.1371/journal.pone.0004774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarthy D.O., Whitney P., Hitt A., Al-Majid S. Indomethacin and ibuprofen preserve gastrocnemius muscle mass in mice bearing the colon-26 adenocarcinoma. Res Nurs Health. 2004;27:174–184. doi: 10.1002/nur.20019. [DOI] [PubMed] [Google Scholar]
- 19.Hisada M., Kamiya S., Fujita K., Belladonna M.L., Aoki T., Koyanagi Y., Mizuguchi J., Yoshimoto T. Potent antitumor activity of interleukin-27. Cancer Res. 2004;64:1152–1156. doi: 10.1158/0008-5472.can-03-2084. [DOI] [PubMed] [Google Scholar]
- 20.Bonetto A., Penna F., Minero V.G., Reffo P., Bonelli G., Baccino F.M., Costelli P. Deacetylase inhibitors modulate the myostatin/follistatin axis without improving cachexia in tumor-bearing mice. Curr Cancer Drug Targets. 2009;9:608–616. doi: 10.2174/156800909789057015. [DOI] [PubMed] [Google Scholar]
- 21.van Halteren H.K., Bongaerts G.P., Verhagen C.A., Kamm Y.J., Willems J.L., Grutters G.J., Koopman J.P., Wagener D.J. Recombinant human erythropoietin attenuates weight loss in a murine cancer cachexia model. J Cancer Res Clin Oncol. 2004;130:211–216. doi: 10.1007/s00432-003-0526-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.al-Majid S., McCarthy D.O. Cancer-induced fatigue and skeletal muscle wasting: the role of exercise. Biol Res Nurs. 2001;2:186–197. doi: 10.1177/109980040100200304. [DOI] [PubMed] [Google Scholar]
- 23.van Norren K., Kegler D., Argiles J.M., Luiking Y., Gorselink M., Laviano A., Arts K., Faber J., Jansen H., van der Beek E.M., van Helvoort A. Dietary supplementation with a specific combination of high protein, leucine, and fish oil improves muscle function and daily activity in tumour-bearing cachectic mice. Br J Cancer. 2009;100:713–722. doi: 10.1038/sj.bjc.6604905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strassmann G., Masui Y., Chizzonite R., Fong M. Mechanisms of experimental cancer cachexia. Local involvement of IL-1 in colon-26 tumor J Immunol. 1993;150:2341–2345. [PubMed] [Google Scholar]
- 25.O'Connell T.M., Ardeshirpour F., Asher S.A., Winnike J., Yin X., George J., Guttridge D.C., He W., Wysong A., Willis M.S., Couch M.E. Metabolomic analysis of cancer cachexia reveals distinct lipid and glucose alterations. Metabolomics. 2008;4:216–225. [Google Scholar]
- 26.Willis M.S., Ike C., Li L., Wang D.Z., Glass D.J., Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–459. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willis M.S., Rojas M., Li L., Selzman C.H., Tang R.H., Stansfield W.E., Rodriguez J.E., Glass D.J., Patterson C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am J Physiol Heart Circ Physiol. 2009;296:H997–H1006. doi: 10.1152/ajpheart.00660.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H.H., Willis M.S., Lockyer P., Miller N., McDonough H., Glass D.J., Patterson C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willis M.S., Schisler J.C., Li L., Rodriguez J.E., Hilliard E.G., Charles P.C., Patterson C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ Res. 2009;105:80–88. doi: 10.1161/CIRCRESAHA.109.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Menyar A.A. Cytokines and myocardial dysfunction: state of the art. J Card Fail. 2008;14:61–74. doi: 10.1016/j.cardfail.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Depre C., Shipley G.L., Chen W., Han Q., Doenst T., Moore M.L., Stepkowski S., Davies P.J., Taegtmeyer H. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med. 1998;4:1269–1275. doi: 10.1038/3253. [DOI] [PubMed] [Google Scholar]
- 32.McMullen J.R., Jennings G.L. Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–262. doi: 10.1111/j.1440-1681.2007.04585.x. [DOI] [PubMed] [Google Scholar]
- 33.Reuter S., Charlet J., Juncker T., Teiten M.H., Dicato M., Diederich M. Effect of curcumin on nuclear factor kappaB signaling pathways in human chronic myelogenous K562 leukemia cells. Ann N Y Acad Sci. 2009;1171:436–447. doi: 10.1111/j.1749-6632.2009.04731.x. [DOI] [PubMed] [Google Scholar]
- 34.Glass D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–1984. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 35.Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K., Pan Z.Q., Valenzuela D.M., DeChiara T.M., Stitt T.N., Yancopoulos G.D., Glass D.J. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 36.Willis M.S., Townley-Tilson W.H., Kang E.Y., Homeister J.W., Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463–478. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez J.E., Schisler J.C., Patterson C., Willis M.S. Seek and destroy: the ubiquitin–proteasome system in cardiac disease. Curr Hypertens Rep. 2009;11:396–405. doi: 10.1007/s11906-009-0069-7. [DOI] [PubMed] [Google Scholar]
- 38.Willis M.S., Schisler J.C., Patterson C. Appetite for destruction: e3 ubiquitin-ligase protection in cardiac disease. Future Cardiol. 2008;4:65–75. doi: 10.2217/14796678.4.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziegelbauer K., Gantner F., Lukacs N.W., Berlin A., Fuchikami K., Niki T., Sakai K., Inbe H., Takeshita K., Ishimori M., Komura H., Murata T., Lowinger T., Bacon K.B. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br J Pharmacol. 2005;145:178–192. doi: 10.1038/sj.bjp.0706176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.May M.J., D'Acquisto F., Madge L.A., Glockner J., Pober J.S., Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 41.Hideshima T., Chauhan D., Richardson P., Mitsiades C., Mitsiades N., Hayashi T., Munshi N., Dang L., Castro A., Palombella V., Adams J., Anderson K.C. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 42.Willis M.S., Patterson C. Into the heart: the emerging role of the ubiquitin-proteasome system. J Mol Cell Cardiol. 2006;41:567–579. doi: 10.1016/j.yjmcc.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 43.Patterson C., Ike C., Willis P.W.t., Stouffer G.A., Willis M.S. The bitter end: the ubiquitin-proteasome system and cardiac dysfunction. Circulation. 2007;115:1456–1463. doi: 10.1161/CIRCULATIONAHA.106.649863. [DOI] [PubMed] [Google Scholar]
- 44.Mearini G., Schlossarek S., Willis M.S., Carrier L. The ubiquitin-proteasome system in cardiac dysfunction. Biochim Biophys Acta. 2008;1782:749–763. doi: 10.1016/j.bbadis.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Willis M.S., Schisler J.C., Portbury A.L., Patterson C. Build it up–tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res. 2009;81:439–448. doi: 10.1093/cvr/cvn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moss N.C., Stansfield W.E., Willis M.S., Tang R.H., Selzman C.H. IKKbeta inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;293:H2248–H2253. doi: 10.1152/ajpheart.00776.2007. [DOI] [PubMed] [Google Scholar]
- 47.Moss N.C., Tang R.H., Willis M., Stansfield W.E., Baldwin A.S., Selzman C.H. Inhibitory kappa B kinase-beta is a target for specific nuclear factor kappa B-mediated delayed cardioprotection. J Thorac Cardiovasc Surg. 2008;136:1274–1279. doi: 10.1016/j.jtcvs.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 48.Soysa N.S., Alles N., Shimokawa H., Jimi E., Aoki K., Ohya K. Inhibition of the classical NF-kappaB pathway prevents osteoclast bone-resorbing activity. J Bone Miner Metab. 2009;27:131–139. doi: 10.1007/s00774-008-0026-6. [DOI] [PubMed] [Google Scholar]
- 49.Long Y.M., Chen K., Liu X.J., Xie W.R., Wang H. Cell-permeable Tat-NBD peptide attenuates rat pancreatitis and acinus cell inflammation response. World J Gastroenterol. 2009;15:561–569. doi: 10.3748/wjg.15.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jimi E., Aoki K., Saito H., D'Acquisto F., May M.J., Nakamura I., Sudo T., Kojima T., Okamoto F., Fukushima H., Okabe K., Ohya K., Ghosh S. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004;10:617–624. doi: 10.1038/nm1054. [DOI] [PubMed] [Google Scholar]
- 51.Shibata W., Maeda S., Hikiba Y., Yanai A., Ohmae T., Sakamoto K., Nakagawa H., Ogura K., Omata M. Cutting edge: the IkappaB kinase (IKK) inhibitor. NEMO-binding domain peptide, blocks inflammatory injury in murine colitis J Immunol. 2007;179:2681–2685. doi: 10.4049/jimmunol.179.5.2681. [DOI] [PubMed] [Google Scholar]
- 52.Tas S.W., Vervoordeldonk M.J., Hajji N., May M.J., Ghosh S., Tak P.P. Local treatment with the selective IkappaB kinase beta inhibitor NEMO-binding domain peptide ameliorates synovial inflammation. Arthritis Res Ther. 2006;8:R86. doi: 10.1186/ar1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waddell D.S., Baehr L.M., van den Brandt J., Johnsen S.A., Reichardt H.M., Furlow J.D., Bodine S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab. 2008;295:E785–E797. doi: 10.1152/ajpendo.00646.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stitt T.N., Drujan D., Clarke B.A., Panaro F., Timofeyva Y., Kline W.O., Gonzalez M., Yancopoulos G.D., Glass D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 55.Sacheck J.M., Ohtsuka A., McLary S.C., Goldberg A.L. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab. 2004;287:E591–E601. doi: 10.1152/ajpendo.00073.2004. [DOI] [PubMed] [Google Scholar]
- 56.Attaix D., Baracos V.E. MAFbx/Atrogin-1 expression is a poor index of muscle proteolysis. Curr Opin Clin Nutr Metab Care. 2010;13:223–224. doi: 10.1097/MCO.0b013e328338b9a6. [DOI] [PubMed] [Google Scholar]
- 57.Hu M.C., Lee D.F., Xia W., Golfman L.S., Ou-Yang F., Yang J.Y., Zou Y., Bao S., Hanada N., Saso H., Kobayashi R., Hung M.C. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 58.Dan H.C., Adli M., Baldwin A.S. Regulation of mammalian target of rapamycin activity in PTEN-inactive prostate cancer cells by I kappa B kinase alpha. Cancer Res. 2007;67:6263–6269. doi: 10.1158/0008-5472.CAN-07-1232. [DOI] [PubMed] [Google Scholar]
- 59.Hutti J.E., Turk B.E., Asara J.M., Ma A., Cantley L.C., Abbott D.W. IkappaB kinase beta phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-kappaB pathway. Mol Cell Biol. 2007;27:7451–7461. doi: 10.1128/MCB.01101-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia Y., Padre R.C., De Mendoza T.H., Bottero V., Tergaonkar V.B., Verma I.M. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci U S A. 2009;106:2629–2634. doi: 10.1073/pnas.0812256106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raffaello A., Milan G., Masiero E., Carnio S., Lee D., Lanfranchi G., Goldberg A.L., Sandri M. JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J Cell Biol. 2010;191:101–113. doi: 10.1083/jcb.201001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferreira R., Neuparth M.J., Vitorino R., Appell H.J., Amado F., Duarte J.A. Evidences of apoptosis during the early phases of soleus muscle atrophy in hindlimb suspended mice. Physiol Res. 2008;57:601–611. doi: 10.33549/physiolres.931272. [DOI] [PubMed] [Google Scholar]
- 63.Siu P.M., Pistilli E.E., Murlasits Z., Alway S.E. Hindlimb unloading increases muscle content of cytosolic but not nuclear Id2 and p53 proteins in young adult and aged rats. J Appl Physiol. 2006;100:907–916. doi: 10.1152/japplphysiol.01012.2005. [DOI] [PubMed] [Google Scholar]
- 64.Sakuma K., Yamaguchi A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr Aging Sci. 2010;3:90–101. doi: 10.2174/1874609811003020090. [DOI] [PubMed] [Google Scholar]
- 65.Mouisel E., Vignaud A., Hourde C., Butler-Browne G., Ferry A. Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in mTOR signaling in skeletal muscles of venerable (18–24-month-old) dystrophic mdx mice. Muscle Nerve. 2010;41:809–818. doi: 10.1002/mus.21624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.