

Abstract
Cannabinoid receptor 2 (CB2) activation is suggested to trigger the peroxisome proliferator-activated receptor-γ (PPARγ) pathway, and agonists of both receptors improve colitis. Recently, the plant metabolite (E)-β-caryophyllene (BCP) was shown to bind to and activate CB2. In this study, we examined the anti-inflammatory effect of BCP in dextran sulfate sodium (DSS)-induced colitis and analyzed whether this effect was mediated by CB2 and PPARγ. Oral treatment with BCP reduced disease activity, colonic macro- and microscopic damage, myeloperoxidase and N-acetylglucosaminidase activities, and levels and mRNA expression of colonic tumor necrosis factor-α, IL-1β, interferon-γ, and keratinocyte-derived chemokine. BCP treatment also inhibited the activation of extracellular signal-regulated kinase 1/2, nuclear factor κB, IκB-kinase α/β, cAMP response element binding and the expression of caspase-3 and Ki-67. Moreover, BCP enhanced IL-4 levels and forkhead box P3 mRNA expression in the mouse colon and reduced cytokine levels (tumor necrosis factor-α, keratinocyte-derived chemokine, and macrophage-inflammatory protein-2) in a culture of macrophages stimulated with lipopolysaccharide. The use of the CB2 antagonist AM630 or the PPARγ antagonist GW9662 significantly reversed the protective effect of BCP. Confirming our results, AM630 reversed the beneficial effect of BCP on pro-inflammatory cytokine expression in IEC-6 cells. These results demonstrate that the anti-inflammatory effect of BCP involves CB2 and the PPARγ pathway and suggest BCP as a possible therapy for the treatment of inflammatory bowel disease.
Inflammatory bowel diseases (IBDs) are a group of chronic diseases that affect the gastrointestinal tract and have been mainly subdivided as ulcerative colitis and Crohn's disease.1 The IBDs are characterized by a strong leukocyte activation and infiltration into the intestinal tissues, the release of proinflammatory cytokines2 and enzymes, and the formation of reactive oxygen species. All of these events can induce an extensive and unbalanced activation of the mucosal immune system, driven by the commensal flora.3
Recent evidence suggests a role for the cannabinoid system in IBD regulation. Cannabinoid receptors 1 and 2 (CB1 and CB2) are expressed in normal human colon4,5 and are up-regulated in IBD colonic tissue. In addition, an enhanced level of endocannabinoids was found in biopsy specimens from patients with ulcerative colitis.5 It is thought that CB1 activation results in a decrease of intestinal hypermotility and hypersecretion, whereas the activation of CB2 results in the inhibition of proinflammatory mediators. In addition, both CB2 and CB1 knockout mice are more susceptible to the development of experimental colitis, and the activation of these receptors is extremely important for the abrogation of intestinal inflammation.6 The role of CB2, however, is directly involved with innate immune system, because CB2 is primarily expressed in immune cells, such as macrophages, CD4+ and CD8+ T cells, monocytes, and polymorphonuclear neutrophils,7,8 which are intimately related to the inflammatory response in IBD.9,10 Although CB2 expression has been recently reported within the central nervous system,11 including the spinal cord, microglial cultures, brainstem, and cortex,12 it has not been associated with the central nervous system side effects of CB1 activation. This observation suggests that CB2 is a more attractive therapeutic target for the treatment of IBDs, such as ulcerative colitis.
Several studies support the notion that endocannabinoids exert their anti-inflammatory properties, at least in part, by the activation of the peroxisome proliferator-activated receptor-gamma (PPARγ) pathway.13,14 PPARγ is a member of the superfamily of nuclear receptors and has important anti-inflammatory activity,15,16 because it inhibits the activation of nuclear factor κB (NFκB) and the expression of the proinflammatory cytokines IL-1β and tumor necrosis factor-α (TNF-α).17–19 The colon expresses a high density of PPARγ,20,21 and much evidence points to the involvement of the PPARγ receptor in the regulation of intestinal inflammation.20,22
Recently, Gertsch et al23 demonstrated that the sesquiterpene (E)-β-caryophyllene (BCP) selectively binds to CB2 and acts as a full agonist. In confirming this hypothesis, the authors demonstrated that oral administration of this compound exerted potent anti-inflammatory effects in wild-type mice but not in CB2 knockout (Cnr2−/−) mice. However, the mechanisms underlying the anti-inflammatory effects of BCP are not completely understood. BCP could inhibit the pathways triggered by the activation of the Toll-like receptor complex CD14/TLR4/MD2, which typically leads to the expression of proinflammatory cytokines (IL-1β, IL-6, IL-8, and TNF-α).24 In addition, BCP suppresses extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase 1/2 (JNK1/2) signaling. All of these effects contribute to the anti-inflammatory effects of the sesquiterpene.23 BCP is present in a number of plant species that have been widely used in folk medicine for their anti-inflammatory and healing properties, including wild sage [Cordia curassavica (Jacq.) Roem. & Schult. (synonym: Cordia verbenacea DC.)] and black pepper (Piper nigrum L.).25,26
Because both PPARγ and the cannabinoid system are relevant targets for the treatment of IBD, we investigated the molecular mechanisms underlying the anti-inflammatory effects of oral BCP in experimental colitis induced by dextran sulfate sodium (DSS) administration in mice. The findings suggest that BCP seems to reduce intestinal inflammation through CB2 activation and acts directly or indirectly with the PPARγ nuclear receptor. We therefore suggest BCP as a possible therapy for the treatment of IBD.
Materials and Methods
Animals
Male CD1 mice (8 to 10 weeks of age) were obtained from the animal housing unit of the Laboratory of Experimental Pharmacology at the Federal University of Santa Catarina (Laboratório de Farmacologia Experimental–LAFEX, Universidade Federal de Santa Catarina Florianópolis, SC, Brazil). Animals were housed in collective cages at 22 ± 1°C under a 12-hour light/dark cycle (lights on at 7:00 a.m.) with free access to laboratory chow and drinking water (tap water). Experiments were performed during the light phase of the cycle. The experimental procedures were previously approved by the Federal University of Santa Catarina Committee on the Ethical Use of Animals and the study was conducted in accordance with Brazilian regulations on animal welfare.
Induction and Assessment of DSS-Induced Colitis
Male CD1 mice (5 to 7 per group) were provided with a solution of filtered water containing 3% (w/v) DSS (mol. wt. 36,000 to 50,000 kDa) ad libitum over a 5-day period. Every other day, the DSS solution was replenished and the amount consumed was measured. At the end of this 5-day period, DSS was replaced by normal drinking water for 2 days and, at the end of day 7, the animals were euthanized. Control mice received only normal drinking water. All animals were examined once a day and the disease activity index (DAI) was assessed as described previously.27,28 Briefly, the DAI combines scores for weight loss, stool consistency, and bleeding. Scores were defined as follows: stool consistency was graded 0 for no diarrhea, 2 for loose stool that did not stick to the anus, and 4 for liquid stool that did stick to the anus. The presence of fecal blood was graded 0 for none, 2 for moderate, and 4 for gross bleeding. For weight loss, a value of 0 was assigned if body weight increased or remained within 1% of baseline, 1 for a 1% to 5% loss, 2 for a 5% to 10% loss, 3 for a 10% to 15% loss, and 4 for weight loss >15%. At the end of the 7-day period, the colons were removed and examined for weight, the consistency of the stool found within, and gross macroscopic appearance and length (which was measured from 1 cm above the anus to the top of the cecum). This macroscopic scoring was performed as described previously29 (for detail, see Supplemental Table S1 at http://ajp.amjpathol.org). For each animal, the three subscores were summed to provide the total macroscopic score.
In another set of experiments, each excised portion of the distal colon was fixed immediately in a 4% (w/v) formaldehyde solution. Tissues were embedded in paraffin, sectioned (5 μm thickness), mounted on glass slides, and deparaffinized. For a general histological analysis, slices were stained using standard H&E techniques. Samples were analyzed by light microscopy and were scored as described previously29 (see Supplemental Table 2 at http://ajp.amjpathol.org). In this case, the experiments were performed in a double-blind manner.
Induction and Assessment of Oxazolone-Induced Colitis
CD1 mice were presensitized by the epicutaneous application of oxazolone [4-ethoxymethylene-2-phenyl-2-oxazolin-5-one, 3% (w/v) in 150 μL 100% ethanol] on day 7. Briefly, 1-day fasted mice were given oxazolone [3% in 100 μL 50% (v/v) ethanol] delivered using a polyethylene PE50 catheter that was inserted into the colon 4 cm proximal to the anus under anesthesia administered with an intraperitoneal injection of a mixture of ketamine and xylazine. Control mice received 100 μL of 50% ethanol under the same conditions. After colonic instillation, the animals were kept in a head-down position for 2 minutes and were kept from food and water for 4 hours (in a separate holding cage) before transfer to their home cages. Body weight was estimated daily for 72 hours after treatment, starting on the day preceding colitis induction. The number of surviving animals in each group was recorded daily to determine survival rates.
Pharmacological Treatments
Animals were orally treated by gavage with 12.5, 25, or 50 mg/kg of BCP twice a day from day 0 to day 7 (preventive treatment) or with 50 mg/kg from day 3 to day 7 (therapeutic treatment). In another set of experiments, animals received an intraperitoneal injection of the CB2 antagonist AM630 (10 mg/kg) or the PPARγ antagonist GW9662 (1 mg/kg) once a day for 7 days, given 30 minutes before the BCP (50 mg/kg). The dose of each drug was chosen based on preliminary studies or previous publications.30–32 The BCP was solubilized in a 0.9% NaCl solution and the AM630 and GW9662 were prepared in 2% dimethyl sulfoxide in saline solution. All-vehicle solutions were used for the respective control animal treatments.
To evaluate the potential effects of BCP on oxazolone-induced colitis, mice were treated twice daily for 3 days with either BCP (50 mg/kg per day, p.o.) or vehicle, starting 24 hours after oxazolone administration. All animals were sacrificed by cervical dislocation at 72 hours after oxazolone administration (ie, 4 hours after receiving the last treatment).
Myeloperoxidase and N-Acetylglucosaminidase Assays
Neutrophil and macrophage infiltration into the colon was assessed indirectly by measuring myeloperoxidase (MPO) and N-acetylglucosaminidase (NAG) activities, respectively. The MPO and NAG assays were performed as described previously.9 On day 7, animals were sacrificed and colon tissue segments were homogenized in 5% EDTA/NaCl buffer (pH 4.7) and centrifuged at 10,000 × g for 15 minutes at 4°C. The pellet was resuspended in 0.5% hexadecyl trimethyl ammonium bromide buffer (pH 5.4), and the samples were frozen in liquid nitrogen and thawed three times. On final thawing, the samples were similarly centrifuged, and 25 μL of the supernatant was used for MPO and NAG assays. The MPO enzymatic reaction was assessed by the addition of 1.6 mmol/L tetramethylbenzidine, 80 mmol/L NaPO4 and 0.3 mmol/L hydrogen peroxide (H2O2). The NAG activity measurement was determined by adding 2.25 mmol/L p-nitrophenyl-2-acetamido-β-glucopyranoside and 100 μL 50 mmol/L citrate buffer (pH 4.5). The absorbances were measured spectrophotometrically at 690 and 405 nm for MPO and NAG, respectively, and the results are expressed in optical density per milligram tissue.
Determination of Cytokine Levels
For determination of cytokine levels, colon segments were homogenized in phosphate buffer containing 0.05% Tween 20, 0.1 mmol/L phenylmethylsulfonyl fluoride, 0.1 mmol/L benzethonium chloride, 10 mmol/L EDTA, and 20 UI aprotinin A. The homogenate was centrifuged at 3000 × g for 10 minutes, and the supernatants were stored at −70°C for further analysis. Levels of TNF-α, IL-1β, keratinocyte-derived chemokine (CXCL1/KC), interferon-γ (INF-γ), IL-4, IL-10, and transforming growth factor-β were evaluated using enzyme-linked immunosorbent assay kits from R&D Systems (Minneapolis, MN) according to the manufacturer's instructions. The amount of protein in each sample was measured using the Bradford method.33
RNA Extraction and Real-Time PCR
Total RNA from colons was extracted using the TRIzol protocol, and its concentration was determined using a NanoDrop 1100 spectrophotometer (NanoDrop Technologies, Wilmington, DE). A reverse transcription assay was performed as described in the M-MLV reverse transcriptase protocol according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). cDNA (300 ng) was amplified in triplicate using the TaqMan universal PCR master mix kit with specific TaqMan gene expression target genes, the 3′ quencher MGB and FAM-labeled probes for mouse TNF-α (Mm00443258_m1), IL-1β (Mm01336189_m1), CXCL1/KC (Mm00433859_m1), IFN-γ (Mm99999071_m1), CB2 (Mm00438286_m1), PPARγ (Mm00440940_m1), forkhead box P3 (Foxp3) (Mm00475165_m1), GAPDH (NM_008084.2), rat TNF-α (Rn99999017_m1), and rat β actin (Rn00667869_m1) that was used as an endogenous control for normalization. The PCR reactions were performed in a 96-well optical reaction plate (Applied Biosystems, Foster City, CA). The thermocycler parameters were as follows: 50°C for 2 minutes, 95°C for 10 minutes, 50 cycles of 95°C for 15 seconds, and 60°C for 1 minute. Expression of the target genes was calibrated against conditions found in control animals (ie, animals that received vehicle).
Mesenteric Lymph Node Macrophage Purification and Stimulation
Mesenteric lymph nodes obtained from naïve mice were macerated in RPMI 1640 medium and filtered through a 220-μm filter. The resulting suspension was centrifuged at 1500 × g for 7 minutes, the supernatant was discarded, and the cell pellet was resuspended in RPMI 1640 medium supplemented with 10% fetal bovine serum, 20 mmol/L HEPES, 3 × 10−5 mol/L 2-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin. Macrophages were purified by adherence to plastic. Briefly, 0.1 ml of the cell suspension (1 × 106 cells/ml) were distributed in 96-well plates and incubated for 2 hours at 37°C in a 5% CO2 enriched atmosphere. Afterward, the cells were washed and the remaining cells were stimulated for 24 hours with lipopolysaccharide (LPS; 10 μg/ml) in the presence or absence of BCP (10 μmol/L) in a final volume of 200 μL/well. After stimulation, the plate was centrifuged (200 × g, 10 minutes), and the cell-free supernatant was collected and stored at −70°C for cytokine determination. The concentration of TNF-α, IL-4, MIP-2, and CXCL1/KC was measured using an enzyme-linked immunosorbent assay kit according to the manufacturer's instructions.
Murine Bone Marrow-Derived Macrophages
CD1 mice were sacrificed by cervical dislocation. Total bone marrow was obtained from mice by flushing the femurs and tibiae with Dulbecco's modified Eagle's medium. Bone marrow–derived macrophages were obtained as described previously.34 Briefly, bone marrow mononuclear phagocyte precursor cells were propagated in suspension by culturing in macrophage medium (Dulbecco's modified Eagle's medium containing glucose, supplemented with 2 mmol/L L-glutamine, 10% fetal calf serum, 10 mmol/L HEPES, 100 μg/ml streptomycin, and 100 U/ml penicillin (all from Sigma-Aldrich, St. Louis, MO) supplemented with 20% L929 cell-conditioned medium (as a source of macrophage colony stimulating factor). Cells were incubated at 37°C in a 5% CO2 enriched atmosphere and were fed on day 5 by replacing the medium supplemented with 20% L929 cell-conditioned medium. Cells were harvested on day 7 and 2 × 105 cells/ml were cultured in a 96-well cell culture plate for 24 hours. Afterward, adherent cells were stimulated for 24 hours with LPS (10 μg/ml) in the presence or absence of BCP (10 μmol/L) in a final volume of 250 μL/well. After stimulation, the plate was centrifuged (200 × g, 10 minutes) and the cell-free supernatant was collected and stored at −70°C for cytokine determination. The concentration of TNF-α and CXCL1/KC was measured using an enzyme-linked immunosorbent assay kit according to the manufacturer's instructions.
Intestinal Epithelial Cell-6 Culture
Intestinal epithelial cell-6 (IEC-6), a nontransformed rat intestinal epithelial-derived cell line, was maintained in Dulbecco's modified Eagle's medium (containing glucose and supplemented with 2 mmol/L L-glutamine, 10 mmol/L HEPES, 100 μg/ml streptomycin, and 100 U/ml penicillin). Cells (2 × 105 cells/well) were distributed in 24-well plates and all experiments were performed after cultures reached confluence (2 to 3 days after plating). After confluence was achieved, the cell culture medium was replaced. Cells were treated with BCP (10 μmol/L) for 30 minutes before stimulation.
In another set of experiments, to assess the participation of the CB2 in the effect of BCP, cells were pretreated with the CB2 selective antagonist AM630 at 1 hour before BCP treatment. Cells were then stimulated with LPS (1 μg/ml) for 4 hours in the presence or absence of BCP, AM630 (5 μmol/L), or vehicle. Control cells were incubated with the appropriate corresponding vehicles. After stimulation, the plate was centrifuged (200 × g, 10 minutes), and the cell-free supernatant was collected and stored at −70°C for determination of the protein levels for the cytokine CINC-1 and mRNA expression for TNF-α, as described above.
Immunohistochemistry Analysis
Immunohistochemical reaction analysis was performed using the colon according to methods described previously.35 Slices (5 μm thickness) were stained using the following primary antibodies and respective dilutions: polyclonal goat anti-phospho-ERK (1:300), monoclonal mouse anti-phospho-p65 NFκB (1:50), polyclonal rabbit anti-phospho-CREB (1:300), polyclonal rabbit anti-phospho-IKKα/β (1:100), polyclonal rabbit anti-cleaved-caspase-3 (1:500), polyclonal rabbit anti-claudin-4 (1:200), and polyclonal rabbit anti-Ki-67 (1:300). High-temperature antigen retrieval was performed by the immersion of the slides in a water bath at 95°C to 98°C in a 10 mmol/L trisodium citrate buffer of pH 6.0 for 45 minutes. The nonspecific binding was blocked by incubating sections for 1 hour with goat normal serum diluted in PBS. After an overnight incubation at 4°C with primary antibodies, the slides were washed with PBS and incubated with the secondary antibody Envision Plus (ready-to-use) for 1 hour at room temperature. After incubation with the appropriate biotinylated secondary antibody, the sections were developed with 3,3′-diaminobenzidine (DakoCytomation, Glostrup, Denmark) in a chromogen solution for an exact amount of time and counterstained with Harris hematoxylin.
Images of colon sections stained with antibodies to phospho-ERK, phospho-p65 NFκB, phospho-CREB, phospho-IKKα/β, cleaved caspase-3, claudin-4, and Ki-67 were acquired using a Sight DS-5M-L1 digital camera connected to an Eclipse 50i light microscope (both from Nikon, Melville, NY) and image acquisition software (Qcapture Pro 5.1; QImaging, Surrey, BC, Canada). All image analysis was performed using NIH ImageJ 1.36b imaging software (NIH, Bethesda, MD). Total pixel intensity was determined for phospho-ERK, phospho-p65 NFκB, phospho-CREB, phospho-IKKα/β, claudin-4, and Ki-67, and data were reported as optical density. To analyze the apoptosis index in the colonic tissue after DSS-administration, cleaved caspase-3 positive cells were assessed by visual inspection by counting the labeled cells in a colon section using a counting grid at ×400 magnification.
Drugs and Reagents
Dextran sulfate sodium (DSS) (mol. wt. 36,000 to 50,000 kDa) was obtained from MP Biomedicals (Solon, OH). Both BCP and oxazolone were obtained from Sigma-Aldrich. GW9662 was obtained from Calbiochem (Merck, Darmstadt, Germany). AM630 was obtained from Tocris Bioscience (Ellisville, MO). Formaldehyde was obtained from Merck (Darmstadt, Germany). Polyclonal goat anti-phospho-ERK (pERK), monoclonal mouse anti-phospho-p65 (NFκB), polyclonal rabbit anti-nuclear phospho-cyclic AMP response element binding protein (CREB), polyclonal rabbit anti-IKKα/β, and polyclonal rabbit anti-cleaved-caspase-3 were purchased from Cell Signaling Technology (Danvers, MA). Polyclonal rabbit anti-claudin-4 was purchased from Spring Bioscience (Pleasanton, CA). Polyclonal rabbit anti-Ki-67 was purchased from Abcam (Cambridge, MA). Secondary antibody Envision Plus, streptavidin–horseradish peroxidase reagent and 3,3′-diaminobenzidine chromogen were purchased from DakoCytomation (Carpinteria, CA). Hydrogen peroxide, Tween 20, Tween 80, EDTA, aprotinin, phosphate-buffered saline, eosin, hematoxylin, tetramethylbenzidine, hydrogen peroxide, and o-phenylenediamine dihydrochloride were purchased from Sigma-Aldrich. Mouse CXCL1/KC, MIP-2, IFN-γ, TNF-α, IL-4, transforming growth factor-β, IL-10, IL-1β/IL-1F2, and rat CINC-1 DuoSet kits were obtained from R&D Systems (Minneapolis, MN). TRIzol and M-MLV reverse transcriptase were purchased from Invitrogen (Carlsbad, CA). Primers and probes for mouse TNF-α (Mm00443258_m1), CXCL1/KC (Mm00433859_m1), IFN-γ (Mm99999071_m1), PPARγ (Mm00440940_m1), FoxP3 (Mm00475165_m1), CB2 (Mm00438286_m1), GAPDH (NM_008084.2), rat TNF-α (Rn99999017_m1), and rat β actin (Rn00667869_m1) TaqMan universal PCR master mix kit were purchased from Applied Biosystems (Foster City, CA).
Data Analysis
All data are expressed as means ± SEM. For nonparametric data, a Kruskal–Wallis test followed by a Dunn test was used. For parametric data, the statistical differences between groups were determined by one-way analysis of variance followed by a Student-Newman-Keuls test. Statistical analyses were performed using GraphPad Prism 4 software (GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant.
Results
β-Caryophyllene Ameliorates DSS- and Oxazolone-Induced Colitis
Mice treated with 3% DSS developed a severe illness characterized by bloody diarrhea and sustained weight loss. Preventive treatment with BCP in three different doses (12.5, 25, or 50 mg/kg, p.o.) was used to assess the potential dose-dependent effects of BCP. At the two higher doses, 25 and 50 mg/kg, BCP resulted in a significant reduction of the DAI score. The highest dose (50 mg/kg, p.o.) was more effective and exhibited an earlier effect (significant from day 4; Figure 1, A and B). For this reason, a dose of 50 mg/kg (p.o.) of BCP was used in subsequent experiments. Mice treated with BCP (50 mg/kg) were protected from marked weight loss and recovered a healthy appearance, similar to that of control mice (Figure 1C). DSS administration resulted in colon inflammation associated with hyperemia, ulceration, and bowel wall thickening leading to an increase of macroscopic colon damage and colon length reduction (Figure 1, D and E). BCP oral treatment markedly reduced macroscopic damage in a dose-dependent manner (Figure 1D) and partially prevented colon length reduction (Figure 1, E and F) on day 7 after colitis induction. Therapeutic treatment with BCP (50 mg/kg, p.o.) also promoted an improvement in clinical parameters analyzed from day 3. Of note, no difference was observed in water or DSS solution consumption between the experimental groups (data not shown). No side effects were observed with BCP alone (50 mg/kg) in acute toxicological studies; the administration of BCP alone did not alter the DAI score, microscopic damage, or MPO activity in animal colons, compared with the control group (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Figure 1.
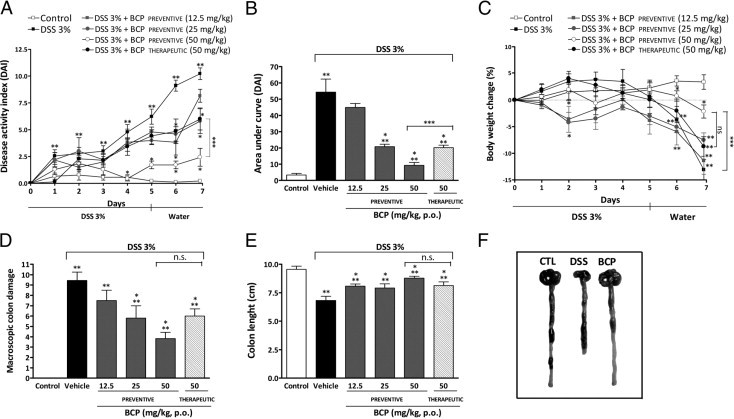
β-Caryophyllene (BCP) ameliorates DSS-induced colitis. Mice received DSS (3%) for 5 days and drinking water for the next 2 days. Animals were orally treated by gavage with 12.5, 25, or 50 mg/kg of BCP twice a day from day 0 to day 7 (preventive treatment) or with 50 mg/kg from day 3 to day 7 (therapeutic treatment). Preventive or therapeutic oral treatment with BCP improved the disease activity index (DAI) score (A and B), reduced body weight loss (C) and colon macroscopic damage (D), and enhanced colon length (E), compared with mice from the DSS group. Data are reported as means ± SEM (n = 5 to 7 mice per group). *P < 0.05 versus the DSS-treated group; **P < 0.05 versus the control group; ***P < 0.05 versus the preventive BCP-treated group (50 mg/kg, p.o.). (F) Representative photographs of colons from control mice (CTL), DSS-treated mice, and preventive BCP-treated mice (50 mg/kg, p.o.).
To investigate if BCP treatment would also prove to be beneficial in another preclinical model of colitis, we tested its effects on some parameters of colitis induced by the hapten reagent oxazolone. The rectal administration of oxazolone dissolved in ethanol induced a severe colitis in CD1 mice that was characterized by weight reduction and diarrhea, which led to high death rates. Preventive treatment with BCP (50 mg/kg, p.o., twice daily) markedly enhanced survival and body weight gain (see Supplemental Figure S2 at http://ajp.amjpathol.org).
β-Caryophyllene Treatment Inhibits Cellular Influx and Decreases Colon Damage
DSS-induced colon damage is associated with an influx of inflammatory cells, such as neutrophils and macrophages, into the intestinal mucosa.36 We therefore assessed whether the protective effect of BCP treatment in DSS-mediated colitis was associated with alterations in the composition of inflammatory cell populations in the intestinal mucosa. At 7 days after the initiation of DSS treatment, mucosal neutrophil and macrophage infiltration into the colon were indirectly assessed by measuring MPO and NAG activities, respectively. DSS-treated mice displayed a relevant increase in colonic MPO levels and a slight increase in NAG activity, compared with control animals (Figure 2, A and B). Preventive treatment with BCP (12.5, 25, or 50 mg/kg, p.o.) significantly prevented both MPO and NAG activities incrementally (Figure 2, A and B). Similar results were observed with the therapeutic treatment of BCP (50 mg/kg, p.o.).
Figure 2.
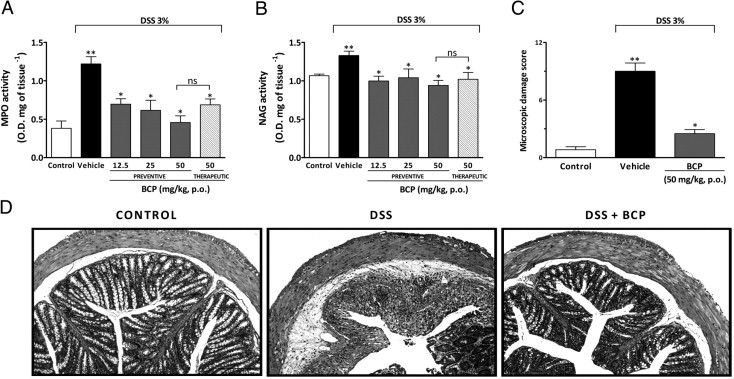
BCP decreases cell influx and reduces microscopic colon damage. At 7 days after BCP oral treatment, colon tissues were processed for histological evaluation or for the measurement of myeloperoxidase (MPO) activity and N-acetylglucosaminidase (NAG) activity. Preventive (12.5, 25, and 50 mg/kg, p.o.) or therapeutic (50 mg/kg, p.o.) treatment with BPC reduced MPO (A) and NAG (B) activity. C: Preventive treatment with BCP (50 mg/kg, p.o.) decreased the microscopic damage score in mouse colon. Data are reported as means ± SEM (n = 5 to 7 mice per group). *P < 0.05 versus the DSS-treated group; **P < 0.05 versus the control group. D: Representative paraffin sections of colons from control mice, DSS-treated, and BCP-treated mice (50 mg/kg, p.o.), stained with H&E. Original magnification, ×200.
To further confirm the MPO and NAG analysis, colons were processed for histological observation. Representative sections with H&E staining revealed a marked cell infiltration into the lamina propria and colonic mucosa in colons from DSS-treated mice. In colons from the control group, no histopathological changes were observed. The colonic tissue from DSS-treated mice appeared thick and sometimes ulcerated, showing a distortion of crypts (Figure 2, C and D). Of note, the histological evaluation of colons from BCP-treated mice revealed a pronounced reduction in the inflammatory response, resulting in a decreased microscopic damage score, compared with colons from DSS-treated mice (Figure 2, C and D).
β-Caryophyllene Treatment Reduces the Production and Expression of Inflammatory Mediators in DSS-Induced Colitis
Accumulating data in the literature suggest that cytokines and chemokines (eg, TNF-α, IL-1β, INF-γ, and CXCL1/KC) are critically involved in inflammatory cell recruitment.9,37 We therefore assessed the mRNA expression and the colonic protein levels of these inflammatory mediators on day 7 after induction of colitis. DSS administration resulted in a pronounced increase in colonic TNF-α, IL-1β, CXCL1/KC, and IFN-γ protein levels (Figure 3, A–D) and mRNA expression (Figure 3, F–I). Preventive treatment with BCP (50 mg/kg) resulted in a striking decrease in these mediators (Figure 3).
Figure 3.
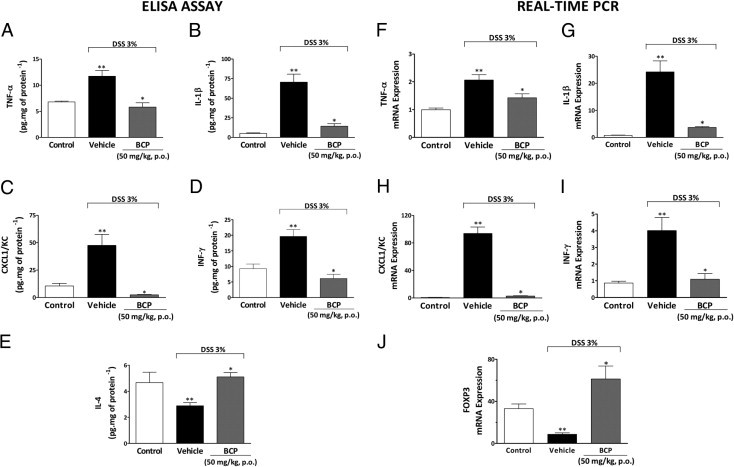
Treatment with BCP changes colonic protein levels and mRNA expression of inflammatory mediators. At the end of 7 days, colon tissue was collected and processed for cytokine levels and mRNA expression. A–E: Enzyme-linked immunosorbent assay. Preventive treatment with BCP (50 mg/kg, p.o.) reduced colonic levels of tumor necrosis factor-α (TNF-α) (A), IL-1β (B), keratinocyte-derived chemokine (CXCL1/KC) (C), and interferon–γ (IFN-γ) (D) and restored levels of IL-4 (E). F–J: Real-time PCR. The same scheme of treatment with BCP also impaired the increase colonic mRNA expression of TNF-α (F), IL-1β (G), CXCL1/KC (H), and IFN-γ (I). In addition, BCP treatment increased colonic forkhead box P3 (Foxp3) mRNA expression (J) as assessed by real-time PCR. The real-time PCR assay was performed in duplicate and GAPDH mRNA was used to normalize the relative amount of mRNA. Data are reported as means ± SEM (n = 5 to 7 mice per group). *P < 0.05 versus the DSS-treated group; **P < 0.05 versus the control group.
The modulation of colonic anti-inflammatory cytokine levels by BCP was also investigated. BCP treatment restored colonic IL-4 levels (Figure 3E), which were reduced in DSS-treated mice, but failed to change IL-10 and transforming growth factor-β levels, (see Supplemental Figure S3 at http://ajp.amjpathol.org). Additionally, preventive BCP treatment significantly increased mRNA for the transcription factor FoxP3, compared with the DSS group (Figure 3J). This result suggests that the anti-inflammatory effect displayed by BCP might be related, at least in part, to an increase in CD4+ FoxP3+ regulatory T cells. Additional experiments are needed to confirm this hypothesis.
β-Caryophyllene Reduces Cytokine Release from LPS-Stimulated Macrophages
Because the inflammatory cells present in the DSS-treated colon produce cytokines and chemokines, the observed reduction of these colonic mediators could simply be a consequence of decreased cell migration. To clarify whether BCP could also diminish the production of inflammatory mediators or modify the profile of the cytokines produced by the inflammatory cells present in colonic tissue after inflammatory stimulus, we performed a mesenteric lymph node macrophage culture and evaluated the influence of BCP on production of CXCL1/KC, macrophage inflammatory protein-2 (MIP-2), TNF-α, and IL-4 after in vitro stimulation with LPS (10 μg/ml, for 24 hours), which is an important component of colitis-induced damage. LPS stimulation markedly increased levels of CXCL1/KC, MIP-2, and TNF-α (Figure 4, A–C). This in vitro treatment with BCP (10 μmol/L) significantly reduced levels of CXCL1/KC, MIP-2, and TNF-α after administration of LPS (Figure 4, A–C). However, BCP (10 μmol/L) failed to increase IL-4 levels in the macrophage culture after LPS administration (see Supplemental Figure S4 at http://ajp.amjpathol.org).
Figure 4.
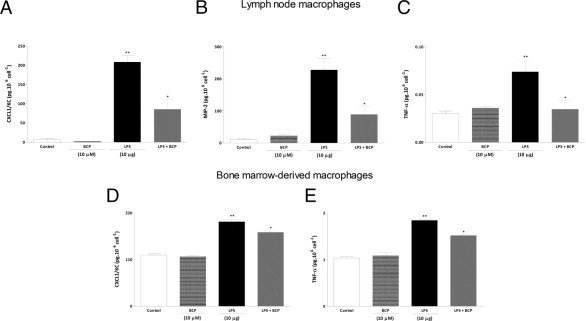
BCP reduces proinflammatory cytokine production in macrophages stimulated with LPS. Macrophages from mesenteric lymph nodes (A–C) and bone marrow (D and E) of naïve mice were stimulated with LPS (10 μg/well) in the presence or absence of BCP (10 μmol/L per well) for 24 hours, and the culture supernatants were analyzed for cytokine levels. BCP incubation reduced production of CXCL1/KC (A and D), macrophage inflammatory protein-2 (MIP-2) (B), and TNF-α (C and E). Data are reported as means ± SEM (n = 5/group). *P < 0.05 versus the LPS-treated group; *P < 0.05 versus the control group.
To investigate whether BCP treatment (10 μmol/L) could also reduce proinflammatory cytokine levels in another macrophage culture, we evaluated its effects on murine bone marrow–derived macrophages. Production of CXCL1/KC and TNF-α by LPS-stimulated bone marrow–derived macrophages was significantly reduced by pretreatment (30 minutes) with BCP (Figure 4, D and E).
β-Caryophyllene Inhibits Colonic NFκB, CREB, ERK1/2, and IKKα/β Activation Induced by DSS
Mitogen-activated protein kinases (MAPK) and transcriptional factors (eg, NFκB and CREB) are among the major proinflammatory signaling pathways involved in colitis.35,38 We therefore further investigated whether BCP treatment could prevent the activation of the p65 NFκB subunit, CREB, ERK1/2, and IκB-kinase α/β (IKKα/β). No significant activation of NFκB, CREB, ERK1/2, or IKKα/β was observed in colons from saline-treated animals (control group). Nonetheless, pronounced phosphorylation of the p65 NFκB subunit, CREB, ERK1/2, and IKKα/β were detected 7 days after beginning the treatment with DSS (Figure 5, A–D; see also Supplemental Figure S5 at http://ajp.amjpathol.org). Of note, oral treatment with BCP consistently impaired activation of this signaling pathway.
Figure 5.
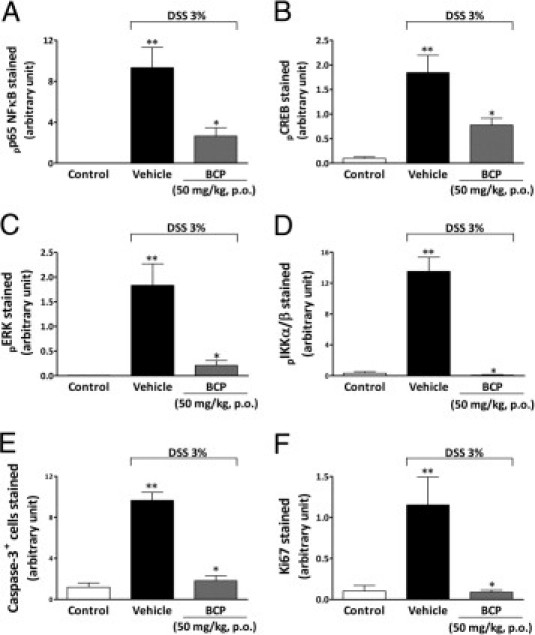
BCP treatment impairs activation of NFκB, CREB, ERK, IKKα/β, caspase-3, and Ki-67 in colonic tissue. After a 7-day BCP treatment, colon samples were processed for immunohistochemical analysis. Preventive treatment with BCP (50 mg/kg, p.o.) significantly reduced phospho-p65, a nuclear factor B subunit (pp65 NFκB) (A), phospho-cAMP response element-binding (pCREB) (B), phospho-extracellular signal-regulated kinase (pERK) (C), phospho-IκB-kinase α/β (pIKKα/β) (D), cleaved caspase-3 (E), and Ki-67 (F) immunostaining. The mean intensity of pp65 NFκB, pCREB, pERK, pIKKα/β, cleaved caspase-3, and Ki-67 staining were determined from image analysis and are represented as arbitrary units. Data are reported as means ± SEM (n = 5 to 7 mice per group). *P < 0.05 versus the DSS-treated group; **P < 0.05 versus the control group.
β-Caryophyllene Inhibits Colonic Caspase-3 and Ki-67 Activation Induced by DSS but Not Claudin-4
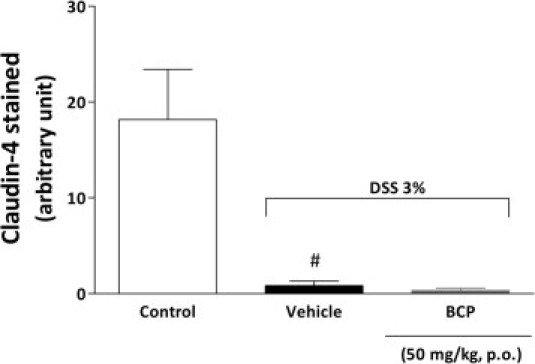
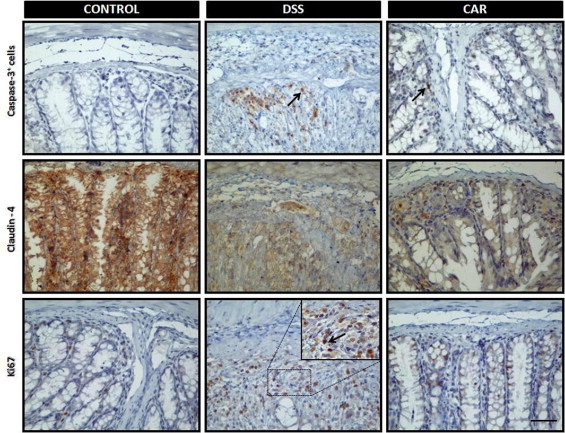
Caspase-3, Ki-67, and claudin-4 play a central role in the inflammatory process. Caspase-3 is important to cell apoptosis.39 Ki-67 is a nuclear protein necessary for cell proliferation,40 and claudin-4 is a fundamental component of tight junctions and adhesion cells.41 To analyze the role of BCP in apoptosis and in cell proliferation and adhesion, we evaluated cleaved caspase-3, Ki-67, and claudin-4 staining in colon tissue after DSS administration. Tissue sections from control mice exhibited very low levels of specific staining for cleaved caspase-3 and Ki-67 in epithelial cells (Figure 5, E and F). In contrast, at 7 days after DSS administration, the DSS group showed intense immunostaining for cleaved caspase-3 and Ki-67 expression in colon tissue; however, treatment with BCP (50 mg/kg, p.o., b.i.d.) during the 7 days after DSS administration significantly inhibited apoptosis and the proliferation index in colon tissue, as measured by monoclonal antibodies against cleaved caspase-3 and Ki-67, respectively (Figure 5, E and F; see also Supplemental Figure S6 at http://ajp.amjpathol.org). In addition, DSS-induced colitis was associated with significant decreases in the colonic expression of the tight junction protein claudin-4, but treatment with BCP did not restore the levels of this protein (see Supplemental Figure S7 at http://ajp.amjpathol.org) or immunostaining (see Supplemental Figure S6 at http://ajp.amjpathol.org).
Beneficial Effects of β-Caryophyllene on DSS-Induced Colitis Are Reversed by CB2 and PPARγ Selective Antagonists
Previous studies have demonstrated that CB2 is up-regulated in inflamed colons of patients with colitis.29,42 The present results corroborate these data by showing a great increase of CB2 mRNA expression in colonic tissue from DSS-treated mice, compared with colons from the control group (Figure 6E). Of note, preventive oral treatment with BCP markedly diminished CB2 expression in colons from mice on day 7 after DSS treatment (Figure 6E).
Figure 6.
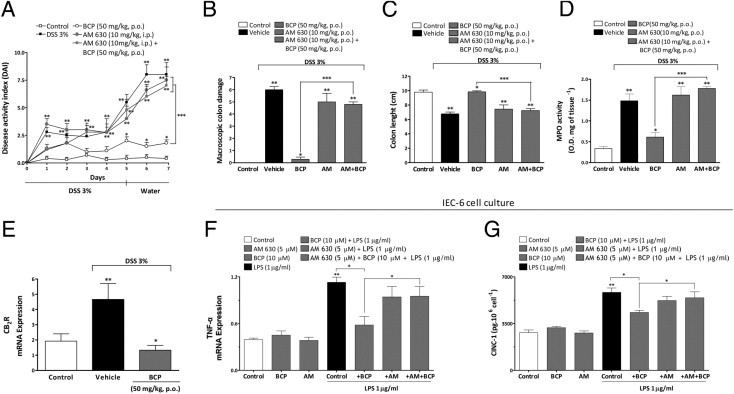
Pharmacological blockade of CB2 receptor reverses BCP anti-inflammatory effects. Mice were given DSS (3%) for 5 days and were treated from day 0 to day 7 with BCP (50 mg/kg, p.o.) alone, with the CB2 selective antagonist AM630 (AM; 10 mg/kg, i.p.) alone, or with AM630 (10 mg/kg, i.p., 30 minutes before) plus BCP (50 mg/kg, p.o.). Preventive oral treatment with BCP ameliorated all of the parameters analyzed, but treatment with AM630 plus BCP impaired the BCP improvement in DAI score (A), BCP macroscopic colon damage amelioration (B), and the BCP protective effect on colon length (C) and on MPO activity (D). At the end of 7 days, DSS enhanced mRNA expression for CB2 on mouse colonic tissue and BCP reduced the mRNA expression of CB2 (E). IEC-6 cells from rats were stimulated with LPS (1 μg/well) in the presence or absence of BCP (10 μmol/L per well) for 24 hours; the cell lysate was analyzed for TNF-α mRNA expression and secreted cytokines in the supernatant were analyzed for CINC-1 levels by enzyme-linked immunosorbent assay. BCP incubation reduced levels of TNF-α mRNA (F) and CINC-1 (G). The real-time PCR assay was performed in duplicate and GAPDH mRNA was used to normalize the relative amount of mRNA. Data are reported as means ± SEM (n = 5 to 7 mice per group). *P < 0.05 versus the DSS-treated group; **P < 0.05 versus the control group; ***P < 0.05 versus the BCP-treated group.
BCP has been shown previously to be a selective agonist of cannabinoid receptor 2.23 Importantly, CB2 activation decreases colon inflammation in different experimental models.29,43 We investigated whether a CB2 selective antagonist could prevent the anti-inflammatory effect of BCP. For this purpose, mice were treated once a day with the selective CB2 antagonist AM630 (10 mg/kg, p.o.), alone or in combination with BCP, for 7 days. Treatment with AM630 alone did not alter the inflammatory parameters observed in the DSS-treated group (Figure 6, A–D); however, AM630 notably abolished the protective effect of BCP against macroscopic colon damage, colon length reduction and MPO activity (Figure 6, A–D).
We investigated how the activation of the CB2 might be implicated in the beneficial effect of BCP in LPS-stimulated IEC-6 cells. The in vitro stimulation of IEC-6 cells with LPS for 4 hours increased mRNA for TNF-α and cytokine CINC-1 levels, which were reduced by preincubation with BCP (10 μmol/L) (Figure 6, F and G). Treatment with AM630 alone did not alter the cytokine levels observed in the LPS-treated group; however, AM630 notably abolished the anti-inflammatory effect of BCP, indicating a functional CB2-dependent mechanism in the production of inflammatory mediators.
Some recent studies have shown a close interaction between the CB2 and PPARγ.13,44 Indeed, PPARγ agonists ameliorate experimental colitis,45–47 and PPARγ expression is reduced in patients with ulcerative colitis.20 To verify whether PPARγ was also involved in colitis improvement after BCP treatment, the PPARγ antagonist GW9662 (1 mg/kg, i.p.) was administered once a day from day 0 to day 7, alone or in combination with BCP. As was observed for CB2 antagonism, the PPARγ antagonist significantly blocked the beneficial effect of BCP in DSS-induced colitis by partially reversing the DAI score, macroscopic colon damage, and colon length and by restoring the activity of MPO (Figure 7, A–D). Furthermore, DSS-treated mice showed a reduction in mRNA expression for PPARγ, but treatment with BCP partially restored PPARγ expression (Figure 7E).
Figure 7.
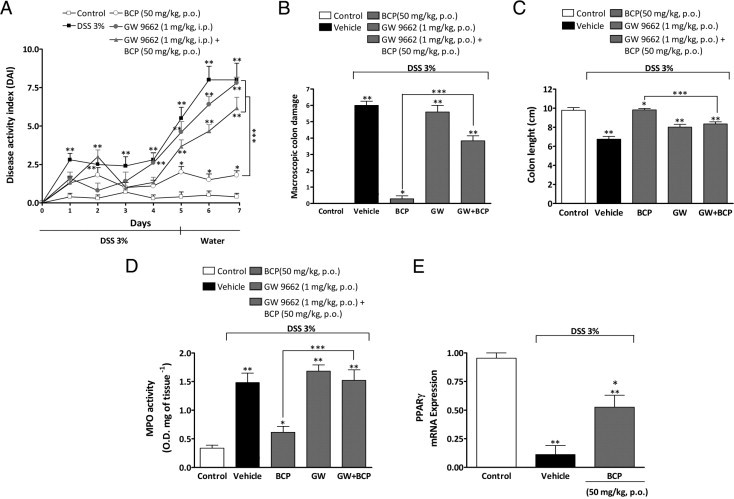
Pharmacological blockade of PPARγ receptor partially reverses BCP anti-inflammatory effects on DSS-induced colitis. Mice were given DSS (3%) for 5 days and were treated from day 0 to day 7 with BCP (50 mg/kg, p.o.) alone, the PPARγ selective antagonist GW9662 (1 mg/kg, i.p.) alone, or with GW9662 (1 mg/kg, i.p., 30 minutes before) plus BCP (50 mg/kg, p.o.). Preventive oral treatment with BCP ameliorated all clinical parameters analyzed, but treatment with GW9662 plus BCP partially reduced BCP improvement in DAI score (A), BCP macroscopic colon damage amelioration (B), and BCP protective effect of on colon length (C) and on MPO activity (D). At the end of 7 days, DSS diminished mRNA expression for PPARγ in the mouse colon and BCP partially recovered the mRNA expression of PPARγ (E). Real-time PCR assay was performed in duplicate; GAPDH mRNA was used to normalize the relative amount of mRNA. Data are reported as means ± SEM (n = 5 to 7 mice per group). *P < 0.05 versus the DSS-treated group; **P < 0.05 versus the control group; ***P < 0.05 versus the BCP-treated group.
Discussion
IBDs are chronic debilitating diseases of the gastrointestinal tract that affect millions of people worldwide. Current therapies for IBDs remain inadequate. In recent years, much effort has gone into identifying promising new targets for IBD treatment. In this context, the use of herbal therapy or active compounds from plants constitutes an attractive approach for IBD treatment.48,49 BCP is a secondary metabolite found in many plant species,50,51 and it exhibits long-lasting oral and topical anti-inflammatory properties in different inflammatory models.49,52
Corroborating a previous report, the present study demonstrated that BCP, given orally, consistently ameliorated the inflammatory signs associated with DSS-induced colitis, such as decreased body weight and colon length and increased colonic tissue damage,53 in both therapeutic and preventive treatment. In all parameters analyzed, preventive treatment with BCP was more effective than therapeutic treatment at the highest dose (50 mg/kg, p.o.), but the significant difference was small, and both treatments are likely to be of use for clinical applications of BCP. In addition, in the present study the improvement in DSS-induced colitis by BCP oral treatment was associated with a direct reduction in colonic cytokines, chemokines, neutrophil migration, and macrophage infiltration and activation. There was also an increase in IL-4 levels and Foxp3 expression. Of great relevance, the present study clearly showed that BCP treatment markedly reduced activation of ERK1/2, p65 NFκB, IKKα/β, CREB, caspase-3, and Ki-67. Notably, the beneficial effect of BCP was dependent on CB2 and PPARγ activation. We also revealed a novel finding, that BCP pretreatment ameliorated oxazolone-induced colitis by decreasing weight loss and enhancing survival, suggesting that the anti-inflammatory effect of BCP is not dependent on the animal model used.
Both macrophages and neutrophils have pathogenic roles in animal models of IBD.9,54 Mononuclear cell infiltration was analyzed indirectly by measuring NAG activity. Colon tissues had a high number of resident mononuclear cells, which was significantly enhanced 7 days after DSS administration. Treatment with BCP prevented the increase in NAG activity, suggesting an impairment of mononuclear cell infiltration. BCP also decreases MPO activity in inflamed colons53 and in rat paws treated with carrageenan49 or LPS.52 The reduction in cellular infiltration observed in the present study was associated with a decrease in colon damage. It has been suggested that the inhibition of migratory mononuclear cells,55 but not resident cells,56 ameliorates colitis. Similarly, the infiltration of neutrophils into the colonic mucosa is believed to play a key role in mediating tissue damage and the clinical symptoms in experimental colitis.9
The pathway underlying the BCP protection of colon damage observed with both preventive and therapeutic treatment could be associated with diminished cleaved caspase-3 and Ki-67, which are fundamental proteins involved in apoptosis and cell proliferation, respectively.39,40 These events are associated with chronic inflammation and colon cancer and are increased under these conditions.57 Furthermore, hyperproliferation and the abnormal distribution of replicating cells along the length of the colonic crypts are a marker of increased susceptibility to colorectal cancer and chronic inflammation.57 In the present study, BCP oral treatment significantly reduced caspase-3 and Ki-67 protein activation in colonic tissue, suggesting that BCP ameliorates acute colitis by reducing cell proliferation and apoptosis. However, these events might be an indirect consequence of a general improvement of inflammation by BCP. In addition, we observed that DSS-induced colitis was associated with significant decreases in the colonic expression of the tight junction protein claudin-4, as described previously by Oshima et al.58 BCP treatment did not, however, restore the levels of this protein. Our data suggest that the anti-inflammatory effects of BCP do not rely on its ability to modify tight-junction protein expression, such as claudin-4.
The prevention of cell infiltration and colon damage observed in mice orally treated with BCP was also associated with reduced protein levels and mRNA expression of TNF-α, IL-1β, INF-γ, and CXCL1/KC, which are soluble mediators involved in cellular migration and adhesion molecule up-regulation.37 The decreased levels of colonic cytokines might therefore explain the reduction of cell infiltration; however, because inflammatory cells produce cytokines and chemokines, the reduction of these mediators in the colon could be simply a consequence of a decreased cell migration. To clarify this hypothesis, we performed a lymph node and bone marrow–derived macrophage culture. We observed that BCP reduced CXCL1/KC, TNF-α, and MIP-2 levels by LPS-induced inflammation and the mRNA expression of CXCL1/KC and TNFα. This indicates that BCP not only decreases cell migration but also modulates cell activation. This finding confirms and extends previous work showing that BCP inhibits IL-1β and TNF-α protein expression in human peripheral whole blood stimulated by LPS.23
The transcription of several cytokines and chemokines are controlled by ERK-NFκB,59 and this transcription factor is activated during experimental colitis.35 We therefore investigated whether the effect of BCP in decreasing the inflammatory mediators was associated with the ERK-NFκB signaling pathway. Our data demonstrated a marked inhibition of the p65 NFκB subunit and ERK-1/2 in the colons of mice treated with BCP. These results are in agreement with previous studies, in which BCP inhibited NFκB activation in the footpad of rats injected with LPS52 and diminished LPS-induced ERK1/2 phosphorylation in human monocytes.23 Moreover, a strong activation of IKKα/β was observed in DSS-treated mice, and this effect was suppressed by BCP treatment. Eckmann et al60 showed that IKKα/β is closely involved in the proinflammatory response in acute colitis induced by DSS, suggesting that blockade of this kinase may be an important target for decreasing the inflammatory response.
Another transcription factor known to be involved in experimental colitis is CREB.35 Genes whose transcription is regulated by CREB include those encoding the T helper type 1 (TH1) cytokines.61,62 The TH1 cytokines encompass the cytokine profile of acute murine DSS-induced colitis (INF-γ, TNF-α, and IL-1β).63 In TH1 cells, the recruitment of phospho-CREB is significantly higher than in TH2 cells.64 In the present study, we demonstrated that BCP treatment decreased DSS-induced CREB activation, which may be linked to decreased TH1 cytokines, such as INF-γ, and the enhancement of TH2 cytokines, namely IL-4. In addition, IL-4 is associated with alternative macrophage activation, polarizing macrophages to the anti-inflammatory phenotype (M2).65 Of note, in vitro macrophage treatment with BCP decreased proinflammatory cytokine production. TH2 regulatory T cells can also produce IL-4.66 The present study demonstrated that BCP treatment enhanced the expression of FoxP3, the most reliable regulatory T-cell marker. Therefore, those cells could also be the source of IL-4. Nonetheless, regulatory T cells are known to produce transforming growth factor-β and IL-10, anti-inflammatory cytokines that were not altered by BCP treatment. The role of these cells in amelioration of BCP-induced colitis should be further examined.
Polarization toward TH2 immunity can also be achieved by cannabinoids in a variety of models. Recently, Gertsch et al23 reported that BCP is a CB2 agonist. CB2 is expressed mainly in immune cells, and its activation limits intestinal inflammation30 and reduces proinflammatory mediator release.67 The present study demonstrated that the anti-inflammatory effect of BCP was almost completely reversed by the selective CB2 antagonist AM630, strongly suggesting that a great part of the anti-inflammatory properties of BCP observed in DSS-induced colitis was mediated by CB2 activation. One of the possible mechanisms of action of BCP is the activation of CB2s and the consequent regulation of the activation and balance of TH1/TH2 cells. To confirm this hypothesis, we performed another experiment in which we measured both the levels and expression of some cytokines in vitro. Our results showed that the incubation with AM630 reversed the inhibitory effect of BCP on levels of proinflammatory chemokine CINC-1/CXCL1 (cytokine chemoattractant for neutrophils) and on TNF-α mRNA expression in the IEC-6 cell line.
We performed another experiment to explore further whether BCP triggers CB2-dependent effects on cytokine levels/expression in vitro. Our results showed that the incubation with the CB2 antagonist AM630 reversed the inhibitory effect of BCP on proinflammatory chemokine CINC-1/CXCL1 levels and TNF-α mRNA expression in the IEC-6 cell line. These results are in agreement with Gertsch et al,23 who found that AM630 reversed the inhibitory effect of BCP on IL-1β and TNF-α protein expression in human peripheral whole blood. The present results provide convincing experimental evidence indicating that the effects of BCP depend on CB2 activation.
A shift toward a TH2 cytokine dominance has been observed also in DSS-induced inflammation after PPARγ ligand treatment. The PPARγ ligands troglitazone, pioglitazone, and rosiglitazone decrease INF-γ and TNF-α levels and increase IL-4 levels and GATA-3 expression.68 Other evidence supports a role of the PPARγ in the regulation of intestinal inflammation: i) there is an impaired PPARγ expression in colonic epithelial cells of patients with ulcerative colitis20; ii) PPARγ agonists dramatically reduce disease severity in mice22; and iii) an increased susceptibility to colitis is observed in PPARγ heterozygous mice (PPARγ+/−).22 A novel finding in the present study was that the anti-inflammatory effects of BCP were markedly reduced by the PPARγ selective antagonist GW9662, demonstrating an important role of this nuclear transcription factor in the mechanism of the anti-inflammatory action of BCP.
Some evidence suggests cross-talk between PPARγ and the cannabinoid pathways. Liu et al13 demonstrated the activation and binding of PPARγ by synthetic cannabinoid. O'Sullivan et al14 demonstrated that Δ9-tetrahydrocannabinol causes vasorelaxation through activation of PPARγ. Both CB2 and PPARγ agonists reduce MPO activity29,69 and increase a TH2 cytokine profile68,70 in experimental colitis in mice. In addition, the activation of CB2 and/or PPARγ significantly reduces CXCL1/KC and MIP-2 tissue levels71,72 and inhibits NFκB, IKKα/β, and CREB activation.45,73–75 As we have noted, all of these effects were observed in the colons from mice treated with BCP. Collectively, therefore, these data reinforce the important role of CB2 and PPARγ activation in BCP anti-inflammatory activity.
Although BCP was suggested to act mainly on CB2, high doses of BCP induce anti-edematogenic effects in CB2 knockout mice,23 indicating that BCP may not act exclusively on this receptor. In fact, some CB2 agonists can directly bind and activate PPARγ.7,13,76 It is noteworthy that BCP is a lipophilic molecule that easily crosses the cell membrane23 and that could, therefore, exert its pharmacological effects by acting directly on intracellular pathways. Indeed, the exact mechanisms by which cannabinoids interact with PPARs remain unclear.
Three potential pathways have been suggested to be involved in the mechanism of the activation of PPARs by cannabinoids: i) direct binding to PPARs; ii) the conversion of cannabinoids into metabolites that in turn activate PPARs; and iii) the interaction of cannabinoids with cell surface receptors, initiating intracellular signaling cascades that lead to the activation of PPARs. The present study demonstrated that the beneficial effects of BCP on DAI score were consistently reversed by CB2 and PPARγ antagonists. Therefore, CB2 activation by BCP may trigger PPARγ activation. We cannot, however, dismiss the possibility of direct action of BCP on the PPARγ pathway. Furthermore, the fact that agonists of both CB2 and PPARγ significantly improved experimental colitis could not be directly related with their expression in inflamed tissue, but it is not fully understood why mRNA levels of CB2 is overexpressed but mRNA levels of PPARγ are underexpressed during DSS-induced colitis.
In conclusion, the present results demonstrate that the plant-derived sesquiterpene BCP, given orally, exhibits both preventive and therapeutic effects in DSS-induced colitis in mice. The mechanisms were associated with activation of CB2 and PPARγ receptors, leading to the inhibition of proinflammatory cytokines and inflammatory cell influx. All of these effects were also associated with the inhibition of nuclear factors NFκB and CREB and a shift toward TH2 cytokines. Taken together, the present findings strongly suggest that BCP could constitute an attractive and apparently safe molecule for development of new anti-inflammatory drugs with therapeutic potential for use in treatment of human IBDs, such as ulcerative colitis and Crohn's disease.
Acknowledgment
We thank Prof. Dr. Patrícia Bozza for providing us with the IEC-6 cell line.
Footnotes
Supported by the National Council for Scientific and Technological Development (CNPq: Conselho Nacional de Desenvolvimento Científico e Tecnológico) (to A.F.B., R.M. and R.C.D., R.F.C., M.C. and D.F.P.L), Coordination for Improvement of Higher Education (CAPES: Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), and the Foundation for Research Support-State of Santa Catarina (FAPESC: Fundação de Apoio à Pesquisa Científica Tecnológica do Estado de Santa Catarina) to A.F.B., R.M., R.C.D., R.F.C., and M.C. (who received grants from CNPq).
A.F.B. and R.M. contributed equally to this study.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.11.052.
Supplementary data
Supplemental Figure S1.
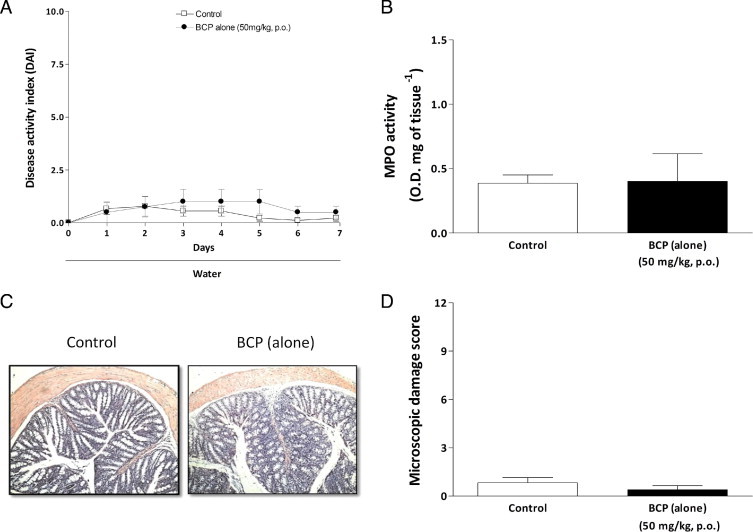
Treatment with BCP alone did not alter the analyzed parameters. Oral treatment (50 mg/kg, p.o., b.i.d.) during 7 days without DSS challenge did not alter the disease activity index (DAI) score (A), MPO activity (B), or colon microscopic damage (D), compared with mice from the control group. C: Representative paraffin sections stained with H&E. Data are reported as means ± SEM (n = 5–7 mice per group).
Supplemental Figure S2.
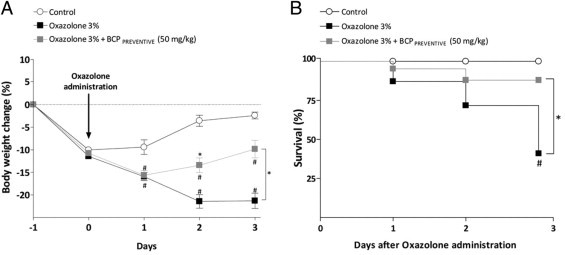
BCP preventive treatment ameliorates oxazolone-induced colitis. Throughout the experiment mice were daily monitored for body weight loss and overall mortality up to 72 hours after oxazolone administration. Preventive treatment with BCP (50 mg/kg, p.o., b.i.d.) recovered mouse weight (A) and increased survival (B), compared with the oxazolone-treated group. Data are reported as means ± SEM (n = 6–8 mice per group). *P < 0.05 versus oxazolone-treated group; #P < 0.05 versus control group.
Supplemental Figure S3.
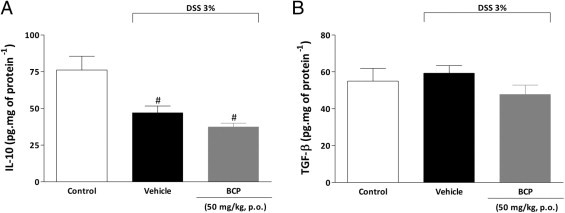
Treatment with BCP did not alter IL-10 and TGF-β protein levels in mouse colon. At the end of the 7-day period, colons were collected and processed for cytokine levels in tissue. DSS decreased colonic levels of IL-10 (A), but did not change levels of transforming growth factor β (TGF-β) (B) as analyzed by enzyme-linked immunosorbent assay. BCP (50 mg/kg, p.o.) preventive treatment did not alter levels of either IL-10 (A) or TGF-β (B). Data are reported as means ± SEM (n = 5–7 mice per group). #P < 0.05 versus control group.
Supplemental Figure S4.
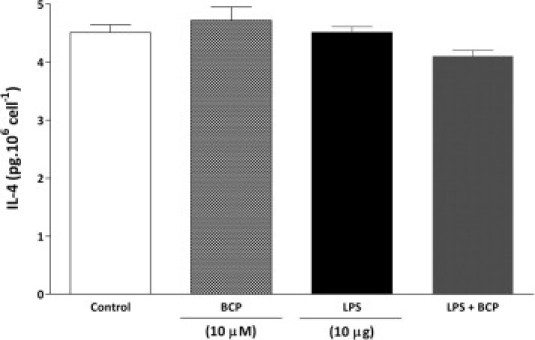
BCP did not alter IL-4 cytokine production induced by LPS in mesenteric lymph node macrophage. Macrophages from mesenteric lymph nodes of naïve mice were stimulated with LPS (10 μg/well) in the presence or absence of BCP (10 μmol/L per well) for 24 hours, and culture supernatants were analyzed for IL-4 cytokine levels. Neither LPS nor BCP incubation changed IL-4 production on mesenteric lymph node macrophages. Data are reported as means ± SEM (n = 5/group).
Supplemental Figure S5.
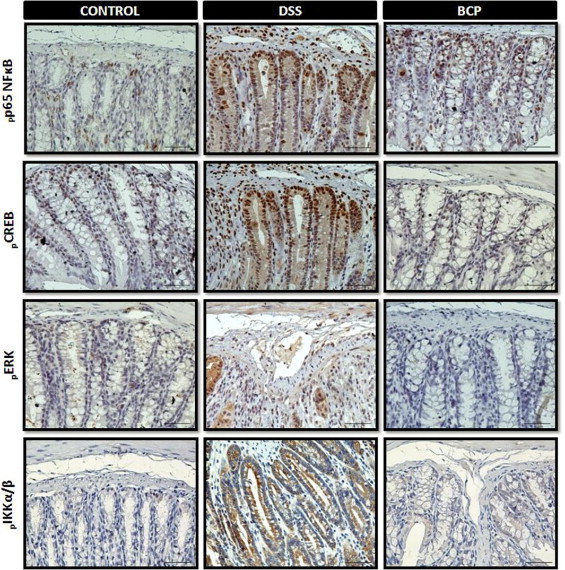
BCP preventive treatment reduced protein activation in the mouse colon with colitis. At 7 days after BCP treatment, colon samples were processed for immunohistochemistry analysis. Preventive treatment with BCP (50 mg/kg, p.o.) significantly reduced phospho-p65 subunit of nuclear factor kappa B (pp65 NFκB), phospho-cAMP response element-binding (pCREB), phospho-extracellular signal-regulated kinase (pERK), and phospho-IκB-kinase α/β (pIKKα/β) immunostaining. Four ocular fields per section and 5–8 mice per group were used in immunohistochemistry assay. Images are representative of 5–7 mice per group mice. Scale bar = 50 μm (all images).
Supplemental Figure S6.
BCP preventive treatment decreased caspase 3 positivity and Ki-67 expression induced by DSS, but was not able to restore claudin-4 levels in colonic tissue. After 7 days of BCP treatment, colon samples were processed for immunohistochemistry analysis. Preventive treatment with BCP (50 mg/kg, p.o.) significantly reduced caspase-3 positivity and Ki-67, but did not restore claudin-4 immunostaining. Four ocular fields per section were used in the immunohistochemistry assay (n = 5–8 mice). Images are representative of 5–7 mice per group. Scale bar = 50 μm (all images).
Supplemental Figure S7.
BCP did not restore claudin-4 levels in colonic tissue. At 7 days after BCP treatment, colon samples were processed for immunohistochemistry analysis. Preventive treatment with BCP (50 mg/kg, p.o.) was not able to restore claudin-4 levels. The mean intensity of claudin-4 staining was determined from image analysis and represented as arbitrary units. Data are reported as means ± SEM (n = 5–7 mice per group). #P < 0.05 versus control group.
References
- 1.Cuzzocrea S. Emerging biotherapies for inflammatory bowel disease. Expert Opin Emerg Drugs. 2003;8:339–347. doi: 10.1517/14728214.8.2.339. [DOI] [PubMed] [Google Scholar]
- 2.Podolsky D.K. The current future understanding of inflammatory bowel disease. Best Pract Res Clin Gastroenterol. 2002;16:933–943. doi: 10.1053/bega.2002.0354. [DOI] [PubMed] [Google Scholar]
- 3.McGuckin M.A., Eri R., Simms L.A., Florin T.H., Radford-Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15:100–113. doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- 4.Roberds S.L., Anderson J., Basi G., Bienkowski M.J., Branstetter D.G., Chen K.S., Freedman S.B., Frigon N.L., Games D., Hu K., Johnson-Wood K., Kappenman K.E., Kawabe T.T., Kola I., Kuehn R., Lee M., Liu W., Motter R., Nichols N.F., Power M., Robertson D.W., Schenk D., Schoor M., Shopp G.M., Shuck M.E., Sinha S., Svensson K.A., Tatsuno G., Tintrup H., Wijsman J., Wright S., McConlogue L. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- 5.D'Argenio G., Valenti M., Scaglione G., Cosenza V., Sorrentini I., Di Marzo V. Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB J. 2006;20:568–570. doi: 10.1096/fj.05-4943fje. [DOI] [PubMed] [Google Scholar]
- 6.Engel MA, Kellermann CA, Burnat G, Hahn EG, Rau T, Konturek PC: Mice lacking cannabinoid CB1-, CB2-receptors or both receptors show increased susceptibility to trinitrobenzene sulfonic acid (TNBS)-induced colitis. J Physiol Pharmacol 61:89–97 [PubMed]
- 7.Galiègue S., Mary S., Marchand J., Dussossoy D., Carrière D., Carayon P., Bouaboula M., Shire D., Le Fur G., Casellas P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 8.Maresz K., Pryce G., Ponomarev E.D., Marsicano G., Croxford J.L., Shriver L.P., Ledent C., Cheng X., Carrier E.J., Mann M.K., Giovannoni G., Pertwee R.G., Yamamura T., Buckley N.E., Hillard C.J., Lutz B., Baker D., Dittel B.N. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med. 2007;13:492–497. doi: 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
- 9.Bento A.F., Leite D.F., Claudino R.F., Hara D.B., Leal P.C., Calixto J.B. The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. J Leukoc Biol. 2008;84:1213–1221. doi: 10.1189/jlb.0408231. [DOI] [PubMed] [Google Scholar]
- 10.Kühl A.A., Kakirman H., Janotta M., Dreher S., Cremer P., Pawlowski N.N., Loddenkemper C., Heimesaat M.M., Grollich K., Zeitz M., Farkas S., Hoffmann J.C. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 11.Morgan N.H., Stanford I.M., Woodhall G.L. Functional CB2 type cannabinoid receptors at CNS synapses. Neuropharmacology. 2009;57:356–368. doi: 10.1016/j.neuropharm.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Van Sickle M.D., Duncan M., Kingsley P.J., Mouihate A., Urbani P., Mackie K., Stella N., Makriyannis A., Piomelli D., Davison J.S., Marnett L.J., Di Marzo V., Pittman Q.J., Patel K.D., Sharkey K.A. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 13.Liu J., Li H., Burstein S.H., Zurier R.B., Chen J.D. Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Mol Pharmacol. 2003;63:983–992. doi: 10.1124/mol.63.5.983. [DOI] [PubMed] [Google Scholar]
- 14.O'Sullivan S.E., Tarling E.J., Bennett A.J., Kendall D.A., Randall M.D. Novel time-dependent vascular actions of Delta9-tetrahydrocannabinol mediated by peroxisome proliferator-activated receptor gamma. Biochem Biophys Res Commun. 2005;337:824–831. doi: 10.1016/j.bbrc.2005.09.121. [DOI] [PubMed] [Google Scholar]
- 15.Rousseaux C., Lefebvre B., Dubuquoy L., Lefebvre P., Romano O., Auwerx J., Metzger D., Wahli W., Desvergne B., Naccari G.C., Chavatte P., Farce A., Bulois P., Cortot A., Colombel J.F., Desreumaux P. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaefer K.L., Denevich S., Ma C., Cooley S.R., Nakajima A., Wada K., Schlezinger J., Sherr D., Saubermann L.J. Intestinal antiinflammatory effects of thiazolidenedione peroxisome proliferator-activated receptor-gamma ligands on T helper type 1 chemokine regulation include nontranscriptional control mechanisms. Inflamm Bowel Dis. 2005;11:244–252. doi: 10.1097/01.mib.0000160770.94199.9b. [DOI] [PubMed] [Google Scholar]
- 17.Jiang C., Ting A.T., Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 18.Ricote M., Li A.C., Willson T.M., Kelly C.J., Glass C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 19.Wahli W. A gut feeling of the PXR, PPAR and NF-kappaB connection. J Intern Med. 2008;263:613–619. doi: 10.1111/j.1365-2796.2008.01951.x. [DOI] [PubMed] [Google Scholar]
- 20.Dubuquoy L., Jansson E.A., Deeb S., Rakotobe S., Karoui M., Colombel J.F., Auwerx J., Pettersson S., Desreumaux P. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology. 2003;124:1265–1276. doi: 10.1016/s0016-5085(03)00271-3. [DOI] [PubMed] [Google Scholar]
- 21.Lefebvre M., Paulweber B., Fajas L., Woods J., McCrary C., Colombel J.F., Najib J., Fruchart J.C., Datz C., Vidal H., Desreumaux P., Auwerx J. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J Endocrinol. 1999;162:331–340. doi: 10.1677/joe.0.1620331. [DOI] [PubMed] [Google Scholar]
- 22.Buisine M.P., Desreumaux P., Leteurtre E., Copin M.C., Colombel J.F., Porchet N., Aubert J.P. Mucin gene expression in intestinal epithelial cells in Crohn's disease. Gut. 2001;49:544–551. doi: 10.1136/gut.49.4.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gertsch J., Leonti M., Raduner S., Racz I., Chen J.Z., Xie X.Q., Altmann K.H., Karsak M., Zimmer A. Beta-caryophyllene is a dietary cannabinoid. Proc Natl Acad Sci USA. 2008;105:9099–9104. doi: 10.1073/pnas.0803601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gertsch J. Anti-inflammatory cannabinoids in diet: towards a better understanding of CB(2) receptor action? Commun Integr Biol. 2008;1:26–28. doi: 10.4161/cib.1.1.6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orav A., Stulova I., Kailas T., Müürisepp M. Effect of storage on the essential oil composition of Piper nigrum L. fruits of different ripening states. J Agric Food Chem. 2004;52:2582–2586. doi: 10.1021/jf030635s. [DOI] [PubMed] [Google Scholar]
- 26.de Carvalho P.M., Jr, Rodrigues R.F., Sawaya A.C., Marques M.O., Shimizu M.T. Chemical composition and antimicrobial activity of the essential oil of Cordia verbenacea D.C. J Ethnopharmacol. 2004;95:297–301. doi: 10.1016/j.jep.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 27.Ghia J.E., Blennerhassett P., Collins S.M. Impaired parasympathetic function increases susceptibility to inflammatory bowel disease in a mouse model of depression. J Clin Invest. 2008;118:2209–2218. doi: 10.1172/JCI32849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandenbark A.A., Chou Y.K., Whitham R., Mass M., Buenafe A., Liefeld D., Kavanagh D., Cooper S., Hashim G.A., Offner H. Treatment of multiple sclerosis with T-cell receptor peptides: results of a double-blind pilot trial [Erratum appeared in Nat Med 1997;3:240] Nat Med. 1996;2:1109–1115. doi: 10.1038/nm1096-1109. [DOI] [PubMed] [Google Scholar]
- 29.Kimball E.S., Wallace N.H., Schneider C.R., D'Andrea M.R., Hornby P.J. Vanilloid receptor 1 antagonists attenuate disease severity in dextran sulphate sodium-induced colitis in mice. Neurogastroenterol Motil. 2004;16:811–818. doi: 10.1111/j.1365-2982.2004.00549.x. [DOI] [PubMed] [Google Scholar]
- 30.Storr M.A., Keenan C.M., Zhang H., Patel K.D., Makriyannis A., Sharkey K.A. Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm Bowel Dis. 2009;15:1678–1685. doi: 10.1002/ibd.20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Napimoga M.H., Vieira S.M., Dal-Secco D., Freitas A., Souto F.O., Mestriner F.L., Alves-Filho J.C., Grespan R., Kawai T., Ferreira S.H., Cunha F.Q. Peroxisome proliferator-activated receptor-gamma ligand, 15-deoxy-Delta12,14-prostaglandin J2, reduces neutrophil migration via a nitric oxide pathway. J Immunol. 2008;180:609–617. doi: 10.4049/jimmunol.180.1.609. [DOI] [PubMed] [Google Scholar]
- 32.Rogerio A.P., Andrade E.L., Leite D.F., Figueiredo C.P., Calixto J.B. Preventive and therapeutic anti-inflammatory properties of the sesquiterpene alpha-humulene in experimental airways allergic inflammation. Br J Pharmacol. 2009;158:1074–1087. doi: 10.1111/j.1476-5381.2009.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 34.Stanley E.R. Murine bone marrow-derived macrophages. Methods Mol Biol. 1997;75:301–304. doi: 10.1385/0-89603-441-0:301. [DOI] [PubMed] [Google Scholar]
- 35.Vitor C.E., Figueiredo C.P., Hara D.B., Bento A.F., Mazzuco T.L., Calixto J.B. Therapeutic action and underlying mechanisms of a combination of two pentacyclic triterpenes, alpha- and beta-amyrin, in a mouse model of colitis. Br J Pharmacol. 2009;157:1034–1044. doi: 10.1111/j.1476-5381.2009.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baumgart D.C., Carding S.R. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 37.Barker J.N., Jones M.L., Swenson C.L., Sarma V., Mitra R.S., Ward P.A., Johnson K.J., Fantone J.C., Dixit V.M., Nickoloff B.J. Monocyte chemotaxis and activating factor production by keratinocytes in response to IFN-gamma. J Immunol. 1991;146:1192–1197. [PubMed] [Google Scholar]
- 38.Arulampalam V., Pettersson S. Uncoupling the p38 MAPK kinase in IBD: a double edged sword? Gut. 2002;50:446–447. doi: 10.1136/gut.50.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrington H.A., Ho K.L., Ghosh S., Tung K.C. Construction and analysis of a modular model of caspase activation in apoptosis. Theor Biol Med Model. 2008;5:26. doi: 10.1186/1742-4682-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerdes J., Lemke H., Baisch H., Wacker H.H., Schwab U., Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 41.Tsukita S., Furuse M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann N Y Acad Sci. 2000;915:129–135. doi: 10.1111/j.1749-6632.2000.tb05235.x. [DOI] [PubMed] [Google Scholar]
- 42.Klein T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 43.Massa F., Marsicano G., Hermann H., Cannich A., Monory K., Cravatt B.F., Ferri G.L., Sibaev A., Storr M., Lutz B. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113:1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burstein S. PPAR-gamma: a nuclear receptor with affinity for cannabinoids. Life Sci. 2005;77:1674–1684. doi: 10.1016/j.lfs.2005.05.039. [DOI] [PubMed] [Google Scholar]
- 45.Su C.G., Wen X., Bailey S.T., Jiang W., Rangwala S.M., Keilbaugh S.A., Flanigan A., Murthy S., Lazar M.A., Wu G.D. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104:383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takagi T., Naito Y., Tomatsuri N., Handa O., Ichikawa H., Yoshida N., Yoshikawa T. Pioglitazone, a PPAR-gamma ligand, provides protection from dextran sulfate sodium-induced colitis in mice in association with inhibition of the NF-kappaB-cytokine cascade. Redox Rep. 2002;7:283–289. doi: 10.1179/135100002125000802. [DOI] [PubMed] [Google Scholar]
- 47.Sánchez-Hidalgo M., Martín A.R., Villegas I., de la Lastra C.A. Rosiglitazone, a PPARgamma ligand, modulates signal transduction pathways during the development of acute TNBS-induced colitis in rats. Eur J Pharmacol. 2007;562:247–258. doi: 10.1016/j.ejphar.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 48.Fernandes E.S., Passos G.F., Campos M.M., Araújo J.G., Pesquero J.L., Avellar M.C., Teixeira M.M., Calixto J.B. Mechanisms underlying the modulatory action of platelet activating factor (PAF) on the upregulation of kinin B1 receptors in the rat paw. Br J Pharmacol. 2003;139:973–981. doi: 10.1038/sj.bjp.0705314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Passos G.F., Fernandes E.S., Campos M.M., Araújo J.G., Pesquero J.L., Souza G.E., Avellar M.C., Teixeira M.M., Calixto J.B. Kinin B1 receptor up-regulation after lipopolysaccharide administration: role of proinflammatory cytokines and neutrophil influx. J Immunol. 2004;172:1839–1847. doi: 10.4049/jimmunol.172.3.1839. [DOI] [PubMed] [Google Scholar]
- 50.Kanokmedhakul S., Kanokmedhakul K., Lekphrom R. Bioactive constituents of the roots of Polyalthia cerasoides. J Nat Prod. 2007;70:1536–1538. doi: 10.1021/np070293a. [DOI] [PubMed] [Google Scholar]
- 51.Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H., Achkar J.P., Brant S.R., Bayless T.M., Kirschner B.S., Hanauer S.B., Nuñez G., Cho J.H. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 52.Medeiros R., Passos G.F., Vitor C.E., Koepp J., Mazzuco T.L., Pianowski L.F., Campos M.M., Calixto J.B. Effect of two active compounds obtained from the essential oil of Cordia verbenacea on the acute inflammatory responses elicited by LPS in the rat paw. Br J Pharmacol. 2007;151:618–627. doi: 10.1038/sj.bjp.0707270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho J.Y., Chang H.J., Lee S.K., Kim H.J., Hwang J.K., Chun H.S. Amelioration of dextran sulfate sodium-induced colitis in mice by oral administration of beta-caryophyllene, a sesquiterpene. Life Sci. 2007;80:932–939. doi: 10.1016/j.lfs.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 54.Ohkawara T., Nishihira J., Takeda H., Hige S., Kato M., Sugiyama T., Iwanaga T., Nakamura H., Mizue Y., Asaka M. Amelioration of dextran sulfate sodium-induced colitis by anti-macrophage migration inhibitory factor antibody in mice. Gastroenterology. 2002;123:256–270. doi: 10.1053/gast.2002.34236. [DOI] [PubMed] [Google Scholar]
- 55.Kanai T., Watanabe M., Okazawa A., Sato T., Yamazaki M., Okamoto S., Ishii H., Totsuka T., Iiyama R., Okamoto R., Ikeda M., Kurimoto M., Takeda K., Akira S., Hibi T. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn's disease. Gastroenterology. 2001;121:875–888. doi: 10.1053/gast.2001.28021. [DOI] [PubMed] [Google Scholar]
- 56.Qualls J.E., Kaplan A.M., van Rooijen N., Cohen D.A. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol. 2006;80:802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- 57.Vetuschi A., Latella G., Sferra R., Caprilli R., Gaudio E. Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig Dis Sci. 2002;47:1447–1457. doi: 10.1023/a:1015931128583. [DOI] [PubMed] [Google Scholar]
- 58.Oshima T., Miwa H., Joh T. Changes in the expression of claudins in active ulcerative colitis. J Gastroenterol Hepatol. 2008;23(Suppl 2):S146–S150. doi: 10.1111/j.1440-1746.2008.05405.x. [DOI] [PubMed] [Google Scholar]
- 59.Granata F., Frattini A., Loffredo S., Del Prete A., Sozzani S., Marone G., Triggiani M. Signaling events involved in cytokine and chemokine production induced by secretory phospholipase A2 in human lung macrophages. Eur J Immunol. 2006;36:1938–1950. doi: 10.1002/eji.200535567. [DOI] [PubMed] [Google Scholar]
- 60.Eckmann L., Nebelsiek T., Fingerle A.A., Dann S.M., Mages J., Lang R., Robine S., Kagnoff M.F., Schmid R.M., Karin M., Arkan M.C., Greten F.R. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci USA. 2008;105:15058–15063. doi: 10.1073/pnas.0808216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cippitelli M., Sica A., Viggiano V., Ye J., Ghosh P., Birrer M.J., Young H.A. Negative transcriptional regulation of the interferon-gamma promoter by glucocorticoids and dominant negative mutants of c-Jun. J Biol Chem. 1995;270:12548–12556. doi: 10.1074/jbc.270.21.12548. [DOI] [PubMed] [Google Scholar]
- 62.Samten B., Ghosh P., Yi A.K., Weis S.E., Lakey D.L., Gonsky R., Pendurthi U., Wizel B., Zhang Y., Zhang M., Gong J., Fernandez M., Safi H., Vankayalapati R., Young H.A., Barnes P.F. Reduced expression of nuclear cyclic adenosine 5′-monophosphate response element-binding proteins and IFN-gamma promoter function in disease due to an intracellular pathogen. J Immunol. 2002;168:3520–3526. doi: 10.4049/jimmunol.168.7.3520. [DOI] [PubMed] [Google Scholar]
- 63.Egger B., Bajaj-Elliott M., MacDonald T.T., Inglin R., Eysselein V.E., Buchler M.W. Characterisation of acute murine dextran sodium sulphate colitis: cytokine profile and dose dependency. Digestion. 2000;62:240–248. doi: 10.1159/000007822. [DOI] [PubMed] [Google Scholar]
- 64.Yano S., Ghosh P., Kusaba H., Buchholz M., Longo D.L. Effect of promoter methylation on the regulation of IFN-gamma gene during in vitro differentiation of human peripheral blood T cells into a Th2 population. J Immunol. 2003;171:2510–2516. doi: 10.4049/jimmunol.171.5.2510. [DOI] [PubMed] [Google Scholar]
- 65.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 66.Duchmann R., Zeitz M. T regulatory cell suppression of colitis: the role of TGF-beta. Gut. 2006;55:604–606. doi: 10.1136/gut.2005.083592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Correa F., Mestre L., Docagne F., Guaza C. Activation of cannabinoid CB2 receptor negatively regulates IL-12p40 production in murine macrophages: role of IL-10 and ERK1/2 kinase signaling. Br J Pharmacol. 2005;145:441–448. doi: 10.1038/sj.bjp.0706215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saubermann L.J., Nakajima A., Wada K., Zhao S., Terauchi Y., Kadowaki T., Aburatani H., Matsuhashi N., Nagai R., Blumberg R.S. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflamm Bowel Dis. 2002;8:330–339. doi: 10.1097/00054725-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 69.Ramakers J.D., Verstege M.I., Thuijls G., Te Velde A.A., Mensink R.P., Plat J. The PPARgamma agonist rosiglitazone impairs colonic inflammation in mice with experimental colitis. J Clin Immunol. 2007;27:275–283. doi: 10.1007/s10875-007-9074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu T., Newton C., Perkins I., Friedman H., Klein T.W. Cannabinoid treatment suppresses the T-helper cell-polarizing function of mouse dendritic cells stimulated with Legionella pneumophila infection. J Pharmacol Exp Ther. 2006;319:269–276. doi: 10.1124/jpet.106.108381. [DOI] [PubMed] [Google Scholar]
- 71.Tschöp J., Kasten K.R., Nogueiras R., Goetzman H.S., Cave C.M., England L.G., Dattilo J., Lentsch A.B., Tschop M.H., Caldwell C.C. The cannabinoid receptor 2 is critical for the host response to sepsis. J Immunol. 2009;183:499–505. doi: 10.4049/jimmunol.0900203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tambe Y., Tsujiuchi H., Honda G., Ikeshiro Y., Tanaka S. Gastric cytoprotection of the non-steroidal anti-inflammatory sesquiterpene, beta-caryophyllene. Planta Med. 1996;62:469–470. doi: 10.1055/s-2006-957942. [DOI] [PubMed] [Google Scholar]
- 73.Konstantinopoulos P.A., Vandoros G.P., Sotiropoulou-Bonikou G., Kominea A., Papavassiliou A.G. NF-kappaB/PPAR gamma and/or AP-1/PPAR gamma 'on/off' switches and induction of CBP in colon adenocarcinomas: correlation with COX-2 expression. Int J Colorectal Dis. 2007;22:57–68. doi: 10.1007/s00384-006-0112-y. [DOI] [PubMed] [Google Scholar]
- 74.Herring A.C., Kaminski N.E. Cannabinol-mediated inhibition of nuclear factor-kappaB, cAMP response element-binding protein, and interleukin-2 secretion by activated thymocytes. J Pharmacol Exp Ther. 1999;291:1156–1163. [PubMed] [Google Scholar]
- 75.Zhao C.Y., Wang Y.D., Zhou J.Y., Jia B., Cui J.F. [An experimental study on the reverse mechanism of PPAR-gamma agonist rosiglitazone in rats with non-alcoholic steatohepatitis] Zhonghua Gan Zang Bing Za Zhi. 2007;15:450–455. Chinese. [PubMed] [Google Scholar]
- 76.O'Sullivan S.E. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.