

Abstract
Loss of function mutations in FGD1 result in faciogenital dysplasia, an X-linked human developmental disorder that adversely affects the formation of multiple skeletal structures. FGD1 encodes a guanine nucleotide exchange factor that specifically activates Cdc42, a Rho family small GTPase that regulates a variety of cellular behaviors. We have found that FGD1 is expressed in human mesenchymal stem cells (hMSCs) isolated from adult bone marrow. hMSCs are multipotent cells that can differentiate into many cell types, including fibroblasts, osteoblasts, adipocytes, and chondrocytes, and are thought to play a role in maintaining musculoskeletal tissues throughout life. We demonstrate an active role of FGD1 in osteogenic differentiation of hMSCs. During osteogenic differentiation of hMSCs in culture, we observed up-regulation of both FGD1 expression and Cdc42 activity. Activating FGD1/Cdc42 signaling by overexpression of either FGD1 or constitutively active Cdc42 promoted hMSC osteogenesis, while inhibiting Cdc42 signaling by either dominant negative mutants of FGD1 or Cdc42 suppressed osteogenesis. These results demonstrate an important role for FGD1/Cdc42 signaling in hMSC osteogenesis and suggest that the defects in bone remodeling in faciogenital dysplasia may persist throughout adult life and serve as a potential pathway that may be targeted for enhancing bone regeneration.
FGD1 was first identified as the gene responsible for faciogenital dysplasia (FGDY), or Aarskog syndrome, an X-linked developmental disorder that adversely affects the formation of a particular set of skeletal structures.1 The cardinal features of this disease include craniofacial dysmorphism, brachydactyly, and disproportionate acromelic short stature. Radiographic abnormalities include maxillary and mandibular hypoplasia, hypoplastic phalanges, delayed bone maturation, and a variety of vertebral anomalies.2
FGD1 encodes a guanine nucleotide exchange factor (GEF) that specifically activates the Rho family small GTPase Cdc42.3 The Rho family small GTPases cycle between the active GTP-bound and inactive GDP-bound states. GEFs activate small GTPases by promoting the exchange of bound GDP for GTP. Like most Rho GEFs, FGD1 contains a Dbl homology domain adjacent to a pleackstrin homology domain that catalyzes the exchange of GDP for GTP on Cdc42, an N-terminal proline-rich domain, a cysteine-rich zinc-finger FYVE (Fab1p, YOTB, Vac1p, and EEA1) domain, and a second C-terminal pleackstrin homology domain.4 Cdc42 has been implicated in multiple signaling pathways that regulate actin cytoskeleton organization, cell polarization, vesicular trafficking, cell cycle progression, and gene expression.5–7 Microinjection of FGD1 into fibroblasts induces actin polymerization and filopodia formation,3,8 which can be blocked by Cdc42 inhibition. FGD1 also activates the c-Jun N-terminal kinase signaling cascade,3,8 promotes G1 cell cycle progression,9 and causes tumorigenic transformation of NIH3T3 fibroblasts.10
In humans, FGD1 seems to be expressed in many tissues during development, including heart, brain, lung, pancreas, and liver.1 However, the exact expression pattern and the functions of FGD1 in adult human tissues have not been well studied. During mouse embryogenesis, FGD1 expression is restricted to regions of incipient and active ossification, including craniofacial bones, vertebrae, ribs, long bones and phalanges, skeletal components derived from neural crest, periaxial mesoderm, and lateral mesoderm.11 This pattern of FGD1 expression directly corresponds to the abnormalities of endochondral and intramembranous bone formation observed in FGDY. FGD1 protein is also expressed in mouse osteoblasts, human osteosarcoma cell lines, and permanent osteoblast-like cell lines, including MC3T3-E1, but not in other mesodermal cells. These accumulated data suggest that FGD1/Cdc42 signaling may play an important role in early skeletal formation and that FGDY is a developmental disorder of dysregulated FGD1/Cdc42 signaling. Postnatally, FGD1 was predominantly expressed in the perichondrium, resting chondrocytes, and joint capsule fibroblasts, with little expression in adult mouse bone.11 These results suggest that, although the role of FGD1/Cdc42 may be restricted to osteoblastogenesis during early developmental processes, FGD1/Cdc42 may play a broader role in adult skeletogenesis.
hMSCs are multipotent cells present in adults that can differentiate into many cell types, including fibroblasts, osteoblasts, adipocytes, chondrocytes, and myocytes.12 Because of their intrinsic ability to self-renew and differentiate into various functional cell types, hMSCs are largely thought to play a role in musculoskeletal tissue maintenance and repair throughout adult life. From an interventional perspective, hMSCs also are being actively pursued as a promising source of cells for cell-based therapeutic strategies in regenerative medicine. Their efficacy in cellular therapy has been demonstrated in treating children with osteogenesis imperfecta,13 hematopoietic recovery,14 and bone tissue regeneration.15 However, the capacity of autologous hMSCs to differentiate into functional bone-forming osteoblasts remains relatively limited for bone regeneration in vivo. An important issue for efficient bone regeneration, therefore, is to identify regulatory pathways that can be targeted to enhance the osteogenic potential of hMSCs.
Given the genetic evidence of the critical role of FGD1 in bone development, we sought to investigate whether there is a role for FGD1/Cdc42 signaling in hMSC osteogenesis. First, we found that both FGD1 expression and Cdc42 activity were up-regulated during hMSC osteogenic differentiation. Furthermore, activating Cdc42 signaling by overexpression of either FGD1 or constitutively active Cdc42 (Cdc42CA) promoted hMSC osteogenesis, whereas inhibiting Cdc42 signaling by either dominant negative FGD1 or Cdc42 suppressed dexamethasone-induced hMSC osteogenesis. Finally, the promoting effect of FGD1 on osteogenesis can be inhibited by dominant negative Cdc42 (Cdc42DN), indicating the pro-osteogenic effect of FGD1 is indeed mediated through activation of Cdc42. These results demonstrate an important role for FGD1/Cdc42 signaling in hMSC osteogenesis, potentially extending the importance of FGD1 function to adult bone remodeling, and suggest a potential target that may be manipulated for bone regeneration.
Materials and Methods
Cell Culture and Reagents
hMSCs were obtained from Lonza (Walkersville, MD) or the Tulane Center for Gene Therapy (New Orleans, LA) and maintained in growth medium (Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 0.3 mg/ml of glutamine, 100 μg/ml of streptomycin, 100 U/ml of penicillin). Only early passage hMSCs (passage 4–6) were used for experimental studies. For osteogenic differentiation, hMSCs were seeded at 3000 cells/cm2 and cultured in osteogenic differentiation media (Lonza) for 9 or 21 days. Cdc42 antibody was obtained from BD (Franklin Lakes, NJ). FGD1 antibody was obtained from Sigma (St. Louis, MO).
Real-Time PCR
Total RNA was isolated using an RNeasy Micro Kit (QIAGEN, Valencia, CA) according to the manufacturer's instructions. cDNA was transcribed with Oligo-dT primers using MMLV reverse transcriptase (Invitrogen, Carlsbad, CA) with 0.5 μg of total RNA per reaction. Quantitative PCR was performed in an ABI 7300 system (Applied Biosystems, Foster City, CA) using TaqMan gene expression assays according to the manufacturer's instructions. Results were analyzed using the relative quantitation method, and all mRNA expression data were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression in the corresponding sample. TaqMan gene expression assays used were as follows: FGD1 (Hs00171676_m1) and GAPDH (Hs99999905_m1).
Lentivirus Production
The mouse cDNAs of wild-type FGD1 (FGD1wt; amino acids 18-960), FGD1DN (amino acids 18-960 with a deletion of amino acids 397-432), Cdc42CA (Q61L mutant), and Cdc42DN (T17N mutant) (gifts from M. Philips, New York University Medical Center, New York, NY) fused to enhanced green fluorescent protein were subcloned into the pRRL-cPPT-CMV-X2-PRE-SIN vector and then co-transfected with pMDLg/pRRE, pCMV-VSVG, and pRSV-REV packaging vectors (gifts from W. Osborne, University of Washington, Seattle, WA) into Ad293 cells using Lipofectamine 2000 (Invitrogen) per the manufacturer's instruction. Viral supernatants were collected after 48 hours, centrifuged at 1500 × g for 5 minutes, filtered through a 0.45-μm filter (Nalgene, Rochester, NY), aliquoted, and stored at −80°C. Viral titer was determined by serial dilution and infection of hMSCs. The titer giving more than 90% transduction efficiency (by examining GFP-positive cells) and minimum cell death was used.
Cdc42 GTPase Assay
Cdc42-GTP levels were measured by pull-down assay. In brief, cells were washed with cold Tris-buffered saline and scraped into 1× MLB buffer (Millipore, Billerica, MA). Cleared lysates were incubated with GST-Pak1-PBD-agarose beads for 60 minutes at 4°C, centrifuged, washed, and eluted by boiling in Laemmli sample buffer. Pak1-PBD beads were made using GST-tagged recombinant Pak1-PBD produced in BL21 cells containing the pGEX-PBD vector (a gift from L. Romer, Johns Hopkins University, Baltimore, MD). Protein levels were detected by Western blot and quantified using Versadoc imaging system (Bio-Rad Laboratories, Hercules, CA).
Alkaline Phosphatase Staining
Cells were fixed with 3.7% formaldehyde, rinsed in PBS, and stained with Fast Blue RR/naphthol (Sigma). For total cell counts, nuclei were stained with DAPI. Cells were photographed and counted using a Nikon Eclipse TE200 (Nikon Instruments Inc, Melville, NY).
Alizarin Red S Staining
Cells were fixed with 4% paraformaldehyde, rinsed in water, and stained with 1% Alizarin Red S solution (VWR, West Chester, PA) for 15 minutes. Samples were washed four times in water and imaged using a Nikon Eclipse TE200 (Nikon Instruments Inc).
Results
In mice, FGD1 transcript is detected in regions of active bone formation during skeletal development. Postnatally, FGD1 is expressed more broadly in skeletal tissues, including perichondrium, resting chondrocytes, and joint capsule fibroblasts.11 To begin to examine whether FGD1 plays a role in hMSC biology, we first tested whether FGD1 is expressed in hMSCs derived from bone marrow. Real-time PCR analysis showed FGD1 mRNA was detected in MSCs from the three different donors examined (Figure 1A). Interestingly, this expression was significantly up-regulated when cells were induced to undergo osteogenesis by culturing in osteogenic induction media (Figure 1A). In contrast, there was little to no up-regulation during hMSC adipogenesis. After confirming that FGD1 is expressed in hMSCs, we focused additional experiments on cells from donor 1. Consistent with mRNA measurements, we observed that levels of FGD1 protein increased during osteogenesis (Figure 1B). Because FGD1 is a Cdc42-specific GEF, we measured Cdc42 activity in hMSCs exposed to osteogenic induction medium. Cdc42 was activated 1 day after osteogenic induction, remained high for several days, and returned to basal level after 5 to 7 days (Figure 1C). This trend in Cdc42 activity is consistent with the FGD1 expression pattern. These data suggest FGD1/Cdc42 signaling may play an important role during hMSC osteogenesis.
Figure 1.
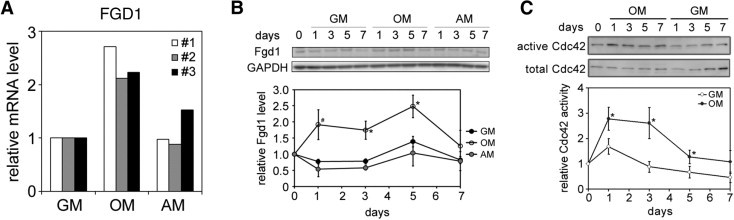
FGD1 expression and Cdc42 activity are both up-regulated during hMSC osteogenesis. A: hMSCs from three different donors (donor 1: Lonza lot 6F4393; donor 2: donor 7025L from the Tulane Center for Gene Therapy; donor 3: Lonza lot 4F1560) were grown in growth medium (GM), osteogenic medium (OM), or adipogenic medium (AM) for 7 days. The mRNA level of FGD1 was analyzed by real-time PCR. B: hMSCs were cultured in GM, OM, or AM for 7 days. FGD1 protein expression was measured by Western blot and normalized to GAPDH on days 1, 3, 5, and 7. Data are represented as mean ± SEM, n = 4. #P = 0.087; *P < 0.05, compared with GM control (by Student's t-test). C: hMSCs were cultured in GM or OM for 7 days and active Cdc42 level was measured by pull-down assay and quantitated on days 1, 3, 5, and 7. Active Cdc42 level was normalized to total Cdc42 level and then to day 0 data. Data are represented as mean ± SD, n = 3. *P < 0.05 with GM control (by Student's t-test).
To decipher the role of FGD1/Cdc42 signaling in hMSC osteogenesis, we constructed lentiviruses encoding FGD1wt, FGD1DN, Cdc42CA, or Cdc42DN16 fused to enhanced green fluorescent protein and infected hMSCs. The FGD1DN construct is a naturally occurring splice variant of the FGD1 gene, which encodes a biologically inactive peptide.4,8,10 It is identical to FGD1wt, except it is missing part of the Dbl homology domain (residues 397 to 432) and therefore lacks GEF activity. When cells were infected with FGD1DN, cultured in osteogenic medium for 9 days, and then assayed for alkaline phosphatase activity as an early osteogenic marker, we found FGD1DN significantly inhibited osteogenesis (Figure 2, A and B). Consistent with this finding, Cdc42DN had the same effect, suggesting FGD1/Cdc42 signaling is required for hMSC osteogenesis. To examine whether activating this signaling pathway might be sufficient to promote osteogenesis, cells infected with different FGD1 and Cdc42 constructs were cultured in nondifferentiating growth medium for 14 days and assayed for alkaline phosphatase activity. Both FGD1wt and Cdc42CA induced osteogenic differentiation in the absence of the induction medium (Figure 2, C and D).
Figure 2.
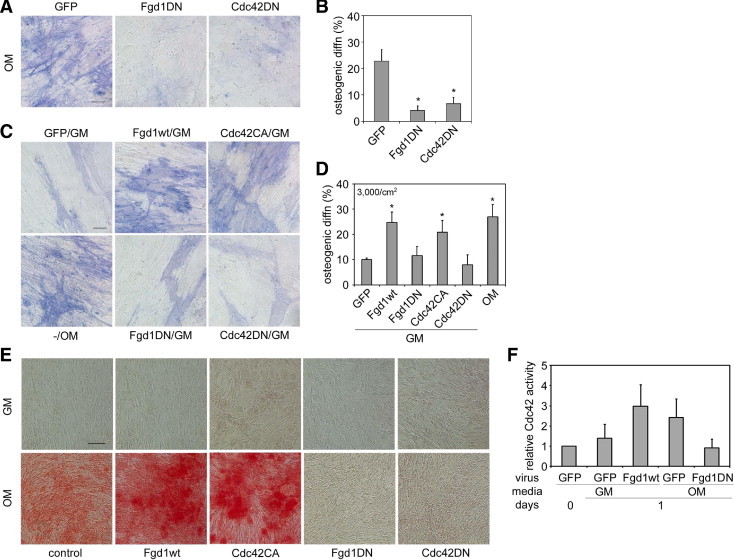
FGD1 and Cdc42 regulate hMSC osteogenesis. A: hMSCs were infected with lentivirus expressing GFP, FGD1DN, or Cdc42DN and cultured in osteogenic medium (OM) for 9 days. Cells were fixed and stained for alkaline phosphatase activity. Scale bar = 50 μm. B: Percentage of hMSC osteogenic differentiation in A. Data are represented as mean ± SD, n = 3. *P < 0.05 with GFP control (by Student's t-test). C: hMSCs were infected with lentivirus expressing different FGD1 or Cdc42 constructs as indicated and grown in growth medium (GM) for 14 days. Cells were fixed and stained for alkaline phosphatase activity. Scale bar = 50 μm. D: Percentage of hMSC osteogenic differentiation in C. Data are represented as mean ± SD, n = 3. *P < 0.05 with GFP control (by Student's t-test). E: hMSCs were infected with lentivirus expressing different FGD1 or Cdc42 constructs and grown in GM or OM for 21 days. Cells were fixed and stained with Alizarin Red S. Scale bar = 200 μm. F: The overexpression of FGD1 constructs changed Cdc42 activity. hMSCs were infected with lentivirus expressing different FGD1 constructs and grown in GM or OM for 1 day. Active Cdc42 level was measured by pull-down assay (as described in Figure 1C). Data are represented as mean ± SD, n = 3.
Although increased alkaline phosphatase activity provides an early indication of osteogenesis, complete differentiation toward the osteoblastic lineage is functionally characterized by the ability of differentiated cells to deposit calcium and mineralize the extracellular matrix. Thus, we performed Alizarin Red S staining for deposited calcium in hMSCs cultured for 21 days in these same conditions. Results showed that enhancing FGD1/Cdc42 signaling in the absence of induction medium was not sufficient to induce mineral deposition (Figure 2E), although increased alkaline phosphatase activity was observed. However, when infected cells were cultured in the osteogenic induction medium, FGD1wt and Cdc42CA further enhanced calcium nodule formation over control, the induction medium alone samples, whereas FGD1DN and Cdc42DN completely inhibited mineralization (Figure 2E). The FGD1 constructs were confirmed to affect Cdc42 signaling as expected: FGD1wt overexpression increased Cdc42 activity, whereas FGD1DN overexpression inhibited Cdc42 activity (Figure 2F). These results demonstrate that FGD1/Cdc42 signaling i) is required for hMSC osteogenesis, ii) by itself is not sufficient to induce differentiation into fully mature osteoblasts, but iii) can potentiate the osteogenesis in the presence of other osteogenic induction signals.
Although FGD1 is thought to be a Cdc42-specific GEF, it might have other unidentified downstream effectors. Conversely, many other GEFs can activate Cdc42. To determine whether the osteogenesis-promoting effect of FGD1 is mediated specifically through Cdc42, we infected hMSCs simultaneously with FGD1wt and Cdc42DN and assayed for osteogenesis after 14 days. Indeed, Cdc42DN completely blocked the pro-osteogenic effect of FGD1wt, confirming that Cdc42 mediates FGD1-induced osteogenesis (Figure 3, A and B). When the same experiment was performed in the presence of osteogenic media, FGD1wt did not further increase osteogenesis after 9 days, and Cdc42DN was still able to partially inhibit osteogenesis (see Supplemental Figure S1 at http://ajp.amjpathol.org), consistent with the model that the FGD1/Cdc42 pathway is involved in osteogenic differentiation.
Figure 3.
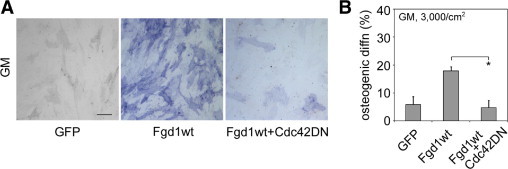
Cdc42DN abrogated the promoting effect of FGD1 on osteogenesis. A: hMSCs were infected with FGD1wt alone or coinfected with FGD1wt and Cdc42DN and grown in growth medium for 14 days. Cells were fixed and stained for alkaline phosphatase activity. Scale bar = 100 μm. Result was quantified in B. Data are represented as mean ± SD, n = 3. *P < 0.05 (by Student's t-test).
Interestingly, the effect of FGD1 or Cdc42 on hMSC osteogenesis is independent of cell density. When hMSCs were seeded at very high density (20,000 cells/cm2), a condition that we have previously demonstrated prevents osteogenic differentiation of hMSCs,17 FGD1wt and Cdc42CA were still able to promote osteogenesis, and the effect of FGD1 was again inhibited by Cdc42DN (see Supplemental Figure S2, A and B, at http://ajp.amjpathol.org).
Discussion
Rho family small GTPases have well-established roles in cytoskeleton remodeling and have been implicated in mediating the signals from extracellular cues to cell fate determination. We have shown previously that changes in hMSC adhesion to the extracellular matrix can modulate RhoA activity, and this RhoA signaling triggers an adipogenic-osteogenic fate switch in hMSCs.17 Similarly, Rac1 GTPase regulates hMSC differentiation into smooth muscle cells or chondrocytes also as a function of cell adhesion.18 Like RhoA and Rac1, FGD1/Cdc42 signaling similarly appears to link the regulation of actin cytoskeletal organization and stem cell fate. FGD1 can directly interact with cortactin and mouse actin-binding protein 1, and these interactions recruit FGD1 to the dynamic cortical actin cytoskeleton, thus regulating actin cytoskeleton remodeling by locally activating Cdc42.19 This link between the parallel regulation of cytoskeletal structure and stem cell differentiation may simply be a hint of the molecular machinery that coordinates differentiated cellular functions with necessary changes in cell morphology. Interestingly, despite the broad similarities among RhoA, Rac, and Cdc42 regulation of hMSC differentiation, the ability of RhoA to drive osteogenesis is blocked when cells are plated at high seeding density or in conditions that prevent cell spreading and flattening on a substrate,17 whereas the effect of FGD1/Cdc42 seems to be independent of such conditions, suggesting that FGD1/Cdc42 regulates osteogenesis through distinct pathways.
Cdc42 activity can be regulated by many other GEFs. Although we cannot exclude the possibility that exogenous expression of other Cdc42 GEFs could also affect hMSC differentiation, it is likely that tissue-specific expression is what distinguishes FGD1. Among the Cdc42-specific GEFs, FGD1 is the only one that has been shown to have strong correlation with skeletogenesis. The genetic basis for FGDY indicates that FGD1 plays an important role in early bone development. However, the role of FGD1 in adult tissues is not well understood. Our results demonstrate that FGD1 continues to be expressed in adult mesenchymal stem cells. hMSCs are crucial for physiologic tissue renewal and regeneration after injury. FGD1 thus may be important in maintaining the bone homeostasis during adulthood. Furthermore, because of the ease of their isolation and their differentiation potential, hMSCs are being explored as a source of cells for use in engineering tissues, including bone for clinical applications. The identification here of a novel mechanism involving FGD1 and Cdc42 for the regulation of osteogenesis thus points to potential molecular handles for enhancing this mechanism for therapeutic applications.
Acknowledgments
We thank Danial Cohen, Ravi Desai, and Yang-Kao Wang for helpful discussions.
Footnotes
Supported by the National Institutes of Health (grants EB00262, HL73305, and GM74048), the University of Pennsylvania Institute for Regenerative Medicine, Center for Musculoskeletal Disorders, Center for Engineering Cells and Regeneration, and March of Dimes-Birth Defects Foundation (grant 6-FY99-425 to J.L.G.). Some of the materials used in this work were provided by the Tulane Center for Gene Therapy through a grant from National Center for Research Resources of the National Institutes of Health (grant P40RR017447).
Supplemental material for this article can be found on http://ajp.amjpathol.org and at doi:10.1016/j.ajpath.2010.11.051.
Supplementary data
Cdc42DN inhibits osteogenesis in the presence of FGD1wt in osteogenic medium (OM). hMSCs were infected with FGD1wt alone or coinfected with FGD1wt and Cdc42DN and grown in OM for 9 days. Cells were fixed, stained for alkaline phosphatase activity, and quantified. Data are represented as mean ± SD, n = 3. *P < 0.05 with GFP control or FGD1wt cells (by Student's t-test).
The effect of FGD1 or Cdc42 on hMSC osteogenesis is independent of cell density. hMSCs were seeded at high density (20,000 cells/cm2), infected with lentivirus expressing different FGD1 or Cdc42 constructs, and grown in growth medium for 14 days. Cells were fixed, stained for alkaline phosphatase activity, and quantified. Data are represented as mean ± SD, n = 3. *P < 0.05 with GFP control or FGD1wt cells (by Student's t-test).
References
- 1.Pasteris N.G., Cadle A., Logie L.J., Porteous M.E.M., Schwartz C.E., Stevenson R.E., Glover T.W., Wilroy R.S., Gorski J.L. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene: a putative Rho/Rac guanine nucleotide exchange factor. Cell. 1994;79:669–678. doi: 10.1016/0092-8674(94)90552-5. [DOI] [PubMed] [Google Scholar]
- 2.Gorski J. FGD1 and faciogenital dysplasia (Aarskog-Scott syndrome) In: Epstein C., Erickson R., Wynshaw-Boris, editors. Inborn Erros in development: the molecular basis of clinical disorders of morphogenesis. Oxford University Press; New York: 2008. pp. 1289–1298. [Google Scholar]
- 3.Zheng Y., Fischer D.J., Santos M.F., Tigyi G., Pasteris N.G., Gorski J.L., Xu Y. The faciogenital dysplasia gene product FGD1 functions as a Cdc42Hs-specific guanine-nucleotide exchange factor. J Biol Chem. 1996;271:33169–33172. doi: 10.1074/jbc.271.52.33169. [DOI] [PubMed] [Google Scholar]
- 4.Pasteris N.G., Buckler J., Cadle A.B., Gorski J.L. Genomic organization of the faciogenital dysplasia (FGD1; Aarskog syndrome) gene. Genomics. 1997;43:390–394. doi: 10.1006/geno.1997.4837. [DOI] [PubMed] [Google Scholar]
- 5.Etienne-Manneville S. Cdc42–the centre of polarity. J Cell Sci. 2004;117:1291–1300. doi: 10.1242/jcs.01115. [DOI] [PubMed] [Google Scholar]
- 6.Cerione R.A. Cdc42: new roads to travel. Trends Cell Biol. 2004;14:127–132. doi: 10.1016/j.tcb.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Rincon S., Coll P.M., Perez P. Spatial regulation of Cdc42 during cytokinesis. Cell Cycle. 2007;6:1687–1691. doi: 10.4161/cc.6.14.4481. [DOI] [PubMed] [Google Scholar]
- 8.Olson M.F., Pasteris N.G., Gorski J.L., Hall A. Faciogenital dysplasia protein (FGD1) and Vav, two related proteins required for normal embryonic development, are upstream regulators of Rho GTPases. Curr Biol. 1996;6:1628–1633. doi: 10.1016/s0960-9822(02)70786-0. [DOI] [PubMed] [Google Scholar]
- 9.Nagata K., Driessens M., Lamarche N., Gorski J.L., Hall A. Activation of G1 progression: JNK mitogen-activated protein kinase, and actin filament assembly by the exchange factor FGD1. J Biol Chem. 1998;273:15453–15457. doi: 10.1074/jbc.273.25.15453. [DOI] [PubMed] [Google Scholar]
- 10.Whitehead I.P., Abe K., Gorski J.L., Der C.J. CDC42 and FGD1 cause distinct signaling and transforming activities. Mol Cell Biol. 1998;18:4689–4697. doi: 10.1128/mcb.18.8.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorski J.L., Estrada L., Hu C., Liu Z. Skeletal-specific expression of Fgd1 during bone formation and skeletal defects in faciogenital dysplasia (FGDY; Aarskog syndrome) Dev Dyn. 2000;218:573–586. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1015>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 12.Pittenger M.F., Mackay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D., Moorman M.A., Simonetti D.W., Craig S., Marshak D.R. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 13.Horwitz E.M., Gordon P.L., Koo W.K.K., Marx J.C., Neel M.D., McNall R.Y., Muul L., Hofmann T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002;99:8932–8937. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koc O.N., Gerson S.L., Cooper B.W., Dyhouse S.M., Haynesworth S.E., Caplan A.I., Lazarus H.M. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18:307–316. doi: 10.1200/JCO.2000.18.2.307. [DOI] [PubMed] [Google Scholar]
- 15.Petite H., Viateau V., Bensaïd W., Meunier A., Pollak Cd, Bourguignon M., Oudina K., Sedel L., Guillemin G. Tissue-engineered bone regeneration. Nature Biotechnol. 2000;18:959–963. doi: 10.1038/79449. [DOI] [PubMed] [Google Scholar]
- 16.Coso O.A., Chiariello M., Yu J.C., Teramoto H., Crespo P., Xu N., Miki T., Gutkind J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 17.McBeath R., Pirone D.M., Nelson C.M., Bhadriraju K., Chen C.S. Cell Shape: Cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 18.Gao L., Rowena M., Christopher S.C. Stem cell shape regulates a chondrogenic versus myogenic fate through Rac1 and N-cadherin. Stem Cells. 2010;28:564–572. doi: 10.1002/stem.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou P., Estrada L., Kinley A.W., Parsons J.T., Vojtek A.B., Gorski J.L. Fgd1, the Cdc42 GEF responsible for faciogenital dysplasia, directly interacts with cortactin and mAbp1 to modulate cell shape. Hum Mol Genet. 2003;12:1981–1993. doi: 10.1093/hmg/ddg209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cdc42DN inhibits osteogenesis in the presence of FGD1wt in osteogenic medium (OM). hMSCs were infected with FGD1wt alone or coinfected with FGD1wt and Cdc42DN and grown in OM for 9 days. Cells were fixed, stained for alkaline phosphatase activity, and quantified. Data are represented as mean ± SD, n = 3. *P < 0.05 with GFP control or FGD1wt cells (by Student's t-test).
The effect of FGD1 or Cdc42 on hMSC osteogenesis is independent of cell density. hMSCs were seeded at high density (20,000 cells/cm2), infected with lentivirus expressing different FGD1 or Cdc42 constructs, and grown in growth medium for 14 days. Cells were fixed, stained for alkaline phosphatase activity, and quantified. Data are represented as mean ± SD, n = 3. *P < 0.05 with GFP control or FGD1wt cells (by Student's t-test).