

Abstract
Molecular chaperones, including the extracellular protein clusterin (CLU), play a significant role in maintaining proteostasis; they have a unique capacity to bind and stabilize non-native protein conformations, prevent aggregation, and keep proteins in a soluble folding-competent state. In this study, we investigated amyloid-infiltrated cardiac tissue for the presence of CLU and measured serum levels of CLU in patients with and without amyloidotic cardiomyopathy (CMP). Cardiac tissues containing amyloid deposits composed of either transthyretin (TTR) or Ig light chain from nine patients with amyloidotic CMP were examined for the presence of CLU using immunohistochemical techniques. CLU staining coincided with the extracellular myocardial amyloid deposits in tissues from patients with familial TTR, senile systemic, and Ig light chain amyloidosis. The association of CLU with cardiac amyloid deposits was confirmed by immunogold electron microscopy. Serum concentrations of CLU were measured in familial TTR, senile systemic, and Ig light chain amyloidosis patient groups and compared with both age-matched healthy controls and with patients with CMP unrelated to amyloid disease. Subset analysis of disease cohorts, based on cardiac involvement, indicated that decreased serum CLU concentrations were associated with amyloidotic CMP. Taken together, these results suggest that CLU may play a pathogenetic role in TTR and Ig light chain amyloidoses and amyloidotic CMP.
Protein misfolding and aggregation are recognized as critical processes in the pathogenesis of a wide range of human diseases. In particular, the deposition of aberrantly folded and self-associated proteins as highly organized β-sheet structured amyloid fibrils is the hallmark of the amyloidoses. Amyloid fibrils bind Congo red dye, producing a characteristic apple-green birefringence when viewed under polarized light. Ex vivo amyloid deposits have been shown to include a variety of accessory proteins and molecules, including serum amyloid P component and glycosaminoglycans. The complexity of amyloid deposits in fat and other tissues has been demonstrated in proteomic studies.1
Two serum proteins that can misfold, aggregate, and form amyloid deposits in the heart and other organs are transthyretin (TTR) and immunoglobulin light chain (LC). Familial TTR-associated amyloidosis (ATTR) is caused by point mutations in the TTR gene that give rise to destabilized mutant proteins. In senile systemic amyloidosis (SSA), amyloid deposits are composed of wild-type TTR and fibrils are mainly found in the hearts of older individuals. Clonal LC monomers and fragments are components of the fibrils found in Ig light chain amyloidosis (AL) or primary amyloidosis, which occurs in association with bone marrow plasma cell disorders. Although ATTR, SSA, and AL amyloidoses are multiorgan diseases, their most pronounced phenotype is a restrictive cardiomyopathy (CMP) that may present clinically as congestive heart failure, arrhythmias, and sudden death.2–5
In vivo, the intra- and extra-cellular maintenance of proper protein folding and the prevention of aggregation is facilitated by chaperones. Clusterin (CLU), also called apolipoprotein J, is a ubiquitous protein that reportedly functions as an extracellular chaperone and may play a role in the pathological mechanism of amyloid precursor protein misfolding. CLU has a remarkable conformational adaptability that is typical of molecular chaperones and is attributed to three large molten globule domains, three amphipathic regions, and two coiled-coil α-helices. This molecular structure is responsible for the unique high affinity, low specificity, binding property of the protein.6–8
CLU has been identified in cerebrospinal and seminal fluids, breast milk, urine, and plasma; gene expression has been demonstrated in heart, testis, liver, stomach, brain, and kidney.9,10 Studies suggest that the regulation of CLU is directly linked to the heat shock response through HSF1-HSF2 heterocomplex binding to the CLU promoter; thus, modulation of CLU transcription occurs in stress- or disease-induced states.11,12 Overexpression of CLU has been reported in Alzheimer's disease (AD) studies demonstrating that the chaperone complexes to soluble amyloid β protein (Aβ) and is present as a component of the amyloid plaques. In addition, CLU has been linked to cardiovascular diseases; it is a constituent of human atherosclerotic plaques, upregulated at both mRNA and protein levels in myocarditis and ischemia models, and localized to damaged myocardium in myocardial infarction.8,10,13,14
Prompted by these observations, we investigated the role of CLU in systemic forms of amyloidosis. We examined the presence of CLU in amyloid deposits from cardiac tissue specimens in cases of SSA, ATTR, and AL amyloidoses. In addition, we quantified serum concentrations of CLU in amyloid and age-matched control specimens, and correlated levels with cardiac amyloid disease.
Materials and Methods
Study Cohorts
Patient information and biological samples were obtained from the Boston University Amyloid Treatment and Research Program repository, with the approval of the Institutional Review Board at the Boston University Medical Campus in accordance with the Declaration of Helsinki. Clinical data included details of history, physical examination, and routine laboratory studies. The diagnosis of amyloidosis was based on histological proof of congophilic fibrillar deposits in fat aspirates or tissue biopsies. Amyloid disease type was determined by a combination of immunochemical, biochemical, and genetic techniques. AL amyloidosis was established with evidence of a plasma cell dyscrasia identified by clonal plasma cells in a bone marrow biopsy and a monoclonal immunoglobulin LC by serum and urine immunofixation electrophoresis and/or free light chain nephelometry. Identification of a pathological TTR gene mutation and/or mutant protein indicated ATTR amyloidosis. In SSA, the diagnosis was made when testing for AL and ATTR were negative and there was immunological or biochemical proof of wild-type TTR in the amyloid deposits.
Cardiac function was assessed by echocardiogram. Left ventricular mass (LVM) was derived from the formula described by Devereux et al.15 Patients were identified as having cardiac involvement if they had clinical symptoms of congestive heart failure (defined as New York Heart Association Functional Class >1) and/or septal or ventricular wall thickening >12 mm on echocardiogram without a history of hypertension. Electrocardiography was also performed, and brain natriuretic peptide (BNP) levels were determined.
Commercial control sera collected from healthy, consented, paid human donors from FDA-licensed and inspected donor centers was purchased from Bioreclamation (Westbury, NY). Sera from patients with ischemic and nonischemic CMP, left ventricular ejection fraction (LVEF) <40%, and no amyloid disease served as a second control group in the study. All sera were age-matched (ie, from individuals aged 60 years or older).
Antibodies and Other Reagents
Polyclonal rabbit anti-human CLU (H-330), monoclonal mouse anti-human CLU (CLI-9), and goat anti-rabbit IgG-horseradish peroxidase (HRP) mouse/human adsorbed antibody solutions were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); polyclonal rabbit anti-human-prealbumin (TTR) antisera from DakoCytomation (Glostrup, Denmark) and polyclonal goat anti-human-kappa light chain antibodies conjugated to HRP from Novus Biologicals (Littleton, CO) were also obtained. Envision+ system-HRP labeled polymer anti-rabbit secondary antibody, proteinase K, Dako antibody diluent, and Tris-buffered saline–Tween (TBST) buffer were from DakoCytomation. Congo red, hematoxylin, Citra Plus, and Power Block solutions were purchased from BioGenex (San Ramon, CA). Polyclonal goat anti-rabbit IgG:10 nm antibody solution (British BioCell International, Cardiff, UK), Lowicryl K4M resin, and Formvar film 150 square mesh nickel grids were all acquired through Electron Microscopy Sciences (Hatfield, PA). All other chemicals were from Thermo Fisher Scientific (Fairlawn, NJ) or Sigma-Aldrich (St. Louis, MO) and were of the highest grade available.
Congo Red Staining and Immunohistochemistry
Formalin fixed paraffin embedded cardiac samples from autopsied tissues (1 nonamyloid, 3 SSA, 3 ATTR, and 3 AL) were serially sectioned to 5 μm thickness and processed for Congo red staining and immunohistochemical analysis. For Congo red staining, deparaffinized sections were counterstained with Mayer's hematoxylin for 1 minute, placed in alkaline 80% alcohol/NaCl for 20 minutes, stained in alkaline Congo red for 20 minutes, rinsed in ethanol and xylene, and observed by polarized light microscopy.
Indirect immunohistochemical techniques with rabbit anti-human primary antibodies and goat anti-rabbit secondary antibodies conjugated to HRP were used for analyses of CLU and TTR in tissue sections. A direct method, performed with anti-human free kappa LC antibodies conjugated to HRP, was used for kappa LC evaluation. For studies of CLU and LC, slides were initially boiled in antigen retrieval Citra Plus solution and cooled for 20 minutes to room temperature; in TTR analyses, slides were pretreated in 6 mol/L guanidine, pH 7.4 for 1 hour. Subsequently, all slides were washed with double distilled H2O and rinsed using TBST. Tissue sections were applied to slides, blocked with fresh Power Block and rinsed with TBST using the Dako Autostainer Plus automated system from DakoCytomation. Using Dako diluent buffer, stock antibody solutions were diluted 1:50 for CLU, 1:1000 for TTR, and 1:100 for kappa LC-HRP antibodies; sections were incubated in 200 μL of appropriate primary antibody dilutions and a corresponding primary immunobuffer (PIB) negative control for 30 minutes. For the TTR studies, sections were treated with proteinase K before incubation in primary antibody. For the analyses of CLU and TTR, and PIB negative control, 200 μL of Dako Envision+ System-HRP labeled polymer anti-rabbit secondary antibody was applied for 30 minutes, washed with TBST, and treated with diaminobenzidine. Sections were counterstained with Harris' modified hematoxylin. Imaging was performed under bright field microscopy using a SPOT Insight camera with SPOT Advanced v4.6 software (Diagnostic Instruments, Sterling Heights, MI).
Immunogold Labeling and Electron Microscopy
Frozen tissues were cut to ≤1 mm thickness and fixed overnight in 4% paraformaldehyde at 4°C. Sections were washed in sodium cacodylate buffer (Electron Microscopy Sciences, Hatfield, PA) and dehydrated in 50%, 75%, and 90% dimethylformamide solutions. Tissues were embedded in fresh Lowicryl resin and block ultrathin sections were placed on nickel grids.
Tissue sections were blocked for 1 hour with 1.0% bovine serum albumin (BSA), 5.0% normal goat serum in 0.05 mol/L Tris, pH 7.4. Stock primary antibody solutions were diluted with 0.5% BSA, 0.1% fish gelatin, 0.05% Tween in 0.05 mol/L Tris, pH 7.4, as follows: 1:5 for CLU, 1:300 for TTR, and 1:4000 for free kappa LC antibodies. Samples were incubated with appropriate primary antibody dilutions, PIB control, and normal rabbit serum control overnight at 4°C, rinsed, and treated with a 1:10 dilution of 10 nm immunogold conjugated to goat anti-rabbit IgG for 1 hour. Grids were negatively stained using 4% uranyl acetate and 0.4% lead citrate. Immunogold labeling was viewed at ×80,000 magnification using a Jem-1011 electron microscope (Jeol, Peabody, MA) at 80 kV. Electron micrographs were acquired using an ES1000W Erlangshen CCD camera and Gatan Microscopy Suite v1.7 software (Gatan Pleasanton, CA).
Tissue Extraction and Immunoblot Analysis
Approximately 100 mg of autopsied cardiac tissue samples were washed extensively (not less than 5×) using cold 12 mmol/L sodium phosphate, 137 mmol/L NaCl, 2.7 mmol/L KCl, pH 7.4, and nuclease-free H2O (Thermo Fisher Scientific) to remove contaminating blood components. Samples were agitated manually and centrifuged for 15 minutes at 4°C and 12,400 rcf. Wash solution was discarded. This was repeated five times. Tissue samples were washed with nuclease-free H2O, centrifuged for 15 minutes at 4°C and 12,400 rcf, and the supernatant was discarded. Subsequently, 7 mol/L urea, 2 mol/L thiourea, 65 mmol/L dithiothreitol, and 4% CHAPS were added at 300 μL/100 mg of tissue. Samples were homogenized using the TissueRuptor (Qiagen, Venlo, Netherlands) and then were centrifuged for 1.5 hours at 4°C and 25,000 rcf. Supernatant protein concentrations were determined and samples were stored at −80°C. Tissue protein extracts were electrophoresed under reducing conditions using 8–16% gradient Tris-HCl, SDS gels; after transfer, membranes were probed using the SNAP i.d. system (Millipore, Billerica, MA). Equal amounts of total protein were loaded in all lanes for individual blots, as follows: CLU, 40 μg; TTR, 2 μg; and kappa LC, 1 μg.
Serum Clusterin Measurement by Indirect Capture Enzyme-Linked Immunosorbent Assay
High-binding 96-well microplates (R&D Systems, Minneapolis, MN) were coated overnight at 4°C with 100 μL of 62.5 ng/ml mouse monoclonal anti-human CLU capture antibody and blocked with Blocker casein in PBS solution (Thermo Fisher Scientific). Serum samples were diluted 1:40 in 0.1% BSA, 10% Blocker casein in PBS, 0.05% Surfact-Amps 20 (Thermo Fisher Scientific) in PBS, and 100 μL applied to microplate wells in triplicate. Samples were incubated for 2 hours with rotation at 90 rpm. Plates were washed and 100 μL of 2.5 μg/ml rabbit polyclonal anti-human CLU primary detection antibody was added to each well. After 2 hours of incubation, 100 μL of 80 ng/ml preadsorbed goat anti-rabbit secondary detection antibody was added; plates were rotated for 2 hours at 90 rpm. Enzyme-linked immunosorbent assay (ELISA) solutions were developed for 20 minutes using 100 μL of an H2O2 and 3,3′,5,5′-tetramethylbenzidine chromogen system (R&D Systems); development was halted with 100 μL of 2 M sulfuric acid. Absorbance values were measured at 450 nm and 570 nm using a PowerWaveX spectrophotometer (BioTek, Winooski, VT). Standard curves were constructed for independent microplates using rCLU (ie, recombinantly generated human CLU) (R&D Systems), and final serum concentrations were determined by a four-parameter logistic curve fit using KC4 v3.4 software (BioTek, Winooski, VT).
Statistical Analysis
Data from ELISA measurements of serum CLU and echocardiography were analyzed using Prism 5 software (Graphpad Software, La Jolla, CA). One-way analysis of variance and Tukey-Kramer multiple comparison postanalysis of variance, and Pearson or Spearman correlation tests were conducted to determine significance. Continuous variables are described as mean ± SE. Statistical significance was assigned by the criterion of P ≤ 0.05.
Results
Cardiac Amyloid Deposits Contain Clusterin
The presence of amyloid deposits in autopsied cardiac specimens from three SSA, three ATTR, and three AL (kappa LC) cases were confirmed by histological treatment with Congo red; tissue from a nonamyloid heart transplant patient served as a control specimen. Light microscopic analysis of the Congo red-stained sections from all nine amyloid cardiac tissues revealed the green birefringence characteristic of amyloid deposits when viewed under polarized light (see Supplemental Figure S1 at http://ajp.amjpathol.org). No staining was evident in the control sections (data not shown).
The presence of CLU in the cardiac amyloid deposits of patients with SSA, ATTR, or AL was initially investigated by immunohistochemistry (Figure 1). The biochemical nature of the deposits, previously identified as amyloid with Congo red, was confirmed with the appropriate antibody treatment in serial sections from each of the nine tissues. TTR was identified in SSA and ATTR sections (Figure 1B and 1C), and kappa LC was verified in the AL samples (Figure 1H). The control sections were not immunoreactive to TTR or LC antibodies (Figure 1, A and E). In addition, there was no evidence of nonspecific primary antibody binding in the amyloid-laden tissues (Figure 1, D, F, and G) or nonspecific secondary antibody binding in the PIB controls (data not shown). Disruption of normal tissue architecture by the amyloid deposits, located adjacent to and surrounding cardiomyocytes, was evident. TTR and LC deposits exhibited pericellular staining patterns surrounding individual cardiomyocytes.
Figure 1.
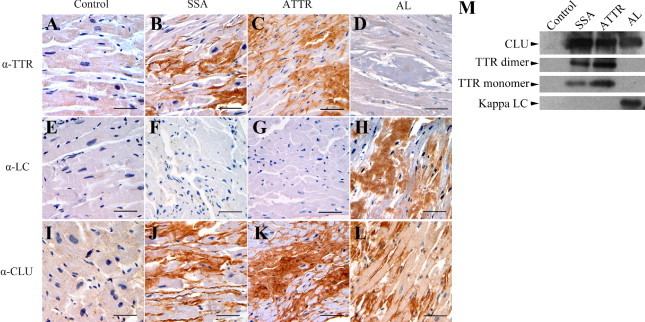
Light microscopic images of immunohistochemical staining for amyloid precursor proteins transthyretin (TTR) and Ig light chain (LC) and for the extracellular protein clusterin (CLU) in serial sections of control (no amyloid), senile systemic amyloidosis (SSA), familial TTR amyloidosis (ATTR), and LC amyloidosis (AL) cardiac tissue specimens. No staining was observed for TTR (A) or LC (E) in the control tissue, though subtle staining for CLU (I) was present. SSA and ATTR sections showed extensive pericellular staining for TTR (B, C) and CLU (J, K); there was no evidence of staining with LC (F, G). In the AL sections, abundant amounts of LC were observed (H) and strong CLU staining (L) was noted; there was no staining for TTR (D). Cardiac tissues from 3 cases in each amyloid group (SSA, ATTR, AL) were analyzed and consistent results were obtained. (Additional tissues are presented in Supplemental Figure S1 at http://ajp.amjpathol.org.) Original magnification, ×40. Scale bars = 50 microns. M: Immunoblot analysis of protein extracts from other portions of the same cardiac samples (control, SSA, ATTR, and AL) analyzed in the immunohistochemical studies.
The immunohistochemical results for CLU indicated that the chaperone was present and highly abundant in all tissue sections that were positive for amyloid deposits (Figure 1, J, K, and L). Moreover, the pericellular staining pattern of CLU in each section was strikingly similar to that observed for the amyloid protein. In AL tissue, extracellular CLU was apparent; additionally, subtle intracellular staining indicated possible cardiomyocyte expression of CLU, as has been reported by Krijnen et al (Figure 1L; Supplemental Figure S1 at http://ajp.amjpathol.org). This observation may be due to the direct cytotoxic nature of free LC proteins on cardiomyocytes as previously observed.16,17 Alternatively, CLU may be internalized through the cell surface receptor megalin. Immunohistochemical results were consistent in all nine amyloid tissues that were analyzed; the results obtained on additional tissues (not shown in Figure 1) are presented in Supplemental Figure S1 (http://ajp.amjpathol.org). Immunoblot analysis of protein extracts from the cardiac tissues confirmed the overabundance of the amyloid protein and presence of CLU in the SSA, ATTR, and AL samples compared with the control (Figure 1M). Lanes containing the control sample (total protein extracted from the nonamyloid cardiac tissue) were loaded at protein concentrations equal to that of the amyloid samples in the individual analysis. Control lanes showed no evidence of amyloid proteins; a low level of CLU was observed.
To confirm the presence of CLU in amyloid deposits composed of TTR or LC, immunogold electron microscopic analysis of the SSA, ATTR, and AL cardiac tissue specimens was performed. Representative results obtained in these studies are presented in Figure 2. Electron-dense, 10 nm gold particles corresponding to TTR-specific (Figure 2, C and E) or LC-specific (Figure 2G) and CLU-specific (Figure 2, D, F, and H) antibodies were clearly visible in areas where the fibrillar amyloid deposits were located (ie, adjacent to the striated cardiomyocytes). Immunogold staining was negative in the control (nonamyloid cardiac tissue) samples and in the amyloid samples where normal cellular and matrix components were visible; occasional, randomly occurring gold particles observed in these regions were attributed to background staining. Furthermore, the amyloid tissues that were treated with PIB or normal rabbit serum (nonimmune) showed little or no staining (see Supplemental Figure S2 at http://ajp.amjpathol.org). These findings indicate that CLU is specifically associated with the amyloid fibrils in cardiac amyloid deposits of patients with SSA, ATTR, or AL.
Figure 2.
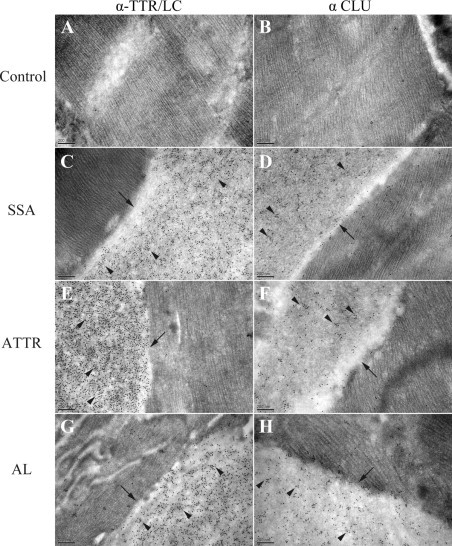
Electron microscopic images of immunogold staining for amyloid precursor proteins, TTR and LC, and CLU in control (A, B), SSA (C, D), ATTR (E, F), and AL (G, H) cardiac tissue specimens. Analysis was performed using secondary (goat anti-rabbit IgG) antibodies conjugated to 10 nm electron dense gold particles. No amyloid deposits were visible in the control sample; no immunogold labeling for TTR, LC, or CLU was noted. In SSA, ATTR, and AL specimens, amyloid deposits (arrows) were present and located adjacent to cardiomyocytes. Extensive gold particle immunolabeling (arrowheads) for TTR was evident in the SSA (C) and ATTR (E) samples; abundant staining for LC was noted in the AL sample (G). Immunogold staining for CLU demonstrated that the protein was present and localized to amyloid deposits in SSA (D), ATTR (F), and AL (H). Original magnification, ×80,000. Abbreviations as in Figure 1.
Serum Levels of Clusterin Are Reduced in Amyloidotic Cardiomyopathy
The mean ages of the study and control groups were not significantly different (Figure 3A). CLU concentrations in SSA, ATTR, and AL sera were measured, and the results were compared with levels in healthy, age-matched controls (Figure 3A). In addition, serum CLU amounts were examined in a cohort of ischemic and nonischemic patients with CMP who did not have amyloidosis. Quantification was accomplished using a monoclonal capture ELISA system developed for this purpose. The mean ± SEM value of serum CLU measured for the healthy control group was 0.659 ± 0.085 mg/ml. CLU levels were significantly lower in SSA (0.174 ± 0.018 mg/ml, P < 0.001). In the ATTR and AL groups, the concentrations were varied, and a subanalysis of patients with and without CMP in these groups was performed. Serum CLU amounts in the ATTR and AL cohorts with no CMP were 0.743 ± 0.038 mg/ml and 0.548 ± 0.062 mg/ml, respectively. Compared with age-matched healthy controls, the ATTR-CMP (0.386 ± 0.026 mg/ml) and AL-CMP (0.291 ± 0.055 mg/ml) groups were significantly lower (P < 0.001). Additionally, the ATTR-CMP and AL-CMP groups were significantly lower than either the ATTR no-CMP group or the AL no-CMP group (all P values <0.01). CLU levels were comparable in the AL and ATTR with no CMP groups, the nonamyloid patients with CMP, and the age-matched healthy controls. A comparison of groups with amyloidotic CMP (SSA, ATTR-CMP, AL-CMP) showed no significant difference.
Figure 3.

Serum concentrations of CLU in patients with, and without, amyloidotic CMP and healthy controls. A: Serum levels of CLU were quantified by enzyme-linked immunosorbent assay in healthy control subjects (white bar), patients with no CMP (AL and ATTR) or with CMP unrelated to amyloid (gray bars), and patients with amyloidotic CMP due to ATTR, AL, or SSA (black bars) ***P < 0.001. B: Correlation analysis of CLU concentrations with echocardiographic measurements of left ventricular mass (LVM) in all amyloidotic CMP groups (Pearson r = −0.2248, one-tailed P < 0.05, n = 59). C: Retrospective analysis of brain natriuretic peptide concentrations in matched serum samples where data were available. The 100 pg/ml threshold for clinical significance is indicated by a dashed line. Abbreviations as in Figure 1. Continuous variable are described as mean ± SE.
Serum Clusterin Correlates with Left Ventricular Mass
In patients with CMP, echocardiography was used to characterize left ventricular mass (LVM), which was correlated with serum CLU. The mean LVM measurements were 299.9 ± 20.99 g for SSA (n = 21), 231.5 ± 15.93 g for AL-CMP (n = 19), and 228.4 ± 16.32 g for ATTR-CMP (n = 19). The nonamyloid group with CMP showed the greatest LVM, at 331.3 ± 21.78 g (n = 15). LVM measurements were significantly greater in the SSA and nonamyloid with CMP groups, compared with the ATTR-CMP or AL-CMP cohorts (P < 0.05). A predicted correlation analysis of serum CLU with LVM was found to have a significant inverse relationship in patients with amyloidotic CMP (Figure 3); that is, greater LVM was associated with lower serum CLU concentration (Pearson r = −0.2248, one-tailed P < 0.05, n = 59). In the nonamyloid with CMP group, there was no significant correlation between CLU and LVM.
When available, serum BNP levels in the tested samples were retrospectively analyzed to confirm the presence of heart failure in patients with CMP. BNP concentrations ≥100 pg/ml were measured in all patients with amyloidotic CMP (SSA, ATTR-CMP, AL-CMP), as well as in patients with CMP unrelated to amyloidosis (Figure 3C). Mean BNP levels were highest in the AL-CMP group (1321 ± 468 pg/ml); mean concentrations were comparable for ATTR-CMP (381 ± 78 pg/ml) and SSA (373 ± 56 pg/ml). Median BNP values are also presented. In the patient groups with no CMP, BNP levels were within the normal range. Correlation analysis showed no significant association of BNP with CLU (Spearman r = −0.2476, two-tailed P > 0.05, n = 37).
Discussion
This study provides evidence that CLU is present in cardiac tissues derived from patients with SSA, ATTR, and AL amyloidosis. Our investigations demonstrated the specific association of CLU with cardiac amyloid fibrillar deposits in these three different forms of systemic amyloid disease. We also showed that increased levels of CLU occur in cardiac tissues from patients with TTR- and LC-associated forms of amyloidotic CMP. Furthermore, our analysis of CLU concentrations in amyloid patient and age-matched, control sera showed an association between amyloidotic CMP and decreased circulating levels of the protein.
It is possible that circulating CLU may be the source of the CLU incorporated into amyloid deposits in the heart; alternatively, local up-regulation of CLU in the heart may occur concomitantly with reduced production of CLU elsewhere. The association of CLU with TTR and LC amyloid deposits is not entirely organ-specific; for example, we have identified CLU in LC deposits of the kidney by immunohistochemistry (data not shown). Further investigation is necessary to determine how frequently this occurs and how it affects serum levels of CLU. Nonetheless, reduced serum concentrations of CLU appear to be more strongly associated with amyloidotic CMP than amyloidosis alone, and do not occur in other forms of cardiomyopathy.
CLU was the first extracellular chaperone to be identified, and extensive investigations over the past decade have shown that it has a chaperoning function similar to small heat shock proteins such as αB-crystallin.7,18–22 The binding of CLU to exposed hydrophobic regions of misfolded proteins has been demonstrated in vitro for various amyloidogenic precursor proteins. Yerbury et al22 demonstrated the ability of CLU to prevent amyloid formation with eight different amyloidogenic proteins through specific binding to prefibrillar amyloid structures; however, TTR and LC were not examined in these studies. In addition, high molecular weight aggregate formation of CLU chaperone-client complexes has been characterized in vitro, with an estimated CLU-to-client stoichiometry of 1:2.21
We note that the ability of CLU to prevent amyloid formation changes in a concentration dependent manner. CLU has been shown to promote fibrillogenesis under appropriate conditions.22 When the amyloidogenic substrate is present in a large molar excess, CLU appears to enhance fibrillogenesis and is a component of the insoluble aggregates.22 Considering this concentration-dependent biphasic effect, it has been posited that CLU can stabilize misfolded protein conformations; by maintaining the exposure of normally buried hydrophobic regions on the amyloidogenic protein, CLU may augment fibril formation.19 Thus, there is no way to predict a priori whether the CLU we have observed in cardiac amyloid deposits is present because of a failed response to prevent aggregation or as part of the pathophysiologic process. Similarly, we cannot as yet determine whether serum CLU concentrations are reduced in the process of developing amyloidotic CMP or whether patients with intrinsically low levels of circulating CLU are at increased risk for cardiac amyloid disease.
Our finding of significantly lower levels of circulating CLU associated with amyloid deposition in the heart across three disease states appears to reflect a unique difference in amyloidosis pathology, dependent on organ involvement. In particular, there is a clear inverse correlation between LVM and CLU. Although this was not assessed in the present study, prolonged and/or robust cardiac tissue remodeling specific to amyloidotic CMP may lead to sequestration of CLU from the circulation as dynamic structural changes take place in both the extracellular matrix and amyloid deposits. Similarly, CLU-bound amyloid may represent not only a failed attempt at removal of circulating amyloidogenic proteins, but also a stabilization effect of fragmented fibrils generated by protease activity during tissue remodeling when the microenvironment equilibrium of CLU:amyloid is decreased. The low serum CLU concentrations observed in SSA may be due to a combination of prolonged subclinical cardiac amyloidosis, significant cardiac remodeling, and an ongoing state of fibril fragmentation and CLU sequestration from circulation.
In summary, our studies suggest a potentially pathological association of CLU with SSA, ATTR, and AL types of amyloidotic CMP. We have demonstrated the presence of CLU in cardiac amyloid deposits composed of TTR and LC by immunohistochemical and immunogold techniques. In addition, serum concentrations of CLU determined by ELISA are significantly lower in patients with amyloidotic CMP, and we find an inverse relationship with LVM. Thus, CLU may be a novel biomarker for amyloidotic CMP. Moreover, further studies are needed to help determine whether serum concentrations of CLU are correlated to amyloid cardiomyopathy disease progression and whether this protein is a useful prognostic indicator.
Acknowledgments
We thank Dr. Saulius Girnius (Boston Medical Center), and Dr. Thomas G. Christensen, Dr. Gheorghe Doros, Dr. Daniel Remick, Jacqueline Bouchard, Bryan Belikoff, and Evan Chiswick (Boston University School of Medicine) for their contributions toward completion of this work.
Footnotes
Supported by National Institutes of Health grant R01AG031804 (L.H.C.) and the Walk for An Angel and Jamieson Funds for Amyloid Research; and by NIH grants HL079099, HL095891, and HL102631 (F.S.).
Financial Disclosure: M.J.G. and L.H.C. are listed in a patent application with the US Patent and Trademark Office as inventors of the use of clusterin in the diagnosis and treatment of amyloidotic cardiomyopathy and the ELISA-based method as described in this publication.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.11.015.
Supplemental material
Light microscopic images of autopsied cardiac tissue specimens from additional cases of senile systemic amyloidosis (SSA), familial TTR amyloidosis (ATTR), and Ig light chain (LC) amyloidosis (AL) stained with Congo Red (A, B, C) and immunohistochemically tested for transthyretin (TTR) (G, H, I), LC (J, K, L), and CLU (M, N, O). The presence of amyloid is demonstrated by an apple-green birefringence when Congo Red-treated sections are viewed under polarized light (D, E, F). In each case, identical tissue structures were observed for the different treatments by using 5-μm-thick serially sectioned samples. Original magnification, ×40.
Electron microscopic images of negative immunogold staining noted in cardiac tissue specimens from control and SSA, ATTR, and AL cases. Primary immunobuffer (PIB) (A, C, E, G) and normal rabbit serum (NRS) (B, D, F, G) treatments are shown. Arrows indicate amyloid fibrils in SSA, ATTR, and AL tissue deposits (C–H) and extracellular matrix components in the control tissue (A and B). Original magnification, ×80,000. Abbreviations as in Supplemental Figure 1.
References
- 1.Lavatelli F., Perlman D.H., Spencer B., Prokaeva T., McComb M.E., Théberge R., Connors L.H., Bellotti V., Seldin D.C., Merlini G., Skinner M., Costello C.E. Amyloidogenic and associated proteins in systemic amyloidosis proteome of adipose tissue. Mol Cell Proteomics. 2008;7:1570–1583. doi: 10.1074/mcp.M700545-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falk R.H., Dubrey S.W. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52:347–361. doi: 10.1016/j.pcad.2009.11.007. [Erratum appeared in Prog Cardiovasc Dis 2010, 52:445–447] [DOI] [PubMed] [Google Scholar]
- 3.Shah K.B., Inoue Y., Mehra M.R. Amyloidosis and the heart: a comprehensive review. Arch Intern Med. 2006;166:1805–1813. doi: 10.1001/archinte.166.17.1805. [DOI] [PubMed] [Google Scholar]
- 4.Sawyer D.B., Skinner M. Cardiac amyloidosis: shifting our impressions to hopeful. Curr Heart Fail Rep. 2006;3:64–71. doi: 10.1007/s11897-006-0004-z. [DOI] [PubMed] [Google Scholar]
- 5.Hassan W., Al-Sergani H., Mourad W., Tabbaa R. Amyloid heart disease: New frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J. 2005;32:178–184. [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey R.W., Dunker A.K., Brown C.J., Garner E.C., Griswold M.D. Clusterin, a binding protein with a molten globule-like region. Biochemistry. 2001;40:11828–11840. doi: 10.1021/bi010135x. [DOI] [PubMed] [Google Scholar]
- 7.Humphreys D.T., Carver J.A., Easterbrook-Smith S.B., Wilson M.R. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J Biol Chem. 1999;274:6875–6881. doi: 10.1074/jbc.274.11.6875. [DOI] [PubMed] [Google Scholar]
- 8.Nuutinen T., Suuronen T., Kauppinen A., Salminen A. Clusterin: a forgotten player in Alzheimer's disease. Brain Res Rev. 2009;61:89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Aronow B.J., Lund S.D., Brown T.L., Harmony J.A., Witte D.P. Apolipoprotein J expression at fluid-tissue interfaces: potential role in barrier cytoprotection. Proc Natl Acad Sci USA. 1993;90:725–729. doi: 10.1073/pnas.90.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg M.E., Silkensen J. Clusterin: physiologic and pathophysiologic considerations. Int J Biochem Cell Biol. 1995;27:633–645. doi: 10.1016/1357-2725(95)00027-m. [DOI] [PubMed] [Google Scholar]
- 11.Michel D., Chatelain G., North S., Brun G. Stress-induced transcription of the clusterin/apoJ gene. Biochem J. 1997;328:45–50. doi: 10.1042/bj3280045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loison F., Debure L., Nizard P., le Goff P., Michel D., le Dréan Y. Up-regulation of the clusterin gene after proteotoxic stress: implication of HSF1-HSF2 heterocomplexes. Biochem J. 2006;395:223–231. doi: 10.1042/BJ20051190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swertfeger D., Witte D., Stuart W., Rockman H., Harmony J. Apolipoprotein J/clusterin induction in myocarditis: a localized response gene to myocardial injury. Am J Pathol. 1996;148:1971–1983. [PMC free article] [PubMed] [Google Scholar]
- 14.Krijnen P.A.J., Cillessen S.A.G.M., Manoe R., Muller A., Visser C.A., Meijer C.J.L.M., Musters R.J.P., Hack C.E., Aarden L.A., Niessen H.W.M. Clusterin: a protective mediator for ischemic cardiomyocytes? Am J Physiol Heart Circ Physiol. 2005;289:H2193–H2202. doi: 10.1152/ajpheart.00355.2005. [DOI] [PubMed] [Google Scholar]
- 15.Devereux R.B., Alonso D.R., Lutas E.M., Gottlieb G.J., Campo E., Sachs I., Reichek N. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450–458. doi: 10.1016/0002-9149(86)90771-x. [DOI] [PubMed] [Google Scholar]
- 16.Brenner D.A., Jain M., Pimentel D.R., Wang B., Connors L.H., Skinner M., Apstein C.S., Liao R. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94:1008–1010. doi: 10.1161/01.RES.0000126569.75419.74. [DOI] [PubMed] [Google Scholar]
- 17.Shi J., Guan J., Jiang B., Brenner D.A., del Monte F., Ward J.E., Connors L.H., Sawyer D.B., Semigran M.J., Macgillivray T.E., Seldin D.C., Falk R., Liao R. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci USA. 2010;107:4188–4193. doi: 10.1073/pnas.0912263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson M.R., Easterbrook-Smith S.B. Clusterin is a secreted mammalian chaperone. Trends Biochem Sci. 2000;25:95–98. doi: 10.1016/s0968-0004(99)01534-0. [DOI] [PubMed] [Google Scholar]
- 19.Wilson M.R., Yerbury J.J., Poon S. Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol BioSyst. 2008;4:42–52. doi: 10.1039/b712728f. [DOI] [PubMed] [Google Scholar]
- 20.Wyatt A.R., Wilson M.R. Identification of human plasma proteins as major clients for the extracellular chaperone clusterin. J Biol Chem. 2010;285:3532–3539. doi: 10.1074/jbc.M109.079566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wyatt A.R., Yerbury J.J., Wilson M.R. Structural characterization of clusterin-chaperone client protein complexes. J Biol Chem. 2009;284:21920–21927. doi: 10.1074/jbc.M109.033688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yerbury J.J., Poon S., Meehan S., Thompson B., Kumita J.R., Dobson C.M., Wilson M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007;21:2312–2322. doi: 10.1096/fj.06-7986com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Light microscopic images of autopsied cardiac tissue specimens from additional cases of senile systemic amyloidosis (SSA), familial TTR amyloidosis (ATTR), and Ig light chain (LC) amyloidosis (AL) stained with Congo Red (A, B, C) and immunohistochemically tested for transthyretin (TTR) (G, H, I), LC (J, K, L), and CLU (M, N, O). The presence of amyloid is demonstrated by an apple-green birefringence when Congo Red-treated sections are viewed under polarized light (D, E, F). In each case, identical tissue structures were observed for the different treatments by using 5-μm-thick serially sectioned samples. Original magnification, ×40.
Electron microscopic images of negative immunogold staining noted in cardiac tissue specimens from control and SSA, ATTR, and AL cases. Primary immunobuffer (PIB) (A, C, E, G) and normal rabbit serum (NRS) (B, D, F, G) treatments are shown. Arrows indicate amyloid fibrils in SSA, ATTR, and AL tissue deposits (C–H) and extracellular matrix components in the control tissue (A and B). Original magnification, ×80,000. Abbreviations as in Supplemental Figure 1.