

Abstract
Pulmonary infection by Yersinia pestis causes pneumonic plague, a necrotic bronchopneumonia that is rapidly lethal and highly contagious. Acute pneumonic plague accompanies the up-regulation of pro-inflammatory cytokines and chemokines, suggesting that the host innate immune response may contribute to the development of disease. To address this possibility, we sought to understand the consequences of neutrophil recruitment during pneumonic plague, and we studied the susceptibility of C3H-HeN mice lacking the CXC chemokine KC or its receptor CXC receptor 2 (CXCR2) to pulmonary Y. pestis infection. We found that without Kc or Cxcr2, disease progression was accelerated both in bacterial growth and development of primary bronchopneumonia. When examined in an antibody clearance model, Cxcr2−/− mice were not protected by neutralizing Y. pestis antibodies, yet bacterial growth in the lungs was delayed in a manner associated with a neutrophil-mediated inflammatory response. After this initial delay, however, robust neutrophil recruitment in Cxcr2−/− mice correlated with bacterial growth and the development of fulminant pneumonic and septicemic plague. In contrast, attenuated Y. pestis lacking the conserved pigmentation locus could be cleared from the lungs in the absence of Cxcr2 indicating virulence factors within this locus may inhibit CXCR2-independent pathways of bacterial killing. Together, the data suggest CXCR2 uniquely induces host defense mechanisms that are effective against virulent Y. pestis, raising new insight into the activation of neutrophils during infection.
The mammalian respiratory tract has limited host defense mechanisms against pathogenic microbes. Upon infection, alveolar macrophages, fibroblasts, and endothelial and epithelial cells produce chemokines to attract immune effector cells, such as neutrophils, macrophages, and NK cells, to the lungs.1 Two families of chemokines, CC and CXC, predominantly signal these cells in response to recognition of invading microbes using pattern recognition receptors or as a response to lung injury induced by the pathogen. In addition to signaling the induction of adhesive factors, such as integrins, necessary for neutrophil chemotaxis, CXC chemokines also signal activation of these cells to enable an increase in intracellular Ca2+, which is necessary for exocytosis and other effector responses that stimulate killing of microbes.2 Antibodies that opsonize the pathogen accelerate the activation of these signaling pathways, as well as additional pathways involving the complement or Fcγ receptors, and improve the efficiency of phagocytic killing by neutrophils.3 These multiple signaling pathways of neutrophil recruitment and activation induced by antibodies ensure a robust response to infection.
CXC chemokines are potent neutrophil attractants and are involved in host defense against extracellular pathogens. In mice, chemokines KC and MIP-2 are part of the CXC family and are considered likely functional homologues of human IL-8.4,5 Secretion of KC and macrophage inflammatory protein-2 induces extravascular migration of neutrophils to sites of infection, cellular apoptosis, and tissue damage by signaling predominantly through CXC receptor 2 (CXCR2).6 Cxcr2 is highly homologous between mouse and human and is expressed by a variety of immune cells, including neutrophils, monocytes, eosinophils, mast cells, basophils, and lymphocytes.7 CXC chemokine secretion can be triggered through recognition of bacterial peptidoglycan or lipopolysaccharide, causing migration of neutrophils to an infection site and the activation of clearance mechanisms.8–11 Likewise, CXC chemokines are also produced in response to apoptosis or host cell damage, resulting in infiltration of neutrophils to injured tissue to clean up dead cells.12 Mice unable to signal through CXCR2 are more susceptible to many bacterial and even some viral infections, both respiratory and nonrespiratory, but less susceptible to inflammatory injury.10,13–19 For many respiratory infections, absence of Cxcr2 results in increased colonization and decreased neutrophil recruitment to the infected site. For example, in response to Streptococcus pneumoniae infection, wild-type mice recruit more neutrophils than Cxcr2−/− mice during 48 to 72 hours, and these numbers decline in time as the mice clear the infection.15 Similarly, in response to Rhinovirus infection, Cxcr2−/− mice recruit fewer neutrophils during at least a 96-hour time period as compared to wild-type mice, and this decrease is associated with an increase in disease in the mutant mice.13 Many other examples of bacterial infections that are cleared by CXCR2-dependent recruitment of neutrophils during the early innate immune response have been described, and in each case, Cxcr2 mutants have decreased neutrophil chemotaxis associated with increased bacterial load.
CXCR2 is also associated with neutrophil-induced host injury, such as what occurs in the lungs during bacterial sepsis.20,21 CXCR2-dependent infiltration and activation of neutrophils to the lungs causes acute injury and pneumonia independent of bacterial colonization of the respiratory tract, suggesting unregulated migration and activation of neutrophils severely damages host tissues.22,23 Small molecule antagonists of CXCR2 can inhibit lipopolysaccharide-induced lung pathology by blocking neutrophil migration to the infection site, suggesting that preventing the activation of CXCR2 can prevent septicemic disease.24
Primary pneumonic plague is a deadly bronchopneumonia that develops following inhalation of Y. pestis. The disease is similar in rodents and man, and is characterized by parenchymal congestion by polymorphonuclear cells, large bacterial colonies, alveolar destruction, and edema.25–27 Y. pestis virulence factors manipulate innate immune responses to avoid detection and promote disease.28 Pattern recognition receptors, such as toll-like receptors 2, 4, or 5, do not appear to be activated during infection due in large part to the presence of a noncanonical lipopolysaccharide, and a lack of flagellin, which helps establish an initial anti-inflammatory response in the host.29 Further immune modulation and cytotoxicity is conferred by a type III secretion system by which extracellular bacteria target macrophages and other immune cells to inject them with proteins that inhibit phagocytosis, alter inflammatory signaling pathways, modulate NF-κB signaling, and result in the death of the target cell.28,30 Late-stage disease involves a large induction of IFN-γ, chemokines, and other systemic pro-inflammatory cytokines.26,31,32 Neutrophil infiltration is prominent in pathological lesions of moribund mice and rats and is associated with bacteria and tissue necrosis.26,27,31–33 These observations suggest that Y. pestis replication strongly promotes neutrophil infiltration during late-stage disease, but this response is ineffective and may even be detrimental to the host.
Immunity to the plague can be conferred by antibodies to low calcium response V-antigen (LcrV), a component of the type III secretion system required for immune evasion and disease.34–37 Neutralizing LcrV antibodies block the type III secretion system and promote phagocytosis, and both activities are required for immunity, suggesting that opsonization of bacteria plays an important role in clearance.38–40 The type III secretion system is a strategy of extracellular bacteria; however Y. pestis is capable of surviving inside activated macrophages in a manner dependent on the pigmentation locus, a 102-kb pathogenicity island required for development of pneumonic and bubonic plague.33,41–46 Furthermore, antibody-mediated phagocytosis of Y. pestis does not lead to its destruction inside activated macrophages in vitro, leaving the mechanism of immunity uncertain.47
In this work, we addressed the role of chemokines and neutrophils in plague, both during disease and in clearance after treatment with protective LcrV antibodies that broadly stimulate innate immune activation. We studied mice defective in signaling through CXC chemokines and characterized host responses to pulmonary Y. pestis challenge. We found that CXCR2 provided protective responses in both models, but had minimal impact on neutrophil recruitment to infected sites. Early containment of the infection was seen in anti-LcrV-treated Cxcr2−/− mice in a manner that was associated with neutrophil recruitment and little to no damaged lung tissue. Despite these seemingly protective responses, however, Cxcr2−/− mice later developed systemic disease and acute bronchopneumonia that were indistinguishable from untreated mice, suggesting that other, seemingly redundant, pathways of neutrophil recruitment and activation may be ineffective against Y. pestis. Systemic ablation of neutrophils also resulted in loss of antibody protection that mimicked the loss of protection seen in Cxcr2−/− mice consistent with CXCR2 as the primary mechanism for inducing bacterial clearance. Deletion of the pigmentation (pgm) locus rendered Y. pestis sensitive to CXCR2-independent clearance, indicating the pgm locus encodes virulence factors that likely inhibit activation of neutrophils. Together the data support a model whereby CXCR2 signaling of neutrophils is necessary to destroy virulent Y. pestis, whereas other chemotactic pathways stimulate recruitment of neutrophils that are inactivated by products of the pgm locus, allowing rapid bacterial replication and fulminant disease.
Materials and Methods
Bacterial Strains
All Y. pestis culture strains used were taken from frozen stocks and streaked for isolation onto heart infusion agar plates. The plates used for Y. pestis CO92 were supplemented with 0.005% Congo red and 0.2% galactose to screen for bacteria that retain the pigmentation locus.48 For the pneumonic plague challenge, a single, red-pigmented colony was used to inoculate heart infusion broth supplemented with 2.5 mmol/L CaCl2 and grown 18 to 24 hours at 37°C at 120 rpm. All handling of samples containing live Y. pestis CO92 was performed in a select agent authorized BSL3 facility under protocols approved by the University of Missouri Institutional Biosafety Committee. Nonpigmented Y. pestis CO92 and KIM D27 were routinely grown fresh from frozen stock on heart infusion agar, followed by aerobic growth at 27°C in heart infusion broth overnight before use in experiments.33,42
Animals
All animal procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Missouri Animal Care and Use Committee. C3H-HeNCrl mice were used for studies on host responses to plague and were the parent strain of the Kc−/− and Cxcr2−/− mice. Wild-type C3H mice were purchased from Charles River Laboratories. Knockout mice were bred and housed in barrier containment facilities at the University of Missouri. Males and females, ranging from 15 to 50 g were used for challenge experiments. During challenge, mice were maintained in select agent-approved animal containment facilities at the University of Missouri. All infected mice were monitored regularly by daily weighing and assignment of health scores. Animals that survived to the end of the 14-day observation period or were identified as moribund (defined by pronounced ataxia, sometimes accompanied by severe dyspnea) were euthanized by CO2 asphyxiation, followed by bilateral pneumothorax; these methods are approved by the American Veterinary Medical Association Guidelines on Euthanasia.
Pneumonic Plague Challenge
Y. pestis CO92, grown as previously described at 37°C, were diluted in sterile PBS to 400, 4000, or 6000 colony forming units (CFU)/0.02 ml just before use for challenge experiments. Actual dose and retention of the pigmentation locus were determined by plating in triplicate on heart infusion agar with Congo red. Where indicated, for some intranasal infections involving nonpigmented Y. pestis strains, mice were given 500 μg FeCl2 by intraperitoneal injection just before challenge. All animals intranasally infected with Y. pestis were first lightly anesthetized by isoflurane inhalation. Animals were observed for recovery from anesthesia and returned to housing.
Purification of Antibody
Rabbit polyclonal LcrV was produced as previously described; serum antibody titer to recombinant LcrV was 105 for all experiments.39 For purification, rabbit serum containing α-LcrV antibodies was applied to a protein G column and purified according to the manufacturer's protocol (Sigma, St. Louis, MO). Samples were then applied to a PD-10 buffer exchange column (GE Healthcare, Buckinghamshire, UK) and eluted in PBS. Total IgG was quantified using bovine IgG as a standard in a BCA protein assay (Pierce, Rockford, IL).
Antibody Treatment
For pneumonic plague challenges, 400 μg/0.4 ml purified antibody was given by intraperitoneal injection 60 minutes before infection. Untreated control mice were given 400 μl sterile PBS by intraperitoneal injection 60 minutes before challenge. In some experiments, mice were given rat anti-mouse Gr-1 (RB6.8C5) monoclonal antibodies (BD Pharmingen, San Jose, CA) intraperitoneally in 100-μg (100 μg/100 μl) or 200-μg (200 μg/200 μl) doses at the times indicated. Control mice in these experiments were given equivalent volumes of PBS.
Quantification of Bacterial Load in Blood and Tissues
Immediately after euthanasia, blood was collected directly from the heart by cardiac puncture. Lungs, spleens, and livers were collected aseptically and homogenized in 1 ml sterile PBS. Serial dilutions of the blood and homogenized tissues were then plated onto heart infusion agar plates for quantification of bacterial load (CFU/ml or CFU/organ, respectively).
Histopathological Evaluation of Tissues
Lungs were perfused with 10% formalin before removal; all other tissues were collected and placed in 10% formalin. All tissues were fixed for a minimum of 48 hours. Fixed tissues were embedded in paraffin and sections were cut and stained with H&E. For histological scoring of neutrophil infiltration (judged by staining positive for Gr-1 by immunohistochemistry), bacterial colonies, congestion, edema, and alveolar destruction, lung lobes were evaluated in a single blind fashion by a veterinarian with expertise in the pathological analysis of disease in rodents.
Immunohistochemistry
Tissues that had been fixed in 10% formalin as previously described were sectioned onto slides for immunohistochemical analysis. Slides were stained with rat anti-mouse F4/80 (Serotec, Oxford, UK) or monoclonal anti-Gr-1 (RB6.8C5),49 and detection was achieved by secondary staining with biotinylated rabbit anti-rat IgG and HRP-streptavidin (Dako, Carpenteria, CA). Staining and detection were carried out according to the manufacturer's guidelines.
Statistical Analysis
Survival and mean time to death were evaluated for statistical significance between groups using the Mann-Whitney rank sum test. Pathological scores were evaluated by one-way ANOVA followed by Dunn's method for multiple comparisons. Percent weight loss compared to pre-challenge weight as well as neutrophil recruitment in lesions with and without bacteria were evaluated by Student's t-test. Significance was concluded if P < 0.05.
Results
CXC Chemokines Contribute to the Clearance of Y. pestis CO92 from the Lung
CXC chemokines, such as KC, are produced by resident macrophages and other phagocytic cells of the mammalian lung in response to bacterial invasion. High-level production of KC is stimulated during pneumonic plague in mice, suggesting it may be active in promoting neutrophil recruitment to infected areas.31 To understand the consequences of KC production, we challenged C3H-HeN mice carrying a homozygous deletion of Kc (Kc−/−) by intranasal instillation of Y. pestis CO92 and followed the development of disease for a 14-day period. At a challenge dose of 4000 CFU, 100% of Kc−/− mice succumbed to infection compared to only 20% of wild-type mice (Figure 1A; P < 0.05). These results strongly suggest that KC serves a protective role in host defense against pulmonary Y. pestis challenge.
Figure 1.
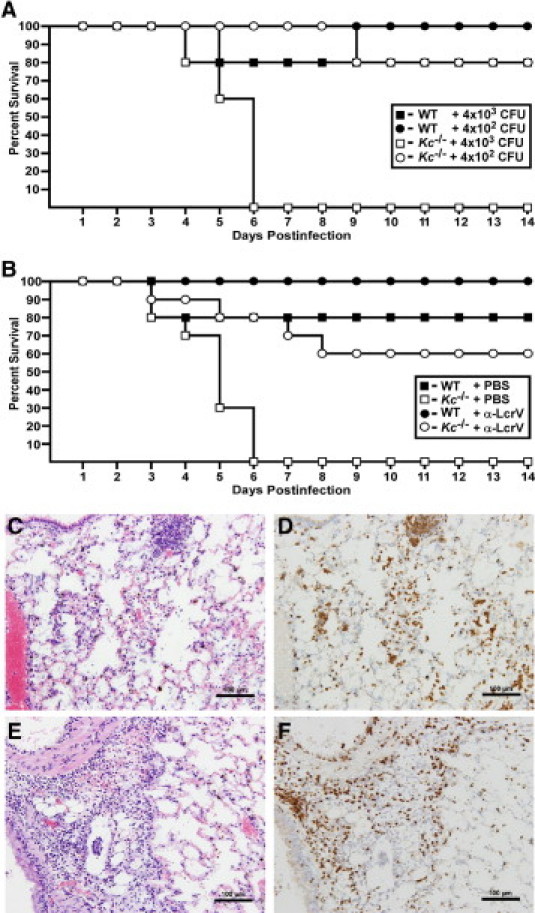
KC contributes to protection against pneumonic plague. A: Wild-type (WT) (n = 5) or Kc−/− C3H mice (n = 5) were challenged by intranasal infection with the indicated doses of Y. pestis CO92 and survival was monitored for 14 days. B: Mice were given 400 μg anti-LcrV polyclonal antibody (n = 3 for wild type; n = 5 for Kc−/−) or PBS (n = 5 for wild type; n = 5 for Kc−/−) by intraperitoneal injection 60 minutes before intranasal challenge with Y. pestis CO92 and survival was monitored for a 14-day period. Data shown were collected in one of two independent experiments with equivalent challenge doses and similar results (n = 10 Kc−/− mice were analyzed for treated and untreated groups). Lungs from moribund mice were inflated with 10% formalin and fixed for 48 hours before staining with H&E (C and E) or anti-Gr-1 (D and F). Both wild-type (C and D) and Kc−/− (E and F) mice developed acute bronchopneumonia with infiltration of neutrophils. Representative images are shown at 20× magnification.
We also tested whether KC was important to antibody-mediated immunity to Y. pestis in a passive transfer of immunity model. In this model, opsonized Y. pestis would be predicted to broadly stimulate the innate immune system, including production of pro-inflammatory cytokines and chemokines, as well as complement and Fcγ receptor (FcR), allowing us to ascertain whether CXCR2 is uniquely required for host defense or if other neutrophil recruitment or activation pathways can be substituted. Anti-LcrV-treated Kc−/− mice appeared only partially protected and 40% died with a mean time to death of 5.75 days compared to 100% survival in wild-type mice (Figure 1B) (P > 0.05). These results are consistent with the involvement of KC in antibody-mediated immunity but suggest that other protective signaling mechanisms also occur. Moribund Kc−/− mice that were not treated with anti-LcrV typically exhibited both severe dypsnea and ataxia. Lungs from these mice were examined for histopathology by staining with H&E and by immunohistochemistry to identify injury, neutrophil infiltration, and bacterial burden to characterize the development of pneumonic plague. Immunohistochemistry with anti-Gr-1, an antibody that stains neutrophils and some monocytes, was also performed to confirm the identity of these cells. Wild-type and Kc−/− mice both developed acute bronchopneumonia, characterized by large bacterial colonies, cellular debris, and infiltration of neutrophils that was indistinguishable from one another (Figure 1, C–F). In each strain, spleens and livers of moribund mice showed evidence of advancing systemic disease, characterized by tissue necrosis and hemorrhage in the spleen and multiple foci of inflammation in the liver (data not shown). Together, these results suggest that KC production, whether during untreated or antibody-treated infection, plays a protective role against pneumonic plague.
Cxcr2 is Essential for Protection Against Pneumonic Plague
In mice, KC and other CXC chemokines signal through CXCR2 to recruit neutrophils during bacterial infections.50 Therefore, we analyzed Cxcr2−/− mice to understand the role of neutrophil infiltration at a more global level. Groups of five to eight wild-type C3H or Cxcr2−/− mice were treated with PBS just before intranasal challenge with 6000 CFU Y. pestis CO92. Wild-type and Cxcr2−/− mice were similarly susceptible to acute disease, which developed within 4 days of infection (Figure 2, P > 0.05). In contrast, while wild-type mice were protected by neutralizing antibodies and did not develop signs of disease, all but one Cxcr2−/− mouse succumbed to the infection (P < 0.05, compared to wild-type mice treated with anti-LcrV). Notably, these mice were initially protected by the antibodies, which provided an extension in the mean time to death (6.3 days for treated mice compared to 4.4 days for untreated mice, P < 0.005). These results suggest that CXCR2-independent responses lead to early containment, but not clearance, of Y. pestis.
Figure 2.
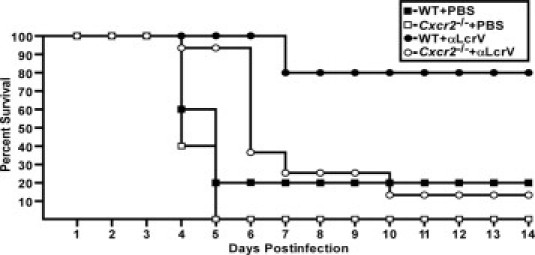
Cxcr2 is essential for protection against pneumonic plague. Wild-type (WT) (n = 5) or Cxcr2−/− (n = 5) C3H mice were given PBS or 400 μg anti-LcrV polyclonal antibody (n = 13 for Cxcr2−/−) by intraperitoneal injection 60 minutes before intranasal infection with 6000 CFU Y. pestis CO92 and survival was monitored for a 14-day period. Data for Cxcr2−/− treated with anti-LcrV were collected in two independent experiments.
Neutrophil Infiltration Correlates with Bacterial Replication and Lung Injury
We next compared disease progression between wild-type and Cxcr2−/− mice from both antibody-treated and PBS-treated groups. In this experiment, wild-type or Cxcr2−/− mice were treated with either PBS or anti-LcrV via intraperitoneal injection 60 minutes before challenge by intranasal instillation of Y. pestis CO92. At 60 hours postinfection, we examined either bacterial load or pathology of the lungs, liver, and spleen of these mice. All PBS-treated Cxcr2 mutant mice had a high titer of bacteria in the lungs, which only occasionally had disseminated to distal tissues, suggestive of primary pneumonic plague (Figure 3A). Analyses of histological sections confirmed these observations, whereas wild-type PBS-treated mice had only mild to moderate infiltration of neutrophils and no visible bacteria (Figure 3, B and C), all Cxcr2−/− mice treated with PBS had severe bronchopneumonia, with neutrophils surrounding foci of bacteria (Figure 3, D and E). The liver and spleen from these mice had developed minor to moderate disease, indicating systemic spread of the infection (Figure 3, H and I). In contrast, we recovered bacteria from only one of four anti-LcrV-treated Cxcr2−/− mice at 60 hours postinfection, indicating that fewer than 100 CFU were generally present in the lungs at this time point. Nevertheless, histological analyses of the lungs from anti-LcrV-treated Cxcr2−/− mice at 60 hours postinfection revealed moderate infiltration of neutrophils in all three animals examined, two of which harbored occasional bacteria (Figure 4, E and F; see Supplemental Table S1 at http://ajp.amjpathol.org). Similarly, inflammatory responses were also seen in the spleen and liver (Figure 4, G and H; see Supplemental Table S2 at http://ajp.amjpathol.org). In contrast, lungs, liver, and spleen from wild-type anti-LcrV-treated mice had few to no disease lesions nor neutrophils at 60 hours postinfection (Figure 4, A–D; see Supplemental Table S3 at http://ajp.amjpathol.org). Together these results suggest that Cxcr2 mutant mice initially contain the infection after treatment with protective antibodies.
Figure 3.
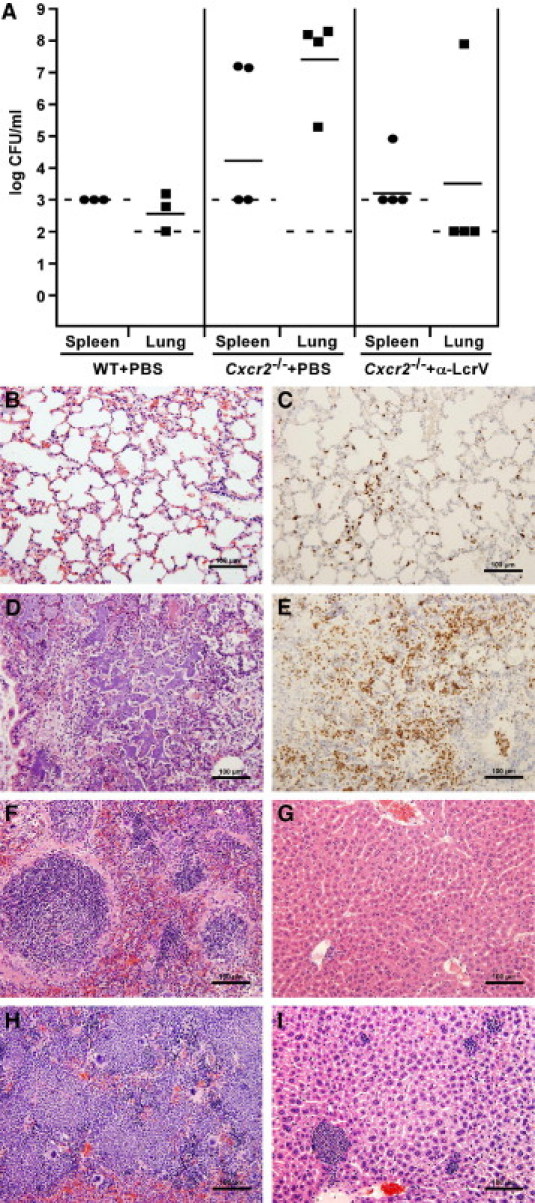
Primary pneumonic plague develops more rapidly in Cxcr2−/− mice. Wild-type (WT) (n = 3) or Cxcr2−/− (n = 4) C3H mice were given PBS IP 60 minutes before intranasal infection with 6000 CFU Y. pestis CO92. Mice were euthanized 60 hours postinfection and tissues were harvested and diluted in PBS to analyze for bacterial burden (A) or fixed in 10% formalin to analyze by histopathology (B–I). WT lungs (B and C), spleen (F), and liver (G); Cxcr2−/− lungs (D and E), spleen (H), and liver (I). Anti-mouse Gr-1 was used in immunohistochemistry for identification of neutrophils (C and E). Dashed lines indicate the limit of detection and solid lines indicate the geometric mean of the samples (A). Images are shown at 20× magnification.
Figure 4.
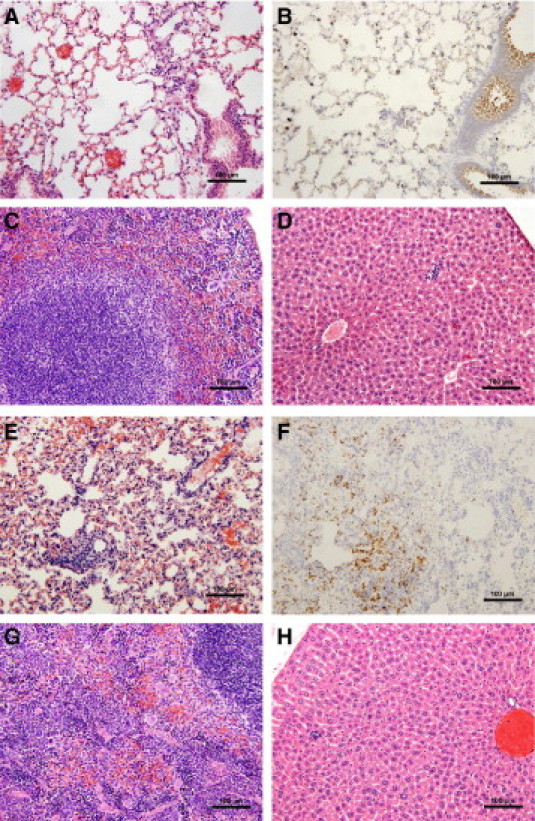
Anti-LcrV antibody treatment slows the progression of disease in Cxcr2−/− mice. Wild-type (WT) and Cxcr2−/− mice, treated with 400 μg anti-LcrV polyclonal antibody IP 60 minutes before intranasal challenge with 6000 CFU Y. pestis CO92 were euthanized 60 hours postinfection, and tissues were harvested and fixed in 10% formalin for histochemical and immunohistochemical analysis. WT lungs (A and B), spleen (C), and liver (D); Cxcr2−/− lungs (E and F), spleen (G), and liver (H). Immunohistochemistry with anti-Gr-1 was used to identify neutrophils (B and F). Images are shown at 20× magnification.
We next evaluated bacterial load and disease severity in antibody-treated Cxcr2−/− mice challenged with Y. pestis CO92 after 90 hours postinfection, when mice in this group showed signs of acute disease. All mice examined harbored a high titer of bacteria in the lungs (Figure 5A). Five of six mice examined also harbored detectable amounts of bacteria in the blood, indicating systemic disease. A second group of four anti-LcrV-treated Cxcr2−/− mice, also euthanized 90 hours postinfection, was examined by histopathology of formalin-fixed tissues. Lungs of antibody-treated mice showed moderate to severe bronchopneumonia with infiltration of neutrophils surrounding large bacterial colonies (Figure 5, B and C; see Supplemental Table S1 at http://ajp.amjpathol.org). This suggests that these mice still developed bronchopneumonia despite the delay in time to disease. However, consistent with systemic bacterial dissemination, spleens of these mice were also necrotic and the livers exhibited hepatitis with multiple foci of inflammation, and thus it appears septicemic plague had also developed (Figure 5, D and E; see Supplemental Table S2 at http://ajp.amjpathol.org).
Figure 5.
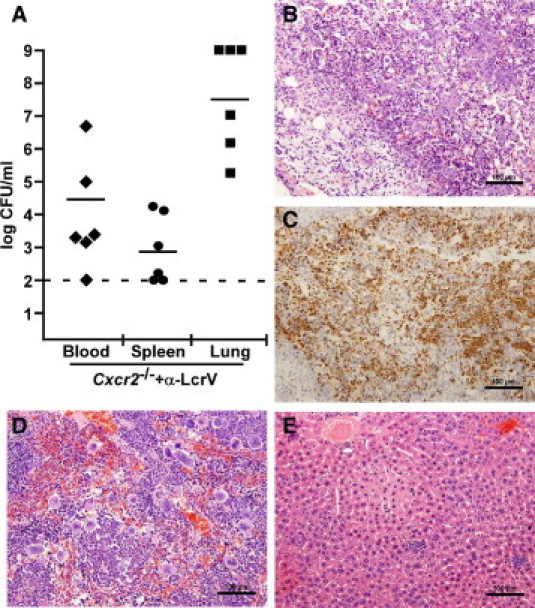
Anti-LcrV antibody treatment initially contains the infection but does not prevent development of pneumonic or septicemic plague. Cxcr2−/− mice (n = 6) were given 400 μg anti-LcrV polyclonal antibodies and challenged intranasally with 6000 CFU Y. pestis CO92. Blood, spleen, and lung were analyzed for bacterial burden at 90 hours postinfection, when death is first seen for this group (A). B–E: A second group Cxcr2−/− mice (n = 4) was challenged with the same dose following anti-LcrV treatment, euthanized 90 hours postinfection and lungs (B and C), spleen (D), and liver (E) were removed, formalin fixed, stained, and analyzed for histopathology. Tissue sections were also stained with anti-Gr-1 to identify neutrophils that had been recruited to the lung (C). Images are shown at 20× magnification.
To quantify the degree of disease in the lungs, severity scores indicative of pneumonic plague at 60 and 90 hours postinfection were determined. Edema, alveolar destruction, neutrophil infiltration, and parenchymal congestion were used as indicators of pneumonia. Assignment of scores corresponding to none through severe (scale of 0 to 3) for each criterion was performed for three lung lobes by examination of slides stained with H&E and anti-Gr-1, and a total score was determined for each mouse (Figure 6). Average scores within each group indicated that PBS-treated Cxcr2−/− mice developed severe pneumonia by 60 hours postinfection, whereas wild-type mice did not (P < 0.05, evaluated by one-way ANOVA). At this time point, anti-LcrV-treated Cxcr2−/− mice showed moderate neutrophil infiltration and few pathological lesions. These observations suggest neutrophil recruitment to the lung occurs before the development of disease lesions and bacterial growth in the absence of CXCR2. Anti-LcrV-treated Cxcr2−/− mice eventually developed pneumonia, but with a delayed time course compared to untreated WT or mutant mice. Bacterial colonies were visible in areas of lung injury surrounded by inflammatory cells, many of which appeared to be neutrophils judged by morphology and positive staining for Gr-1 (Table 1). Together these data suggest that after anti-LcrV treatment, neutrophils are attracted to Y. pestis through multiple, perhaps redundant, chemotactic pathways. CXCR2-expressing cells are able to control the infection, while signaling through other chemotaxis receptors, perhaps CC or C5a receptors, may allow recruitment to the infection site but little to no effector function that could result in bacterial clearance.
Figure 6.
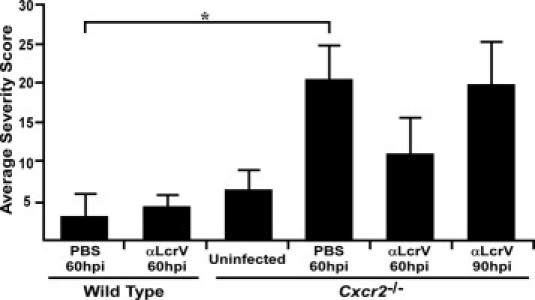
Quantitative histochemistry shows efficient neutrophil recruitment to the lungs of Cxcr2−/− mice despite the failure to prevent bacterial growth and disease. Disease severity scores, based on parenchyml congestion, neutrophil recruitment, edema, and alveolar destruction were determined by analyzing three lung lobes for each mouse and the average score per group was plotted. Group sizes ranged from 2 to 5 [n = 5, uninfected Cxcr2−/−; n = 4 wild type (WT) + PBS, 60 hours; n = 2 WT + anti-LcrV, 60 hours; n = 3 Cxcr2−/− + PBS, 60 hours; n = 3 Cxcr2−/− + anti-LcrV, 60 hours; n = 4 Cxcr2−/− + anti-LcrV, 90 hours]. *P < 0.05 as determined by one-way analysis of variance; hpi indicates hours postinfection.
Table 1.
Average Severity Score at 60 Hours Postinfection per Lung Sample
| Bacteria | Pathological lesion⁎ | Wild type + PBS | Cxcr2-/- + PBS | Cxcr2-/- + anti-LcrV |
|---|---|---|---|---|
| − | Neutrophil infiltration† | 1 (±1.00) | 1.67 (±0.44) | 0.33 (±0.44) |
| Alveolar destruction | 0 | 0 | 0 | |
| Pulmonary edema | 0 | 0 | 0 | |
| Parenchymal congestion | 0.13 (±0.19) | 1.5 (±1.00) | 0.67 (±0.44) | |
| + | Neutrophil infiltration† | NA | 2.50 (±0.67) | 2.00 (±0.00)‡ |
| Alveolar destruction | NA | 2.58 (±0.39)§ | 2.25 (±0.17)§ | |
| Pulmonary edema | NA | 1.17 (±0.56) | 1.50 (±0.33)‡ | |
| Parenchymal congestion | NA | 2.50 (±0.67) | 2.50 (±0.33)‡ |
Data are given ± the standard deviation from the mean; LcrV, low calcium response V-antigen; NA, not applicable; bacteria were not seen in any lobe.
Lesions observed and scored from H&E stain of formalin-fixed tissues.
Neutrophil enumeration from immunohistochemistry anti-Gr1 stain of formalin-fixed tissues.
P<0.05;
P <0.001 compared to no bacteria of the same group; evaluated by Student's t-test.
Temporary Ablation of Neutrophils Slows Progression of Pneumonic Plague
Our experiments thus far suggest that CXCR2 signaling provides protective responses but does not abrogate neutrophil migration to the infected sites of the lungs. To address the contribution of CXCR2-independent neutrophil chemotaxis to the lungs, we first tested wild-type mice, and compared antibody immunity after pretreatment with either PBS or anti-Gr-1 (RB6.8C5), an antibody that ablates circulating neutrophils and some monocytes with a half-life of approximately 1.5 days in C3H-HeN mice.49 RB6.8C5 or PBS was delivered to wild-type C3H mice by intraperitoneal injection of 100 μg on days −1 and +1 relative to intranasal Y. pestis challenge, with anti-LcrV administered 60 minutes before infection. All mice treated with anti-LcrV and RB6.8C5 were initially protected, suggesting that neutrophils are not required for early containment of the infection in the presence of protective antibodies. However, bacteria were not eliminated, and all of these mice eventually developed weight loss at higher levels than PBS-treated mice. Only 40% of RB6.8C5-treated mice progressed to lethal disease by day 7, while the others resolved the infection (Figure 7, A and B). These results suggest that early neutrophil ablation decreases immunity to plague, which is in agreement with recently reported observations of RB6.8C5 treatment in a mouse pneumonic plague model.51
Figure 7.
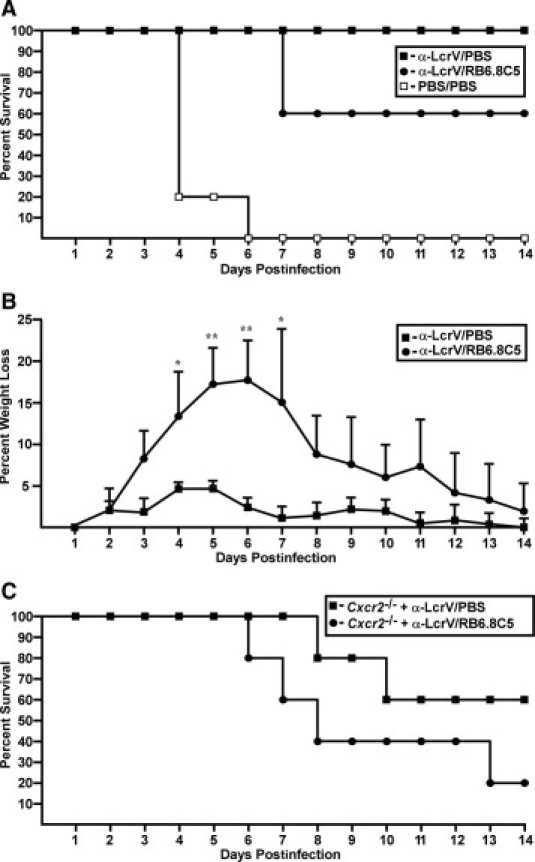
Extended time to death but no protection after anti-LcrV treatment and neutrophil ablation in wild-type and Cxcr2−/− mice. A: Wild-type mice (n = 5) were given 100 μg anti-Gr-1 monoclonal antibody or PBS on days −1 and +1 postinfection and 400 μg anti-LcrV polyclonal antibody or PBS 60 minutes before intranasal infection with 4000 CFU Y. pestis CO92, and survival was monitored for a 14-day period. B: Percent daily weight loss compared to prechallenge weight is shown for all surviving mice in each group for the experiment shown in A. C:Cxcr2−/− mice (n = 5) were given 200 μg anti-Gr-1 or PBS 60 hpi and 400 μg anti-LcrV 60 minutes before inranasal infection with 4000 CFU Y. pestis CO92 and survival was monitored. *P < 0.05, **P < 0.005 as determined by Student's t-test.
We next treated Cxcr2−/− mice with anti-LcrV followed by either PBS or 200 μg RB6.8C5 at 60 hours postinfection, approximately 24 to 36 hours before acute disease appears in the absence of RB6.8C5 treatment. Both untreated and RB6.8C5-treated Cxcr2−/− mice were initially protected from disease similar to wild-type treated mice. On day 5, disease symptoms began in 80% of RB6.8C5-treated animals, and disease progressed to lethality in all of these mice (Figure 7C). Cxcr2−/− mice treated with anti-LcrV and PBS behaved similarly and 40% developed lethal disease (P > 0.05 for RB6.8C5 compared to PBS-treated mice). Together, these results are consistent with CXCR2 signaling as the primary mechanism whereby host neutrophils control Y. pestis infection but also suggest that initial containment of disease after treatment with protective antibodies is not mediated by Gr-1+ cells.
Cxcr2-Independent Signaling Clears Mutant Y. pestis
To address whether Cxcr2 is a general necessity for host defense against bacterial infections of the lung, we challenged Cxcr2−/− mice with a nonpigmented mutant strain of Y. pestis CO92 (Δpgm) by intranasal infection. In this model, Yersinia colonize the lung and do not cause inflammation or pneumonia, but nevertheless cause septicemic plague, and the infection is lethal to mice.33 Pretreatment of mice with iron accelerates bacterial access to the blood, resulting in a more uniform and rapid disease. Intranasal challenge of wild-type mice with 1 × 106 CFU Y. pestis CO92Δpgm resulted in approximately 90% lethality within 10 days, consistent with our previous observations33 (Figure 8A). Strikingly, Cxcr2−/− mice were significantly less susceptible to infection by Y. pestis CO92Δpgm and only one of these mice succumbed to infection after 10 days (P < 0.005, evaluated by the Mann-Whitney rank sum test). We also tested another nonpigmented strain (KIM D27), which is a laboratory strain first isolated in 1965 from a lineage distinct from CO92 for its ability to cause acute disease in Cxcr2 mutant mice.42 In this model, 40% of wild-type C3H-HeN mice succumbed to intranasal challenge with 1 × 106 CFU Y. pestis KIM D27 (Figure 8B). Treatment with iron before challenge increased overall susceptibility and 100% succumbed to the infection (Figure 8C). Both PBS-treated and iron-treated Cxcr2−/− mice exhibited similar, if not less, susceptibility to Y. pestis KIM D27 in both models, with 10% and 60% lethality, respectively, although these differences may not be significant (Figure 8, B and C) (P > 0.05 compared to wild type with or without iron treatment). Together these results strongly suggest that bacterial virulence factors within the pgm locus may prevent neutrophil activation through CXCR2-independent pathways. Furthermore, Cxcr2−/− mice are capable of mounting an effective response against a highly similar bacterial infection in the lungs, indicating that CXCR2 is not broadly required for host defense against bacterial pathogens in the lungs.
Figure 8.
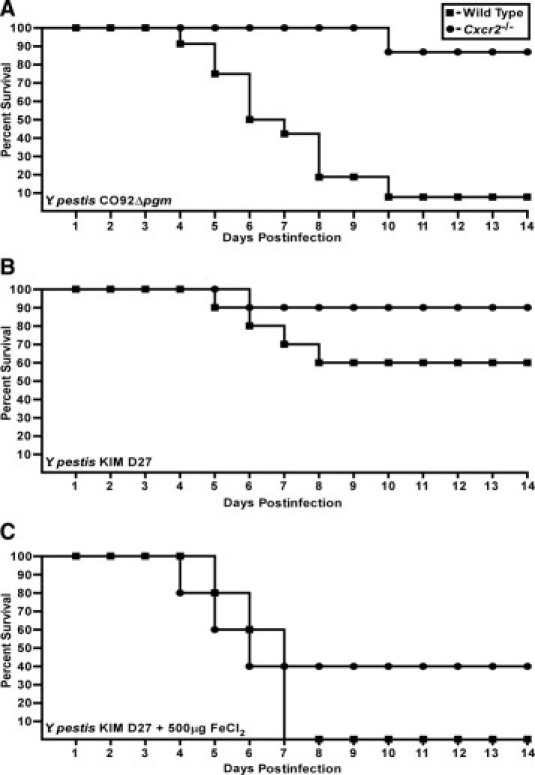
CXCR2 increases sensitivity to nonpigmented Y. pestis strains. A: WT (n = 12) and Cxcr2−/− mice (n = 7) were challenged by intranasal infection with 1 × 106 CFU Y. pestis CO92Δpgm and survival was monitored over a 14-day period; P < 0.005 between wild-type and Cxcr2 mutant mice, evaluated by Mann-Whitney rank sum test. B: Wild-type or Cxcr2−/− mice (n = 10) were given PBS before intranasal infection with 1 × 106 CFU Y. pestis KIM D27 and survival was monitored for 14 days. C: 500 μg inorganic iron (FeCl2) in sterile H2O was injected IP into WT or Cxcr2−/− mice (n = 5) before infection with 1 × 106 CFU Y. pestis D27 and survival was monitored over a 14-day period; P > 0.05 between wild-type and mutant mice, evaluated by Mann-Whitney rank sum test.
Discussion
CXCR2 is known to be critical in mediating effective neutrophil responses to invading bacterial and even viral pathogens. In the case of pneumonic plague (an acute disease), CXCR2 activation is unable to keep pace with bacterial replication and a robust but ineffective inflammatory response occurs that may exacerbate the disease. In this work, we used polyclonal anti-LcrV antibodies, which opsonize the bacteria, as an alternative means to induce multiple pathways of neutrophil recruitment. As with untreated mice, antibody-treated Cxcr2−/− mice mounted a neutrophil-mediated inflammatory response. However, this only delayed and did not prevent lethal disease, suggesting that while there may be redundancy in neutrophil recruitment, CXCR2 provides unique activation that is necessary for controlling the infection. Because pulmonary infection by the nonpigmented strain could be cleared in the absence of Cxcr2, it is likely that bacterial virulence factors within the pigmentation locus contribute to the resistance of Y. pestis to neutrophils or other Gr-1+ monocytes. Recently, CXCR2-dependent activation of neutrophil extracellular traps was shown to be independent of chemotaxis, as small molecule inhibitors of CXCR2 could prevent secretion of neutrophil extracellular traps without affecting neutrophil recruitment.52 Thus, it is likely that similar chemotaxis-independent pathways involving CXCR2 may be required for host defense against Y. pestis.
Neutralizing antibodies aid in bacterial clearance mechanisms involving neutrophils and other phagocytic cells, as well as the complement system. Antibodies that opsonize bacteria promote FcR-dependent uptake and subsequent secretion of the pro-inflammatory CXC and CC chemokines, as well as activation of complement.53 When activated, FcR signaling results in enhanced killing of the bacterial pathogens, which is an important part of antibody-mediated clearance.54,55 In parallel, antibodies also stimulate activation of complement C3a and C5a signaling molecules. Although the classical complement pathway in mice is ineffective against Y. pestis, C5a may still be effective in signaling neutrophil recruitment through binding of the C5a receptor, which is present on the surface of neutrophils.56,57 Moreover, because excess C5a, such as may occur during sepsis, can paralyze neutrophil activation, it is conceivable that the inability of Cxcr2−/− mice to clear the infection may be due to this or another aberrant host response that can promote rather than prevent disease.58
Pneumonic plague in both rodents and humans is not uniform throughout the lungs or between lung lobes, making it difficult to ascertain mechanisms of disease from whole lung cellular samples or cytokine measurements of bronchoalveolar lavage fluid.25,27,31,59 Our quantitative assessments of histopathology revealed either more pronounced or more rapid neutrophil recruitment in Cxcr2 mutants than wild-type mice. Although neutrophils from Cxcr2 knockout mice cannot respond to macrophage inflammatory protein-2 or KC, they have been shown to exhibit normal migration to C5a and CC chemokines, as well as to induce phagocytic processes through FcR.60 Because Y. pestis infection elicits very strong induction of pro-inflammatory cytokines and chemokines, in addition to KC and macrophage inflammatory protein-2, and opsonizing antibodies are also strong inducers of inflammatory responses, it is likely that neutrophil recruitment in Cxcr2−/− mice proceeds by one or more of these alternative, seemingly redundant pathways.15,26,31 In contrast, defense against other bacterial infections that elicit a more limited inflammatory response in the host, may rely more heavily on CXCR2 for neutrophil recruitment.9–11 Thus, although mice require CXCR2 to control Y. pestis as with other bacterial pathogens, our results suggest a novel host pathogen interaction that regulates the activity of these phagocytic cells, independent of their recruitment.
Anti-LcrV antibody neutralizes the type III secretion system, opsonizes the bacteria for efficient uptake, and is effective in promoting protection in the lungs.36,38–40 A model for how macrophages and neutrophils might respond to these antibodies is shown in Figure 9. In this model, LcrV antibody-opsonized bacteria are likely taken up by the lung or alveolar macrophages or neutrophils via FcγR-mediated phagocytosis, and although they may survive inside the phagosome, they may also stimulate production of CC and CXC chemokines for neutrophil recruitment.44,45,47 In parallel, complement recognition of antibody-opsonized bacteria would be expected to activate C5 convertase, resulting in the circulation of C5a and recruitment of neutrophils via the C5a receptor.56 While each may lead to neutrophil recruitment to infected areas, perhaps only CXCR2 also activates specific effector functions necessary for bacterial killing, such as degranulation or extracellular traps (neutrophil extracellular traps), whereas other pathways may be disabled by Yersinia.
Figure 9.
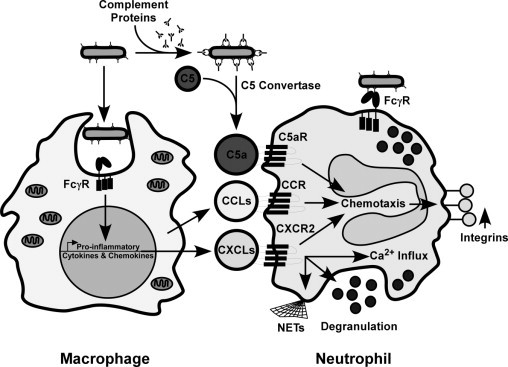
Model for the role of CXCR2 in antibody-mediated immunity to pneumonic plague. Antibody-opsonized bacteria are expected to be taken up by macrophages and neutrophils via the FcγR, and although this may not lead to bacterial clearance by either cell type, chemokines may still be secreted. Simultaneously, activation of C5a convertase leads to increased C5a, which also acts as a chemoattractant for neutrophils and other cells that express the C5a receptor (C5aR). C5aR and CC receptors would be expected to promote neutrophil chemotaxis but may be prevented from activation because of Yersinia virulence factors encoded within the pgm locus. In contrast, CXC chemokine signaling may be required to activate effector functions independent of neutrophil chemotaxis, such as increased intracellular Ca2+, stimulation of granuole release, or secretion of neutrophil extracellular traps (NETs) that promote bacterial clearance, and protect the host from disease.
Because Cxcr2−/− mice treated with anti-LcrV had a significant delay in the development of disease compared to no treatment, it appears that phagocytosis in the lungs without subsequent CXCR2 signaling slows bacterial growth but does not lead to killing of the engulfed bacterium. Furthermore, because neutrophil ablation resulted in the same phenotype, it is likely that this initial containment is not mediated by neutrophils. In vitro, activated macrophages that engulf anti-LcrV-treated bacteria are unable to kill them, while deletion of the pgm locus renders Y. pestis sensitive to activated macrophages.46,47 Our results are consistent with this observation but suggest that bacterial growth may be reduced in this intracellular environment in vivo. CXCR2−/− neutrophils also express FcR; thus it may be that this signaling pathway may be targeted by Yersinia in both cell types.
Recently, human neutrophils were shown to be able to kill intracellular Y. pestis in vitro but were sensitive to killing by extracellular bacteria expressing a functional type III secretion system.61 Our model suggests that this activity may be influenced by CXCR2 and that other activation mechanisms may be silenced by virulence factors of the pigmentation locus. Future experiments will aim to distinguish whether Fcγ, C5a, or CC receptor signaling is silenced by bacterial or host responses and whether human neutrophils respond similarly to antibody-opsonized Y. pestis.
Acknowledgments
We are grateful to members of our laboratory for helpful discussions and assistance with the BSL3 experiments and to Dr. Craig Franklin for assistance with statistical analyses. Histology and immunohistochemistry were performed by the Research Animal Diagnostics Laboratory (RADIL; University of Missouri). The Kc−/− mice were a kind gift from Dr. Sergio Lira (Mount Sinai School of Medicine).
Footnotes
Supported by a grant from the National Institutes of Health/NIAID Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases to D.M.A. (U54 AI157160). N.A.E. was supported by NIH/NIGMS Cellular and Molecular Biology training grant (T32 GM008396); H.L.L. was supported by NIH/NCRR Comparative Medicine training grant (T32 RR007004).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2010.11.067.
Supplementary data
References
- 1.Hamilton T., Novotny M., Datta S., Mandal P., Hartupee J., Tebo J., Li X. Chemokine and chemoattractant receptor expression: post-transcriptional regulation. J Leuk Biol. 2007;82:213–219. doi: 10.1189/jlb.1206754. [DOI] [PubMed] [Google Scholar]
- 2.Nasser M., Raghuwanshi S., Grant D., Venkatakrishna R., Rajarathnam K., Richardson R. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J Immunol. 2009;183:3425–3432. doi: 10.4049/jimmunol.0900305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith K., Clatworthy M. FcgRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oquendo P., Alberta J., Wen D., Graycar J., Derynck R., Stiles C. The platelet-derived growth factor-inducible KC gene encodes a secretory protein related to platelet alpha-granule proteins. J Biol Chem. 1989;264:4133–4137. [PubMed] [Google Scholar]
- 5.Tekamp-Olson P., Gellegos C., Bauer D., McClain J., Sherry B., Fabre M., Van Deventer S., Cerami A. Cloning and characterization of cDNAs for murine macrophage inflammatory protein-2 and its human homologues. J Exp Med. 1990;172:911–919. doi: 10.1084/jem.172.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X., Liu Q., Wang Y., Thorlacius H. CXC chemokines: MIP-2 and KC induce P-selectin-dependent neutrophil rolling and extravascular migration in vivo. Br J Pharmacol. 2001;133:413–421. doi: 10.1038/sj.bjp.0704087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman R., Phillips J., Hipkin R., Curran A., Lundell D., Fine J. CXCR2 antagonists for the treatment of pulmonary disease. Phamacol Ther. 2009;121:55–68. doi: 10.1016/j.pharmthera.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Mehrad B., Strieter R., Moore T., Tsai W., Lira S., Standiford T. CXC chemokine receptor-2 ligands are necessary components of neutrophil mediated host defense in invasive pulmonary aspergillosis. J Immunol. 1999;163:6086–6094. [PubMed] [Google Scholar]
- 9.Moore T., Newstead M., Strieter R., Mehrad B., Beaman B., Standiford T. Bacterial clearance and survival are dependent on CXC chemokine receptor-2 ligands in a murine model of pulmonary Norcardia asteroides infection. J Immunol. 2000;164:908–915. doi: 10.4049/jimmunol.164.2.908. [DOI] [PubMed] [Google Scholar]
- 10.Tsai W., Strieter R., Mehrad B., Newstead M., Zeng X., Standiford T. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun. 2000;68:4289–4296. doi: 10.1128/iai.68.7.4289-4296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tateda K., Moore T., Newstead M., Tsai W., Zeng X., Deng J., Chen G., Reddy R., Yamaguchi K., Standiford T. Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect Immun. 2001;69:2017–2024. doi: 10.1128/IAI.69.4.2017-2024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hotchkiss R., Nicholson D. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–822. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 13.Nagarkar D., Wang Q., Shim J., Zhao Y., Tsai W., Lukacs N., Sajjan U., Hershenson M. CXCR2 is required for neutrophilic airway inflammation and hyperresponsiveness in a mouse model of human Rhinovirus infection. J Immunol. 2009;183:6698–6707. doi: 10.4049/jimmunol.0900298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spehlmann M., Dann S., Hruz P., Hanson E., McCole D., Eckmann L. CXCR2-dependent mucosal neutrophil influx protects against colitis-associated diarrhea caused by an attaching/effacing lesion-forming bacterial pathogen. J Immunol. 2009;183:3332–3343. doi: 10.4049/jimmunol.0900600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbold W., Maus R., Hahn I., Ding N., Srivastava M., Christman J., Mack M., Reutershan J., Briles D., Paton J., Winter C., Welte T., Maus U. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to Pneumococcal lung infection. Infect Immun. 2010;78:2620–2630. doi: 10.1128/IAI.01169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobs J., Ortiz-Lopez A., Campbell J., Gerard C., Mathis D., Benoist C. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 2010;62:1921–1932. doi: 10.1002/art.27470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mei J., Liu Y., Dai N., Favara M., Greene T., Jeyaseelan S., Poncz M., Lee J., Worthen G. CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity. 2010;33:106–117. doi: 10.1016/j.immuni.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ritzman A., Hughes-Hanks J., Blaho V., Wax L., Mitchell W., Brown C. KC (CXCL1)/ CXCR2 mediates neutrophil recruitment and is critical for development of experimental Lyme arthritis and carditis. Infect Immun. 2010;78:4593–4600. doi: 10.1128/IAI.00798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seki M., Kohno S., Newstead M., Zeng X., Bhan U., Lukacs N., Kunkel S., Standiford T. Critical role of IL-1 receptor-associated kinase-M in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J Immunol. 2010;184:1410–1418. doi: 10.4049/jimmunol.0901709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanderbilt J., Mager E., Allen L., Sawa T., Wiener-Kronish J., Gonzalez R., Dobbs L. CXC chemokines and their receptors are expressed in Type II cells and upregulated following lung injury. Am J Respir Cell Mol Biol. 2003;29:661–668. doi: 10.1165/rcmb.2002-0227OC. [DOI] [PubMed] [Google Scholar]
- 21.Neff T., Guo R., Neff S., Sarma J., Speyer C., Gao H., Bernacki K., Huber-Lang M., McGuire S., Hoesel L., Riedemann N., Beck-Schimmer B., Zetoune F., Ward P. Relationship of acute lung inflammatory injury to Fas/FasL system. Am J Pathol. 2005;166:685–694. doi: 10.1016/S0002-9440(10)62290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmal H., Shanley T., Jones M., Friedl H., Ward P. Role for macrophage inflammatory protein-2 in lipopolysaccharide-induced lung injury in rats. J Immunol. 1996;156:1963–1972. [PubMed] [Google Scholar]
- 23.Wang Z., Rui T., Yang M., Valiyeva F., Kvietys P. Alveolar macrophages from septic mice promote polymorphonuclear leukocyte transendothelial migration via an endothelial cell Src Kinase/ NADPH oxidase pathway. J Immunol. 2008;181:8735–8744. doi: 10.4049/jimmunol.181.12.8735. [DOI] [PubMed] [Google Scholar]
- 24.Gordon J., Li F., Zhang X., Wang W., Zhao X., Nayyar A. The combined CXCR1/CXCR2 antagonist CXCL8(3–74)K11R/G31P blocks neutrophil infiltration, pyrexia, and pulmonary vascular pathology in endotoxemic animals. J Leukoc Biol. 2005;78:1265–1272. doi: 10.1189/jlb.0805458. [DOI] [PubMed] [Google Scholar]
- 25.Pollitzer R. World Health Organization; Geneva, Switzerland: 1954. Plague. [Google Scholar]
- 26.Lathem W., Crosby S., Miller V., Goldman W. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A. 2005;102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson D., Ciletti N., Lee-Lewis H., Elli D., Segal J., Overheim K., DeBord K., Tretiakova M., Brubaker R., Schneewind O. Pneumonic plague pathogenesis and immunity in Brown Norway rats. Am J Pathol. 2009;174:910–921. doi: 10.2353/ajpath.2009.071168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang X., Nikolich M., Lindler L. Current trends in plague research: from genomics to virulence. Clin Med Res. 2006;4:189–199. doi: 10.3121/cmr.4.3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montminy S., Khan N., McGrath S., Walkowicz M., Sharp F., Conlon J., Fukase K., Kusumoto S., Sweet C., Miyake K., Akira S., Cotter R., Goguen J., Lien E. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol. 2006;7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 30.Auerbuch V., Golenbock D., Isberg R. Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog. 2009;5:1000686. doi: 10.1371/journal.ppat.1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agar S., Sha J., Foltz S., Erova T., Walberg K., Parham T., Baze W., Suarez G., Peterson J., Chopra A. Characterization of a mouse model of plague after aerosolization of Yersinia pestis CO92. Microbiology. 2008;154:1939–1948. doi: 10.1099/mic.0.2008/017335-0. [DOI] [PubMed] [Google Scholar]
- 32.Agar S., Sha J., Foltz S., Erova T., Walberg K., Baze W., Suarez G., Peterson J., Chopra A. Characterization of the rat pneumonic plague model: Infection kinetics following aerosolization of Yersinia pestis CO92. Microbes Infect. 2009;11:205–214. doi: 10.1016/j.micinf.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Lee-Lewis H., Anderson D. Absence of inflammation and pneumonia during infection with non-pigmented Yersinia pestis reveals new role for the pgm locus in pathogenesis. Infect Immun. 2010;78:220–230. doi: 10.1128/IAI.00559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skrzypek E., Straley S. Differential effects of deletions in lcrV on secretion of V antigen, regulation of the low-Ca2+ response, and virulence of Yersinia pestis. J Bacteriol. 1995;177:2530–2542. doi: 10.1128/jb.177.9.2530-2542.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill J., Leary S., Griffin K., Williamson E., Titball R. Regions of Yesinia pestis V antigen that contribute to protection against plague identified by passive and active immunization. Infect Immun. 1997;65:4476–4482. doi: 10.1128/iai.65.11.4476-4482.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weeks S., Hill J., Friedlander A., Welkos S. Anti-V antigen antibody protects macrophages from Yersinia pestis-induced cell death and promotes phagocytosis. Microb Path. 2002;32:227–237. doi: 10.1006/mpat.2002.0498. [DOI] [PubMed] [Google Scholar]
- 37.Fetherston J., Perry R. Yersinia pestis – etiologic agent of plague. Clin Microbiol Rev. 1997;10:35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cowan C., Philipovskiy A., Wulff-Strobel C., Ye Z., Straley S. Anti-LcrV antibodies inhibits delivery of Yops by Yersinia pestis KIM5 by directly promoting phagocytosis. Infect Immun. 2005;73:6127–6137. doi: 10.1128/IAI.73.9.6127-6137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisele N., Anderson D. Dual-function antibodies to Yersinia pestis LcrV required for pulmonary clearance of plague. Clin Vacc Immunol. 2009;16:1720–1727. doi: 10.1128/CVI.00333-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quenee L., Berube B., Segal J., Elli D., Ciletti N., Anderson D., Schneewind O. Amino acid residues 196–225 of LcrV represent a plague protective epitope. Vaccine. 2010;28:1870–1876. doi: 10.1016/j.vaccine.2009.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burrows T., Jackson S. The virulence-enhancing effect of iron on nonpigmented mutants of virulent strains of Pasteurella pestis. Br J Exp Path. 1956;37:577–583. [PMC free article] [PubMed] [Google Scholar]
- 42.Brubaker R., Beesley E., Surgalla M. Pasteurella pestis: Role of Pesticin I and iron in experimental plague. Science. 1965;149:422–424. doi: 10.1126/science.149.3682.422. [DOI] [PubMed] [Google Scholar]
- 43.Fetherston J., Schuetze P., Perry R. Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102kb of chromosomal DNA which is flanked by a repetitive element. Mol Microbiol. 1992;6:2693–2704. doi: 10.1111/j.1365-2958.1992.tb01446.x. [DOI] [PubMed] [Google Scholar]
- 44.Pujol C., Bliska J. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect Immun. 2003;71:5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grabenstein J., Marceau M., Pujol C., Simonet M., Bliska J. The response regulator PhoP of Yersinia pseudotuberculosis is important for replication in macrophages and for virulence. Infect Immun. 2004;72:4973–4984. doi: 10.1128/IAI.72.9.4973-4984.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pujol C., Grabenstein J., Perry R., Bliska J. Replication of Yersinia pestis in interferon γ-activated macrophages requires ripA, a gene encoded in the pigmentation locus. Proc Natl Acad Sci U S A. 2005;102:12909–12914. doi: 10.1073/pnas.0502849102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noel B., Lilo S., Capurso D., Hill J., Bliska J. Yersinia pestis can bypass protective antibodies to LcrV and activation with gamma interferon to survive and induce apoptosis in murine macrophages. Clin Vaccine Immunol. 2009;16:1457–1466. doi: 10.1128/CVI.00172-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Surgalla M., Beesley E. Congo red-agar plating medium for detecting pigmentation in Pasteurella pestis. Appl Microbiol. 1969;18:834–837. doi: 10.1128/am.18.5.834-837.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown C., Blaho V., Loiacono C. Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental lyme arthritis via the recruitment of Gr-1 polymorphonuclear leukocyte-like cells. Infect Immun. 2004;72:4956–4965. doi: 10.1128/IAI.72.9.4956-4965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stillie R., Stadnyk A. The functional signficance behind expressing two IL-8 receptor types on PMN. J Leuko Biol. 2009;86:529–543. doi: 10.1189/jlb.0208125. [DOI] [PubMed] [Google Scholar]
- 51.Laws T., Davey M., Titball R., Lukaszewski R. Neutrophils are important in early control of lung infection by Yersinia pestis. Microbes Infect. 2010;12:331–335. doi: 10.1016/j.micinf.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 52.Marcos V., Zhou Z., Yildirim A., Bohla A., Hector A., Vitkov L., Wiedenbauer E., Krautgartner W., Stoiber W., Belohradsky B., Rieber N., Kormann M., Koller B., Roscher A., Roos D., Griese M., Eickelberg O., Doring G., Mall M., Hartl D. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med. 2010;16:1018–1023. doi: 10.1038/nm.2209. [DOI] [PubMed] [Google Scholar]
- 53.Nimmerjahn F., Ravetch J. Rcg receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 54.Tian H., Weber S., Thorkildson P., Kozel T., Pirofski L. Efficacy of opsonic and nonopsonic serotype 3 Pneumococcal capsular polysaccharide-specific monoclonal antibodies against intranasal challenge with Streptococcus pneumoniae in mice. Infect Immun. 2009;77:1502–1513. doi: 10.1128/IAI.01075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gondwe E., Molyneux M., Goodall M., Graham S., Mastroeni P., Drayson M., MacLennan C. Importance of antibody and complement for oxidative burst and killing of invasive nontyphoidal Salmonella by blood cells in Africans. Proc Natl Acad Sci U S A. 2010;107:3070–3075. doi: 10.1073/pnas.0910497107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo R., Riedemann N., Sun L., Gao H., Shi K., Reuben J., Sarma V., Zetoune F., Ward P. Divergent signaling pathways in phagocytic cells during sepsis. J Immunol. 2006;c177:1306–1313. doi: 10.4049/jimmunol.177.2.1306. [DOI] [PubMed] [Google Scholar]
- 57.Kolodziejek A., Schnider D., Rohde H., Wojtowicz A., Bohach G., Minnich S., Hovde C. Outer membrane protein X (Ail) contributes to Yersinia pestis virulence in pneumonic plague and its activity is dependent on LPS core length. Infect Immun. 2010;78:5233–5243. doi: 10.1128/IAI.00783-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Riddirisch D., Flierl M., Nadeau B., Day D., Huber-Lang M., Mackay C., Zetoune F., Gerard N., Cianflone K., Köhl J., Gerard C., Sarma J., Ward P. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lien-Teh W. League of Nations Health Organization; Geneva, Switzerland: 1926. A Treatise on Pneumonic Plague. [Google Scholar]
- 60.Lee J., Cacalano G., Camerato T., Toy K., Moore M., Wood W. Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol. 1995;155:2158. [PubMed] [Google Scholar]
- 61.Spinner J.L., Cundiff J.A., Kobayashi S.D. Yersinia pestis type III secretion system-dependent inhibition of human polymorphonuclear leukocyte function. Infect Immun. 2008;76:3754–3760. doi: 10.1128/IAI.00385-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.