

Abstract
Molecular pathways associated with pathogenesis of sporadic papillary renal cell carcinoma (PRCC), the second most common form of kidney cancer, are poorly understood. We analyzed primary tumor specimens from 35 PRCC patients treated by nephrectomy via gene expression analysis and tissue microarrays constructed from an additional 57 paraffin-embedded PRCC samples via immunohistochemistry. Gene products were validated and further studied by Western blot analyses using primary PRCC tumor samples and established renal cell carcinoma cell lines, and potential associations with pathologic variables and survival in 27 patients with follow-up information were determined. We show that the expression of E2-EPF ubiquitin carrier protein, which targets the principal negative regulator of hypoxia-inducible factor (HIF), von Hippel-Lindau protein, for proteasome-dependent degradation, is markedly elevated in the majority of PRCC tumors exhibiting increased HIF1α expression, and is associated with poor prognosis. In addition, we identified multiple hypoxia-responsive elements within the E2-EPF promoter, and for the first time we demonstrated that E2-EPF is a hypoxia-inducible gene directly regulated via HIF1. These findings reveal deregulation of the oxygen-sensing pathway impinging on the positive feedback mechanism of HIF1-mediated regulation of E2-EPF in PRCC.
Renal cell carcinoma (RCC) accounts for 3% of all adult malignancies and its incidence continues to rise at an approximate rate of 2.5% per year.1 RCC is a heterogeneous disease consisting of various subtypes with diverse genetic and morphological features that can be segregated into clear cell (conventional), papillary (chromophil), chromophobe, collecting duct, and unclassified subtypes.2 Papillary RCC (PRCC) is the second most common subtype comprising 10% to 15% of kidney cancer with an estimated annual incidence of 3500 to 5000 cases in the United States.3 Although PRCCs are genetically heterogeneous with complex numerical chromosomal anomalies, PRCC can be morphologically classified into two subtypes: Type 1, which is twice as common as type 2, is characterized by the presence of small cuboidal cells covering thin papillae with a single line of small uniform nuclei and basophilic cytoplasm, whereas type 2 is characterized by the presence of large pseudostratified tumor cells with eosinophilic cytoplasm.4 In general, type 2 patients have poorer prognosis than patients with type 1 PRCC.5 However, the molecular basis underlying differential prognosis of PRCC is largely unknown.
The vast majority of RCC, including PRCC, is sporadic or nonhereditary, whereas approximately 4% of all renal cancers are attributed to familial RCC syndromes.6 Hereditary PRCC is an autosomal dominant disease characterized by multi-focal bilateral type 1 PRCC and is caused by “gain-of-function” mutations on MET, which encodes the hepatocyte growth factor receptor, c-Met. Moreover, MET mutations have also been associated with the vast majority of sporadic type 1 PRCC.7 Hereditary leiomyomatosis renal cell cancer is a rare cancer syndrome characterized by the development of cutaneous and uterine leiomyomas and low-penetrant type 2 PRCC.6,7 Hereditary leiomyomatosis renal cell cancer is caused by mutations of fumarate hydratase, which encodes a Krebs cycle enzyme that catalyzes the conversion of fumarate to malate.8
Biallelic inactivation of von Hippel-Lindau (VHL) tumor suppressor gene is associated with sporadic clear-cell RCC and hereditary VHL disease-associated clear-cell renal cell carcinoma (CCRCC).9 VHL is the substrate-conferring component of an E3 ubiquitin ligase that targets α subunit of hypoxia-inducible factor (HIF), the master transcriptional regulator of the ubiquitous oxygen-sensing pathway, for ubiquitin-mediated destruction.10 In the presence of oxygen, HIFα is hydroxylated on conserved prolines within the oxygen-dependent degradation domain by a family of prolyl-hydroxylases, which enables direct binding to VHL.11 Under hypoxia, HIFα remains unmodified, escapes VHL recognition, and subsequent ubiquitin-mediated destruction, and partners with HIFβ [also known as aryl hydrocarbon receptor nuclear translocator (ARNT)] to form an active heterodimeric transcription factor that engages hypoxia-responsive elements (HREs) in the promoters of numerous hypoxia-inducible genes to trigger various adaptive responses to reduced oxygen tension.12,13 Notably, several tyrosine kinase inhibitors (TKIs) are currently being used with promising results to treat patients with metastatic CCRCC since the loss of VHL leads to the stabilization of HIF and transactivation of HIF-induced receptor tyrosine kinases (RTKs), such as vascular endothelial growth factor receptor and platelet-derived growth factor receptor.14
Whereas the tumorigenesis of sporadic CCRCC is mainly associated with the loss of VHL function, the molecular pathways associated with the pathogenesis of sporadic PRCC are poorly understood. We show that the oxygen-sensing pathway in a significant fraction of sporadic PRCC is compromised, due at least in part to the overexpression of E2-EPF (also known as UBE2S) ubiquitin carrier protein. E2-EPF has been shown previously to target VHL for 26S proteasome-mediated degradation.15 In addition, we identified multiple HREs within E2-EPF promoter that enabled HIF1-mediated E2-EPF transactivation, thereby revealing a positive feedback mechanism of HIF1-mediated regulation of E2-EPF in PRCC. Consistent with this notion, elevated E2-EPF level was associated with increased HIF1α expression profile in primary PRCC samples and a trend associated with worse patient outcome, suggesting that E2EPF could serve as a potential bio/prognostic marker and exploited as a novel molecular target for the management of PRCC.
Materials and Methods
Patients and Specimens
Tissue samples were obtained under sterile conditions from 91 patients with primary papillary RCC from two participating centers (Van Andel Research Institute, Grands Rapids, MI; and Department of Urology of the University Medical Center of the Johannes Gutenberg University, Mainz, Germany). The diagnosis of PRCC was based on H&E sections and classified as PRCC, according to the criteria previously described.4 Subtyping of PRCC and re-evaluation of the diagnosis were performed from two pathologists in 65 tissue samples coming from Van Andel Research Institute, and in 26 samples collected from the University Medical Center of the Johannes Gutenberg University, by respective institute pathologists. After re-evaluation, all patients with non-PRCC diagnosis (Van Andel Research Institute, n = 8 and University Medical Center of the Johannes Gutenberg University, n = 1) were excluded from further analysis. Clinicopathologic features and information on oncologic follow-up were available from 27 PRCC patients presenting with PRCC (Table 1). The tumor size was based on the pathologic tumor size, according to the maximum tumor diameter from the surgical specimens. The tumor staging was revised according to the 2003 TNM classification system, and grading was based on the Fuhrman classification system. Institutional review board approval was obtained from the two participating institutions and informed consent was obtained from all participants.
Table 1.
Clinicopathologic Features and E2EPF Expression Profile in PRCC Patients with Oncologic Follow-Up
| Code | Institute | Method | Sex | Size | Subtype | T | N | MT | Grading | E2-EPF | HIF1α | Status | Survival |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2883 | MZ | TMA, WB | M | 30 | 1 | 1 | 0 | 0 | 3 | Low | Low | NED | 83,0 |
| 2581 | MZ | WB | F | 53 | 1 | 1 | 0 | 0 | 2 | Low | Low | NED | 120,0 |
| 2873 | MZ | TMA | M | 80 | 1 | 3 | 2 | Low | NA | NED | 85,0 | ||
| 2900 | MZ | TMA, WB | F | 120 | 1 | 2 | 0 | 2 | Low | Low | NED | 78,0 | |
| 2540 | MZ | TMA | M | 25 | 1 | 3 | 2 | High | NA | NED | 181,8 | ||
| 2559 | MZ | TMA, WB | F | 50 | 1 | 1 | 2 | Low | Low | NED | 120,0 | ||
| 2807 | MZ | TMA, WB | M | 60 | 1 | 1 | 0 | 0 | 1 | Low | Low | DOD | 34,0 |
| 2835 | MZ | TMA | M | 35 | 1 | 3 | 1 | 1 | 4 | High | NA | DOD | 5,0 |
| 76864 | VA | TMA | M | 35 | 1 | 1 | 0 | 0 | 2 | Low | NA | NED | 13,0 |
| 76886 | VA | TMA | F | 25 | 1 | 1 | 0 | 0 | 2 | Low | NA | NED | 4,0 |
| 2818 | MZ | TMA, WB | F | 105 | 2 | 2 | 0 | 2 | Low | Low | NED | 102,0 | |
| 2749 | MZ | TMA, WB | M | 90 | 2 | 2 | 0 | 2 | Low | High | NED | 42,0 | |
| 2659 | MZ | WB | M | 65 | 2 | 1 | 0 | 3 | High | High | DOD | 63,7 | |
| 2799 | MZ | TMA | M | 53 | 2 | 1 | 0 | 0 | 2 | High | NA | NED | 109,0 |
| 2667 | MZ | WB | M | 55 | 2 | 3 | 0 | 0 | 2 | High | Low | DOD | 101,6 |
| 2899 | MZ | TMA, WB | M | 45 | 2 | 3 | 3 | High | High | DOD | 24,6 | ||
| 2760 | MZ | WB | F | 90 | 2 | 2 | 0 | 0 | 2 | High | High | NED | 118,7 |
| 2701 | MZ | TMA, WB | F | 25 | 2 | 1 | 1 | High | High | NED | 120,0 | ||
| 2703 | MZ | TMA | M | 140 | 2 | 2 | 0 | 2 | High | NA | DOD | 26,0 | |
| 2855 | MZ | WB | F | 95 | 2 | 2 | 0 | 0 | 3 | High | Low | NED | 104,0 |
| 2674 | MZ | TMA | F | 70 | 2 | 1 | 0 | 2 | Low | NA | DOD | 6,0 | |
| 2858 | MZ | TMA | M | 37 | 2 | 3 | 1 | 2 | High | NA | DOD | 28,0 | |
| 2781 | MZ | TMA, WB | M | 40 | 2 | 3 | 2 | Low | Low | NED | 114,0 | ||
| 2554 | MZ | TMA | M | 55 | 2 | 2 | 3 | High | NA | NED | 108,0 | ||
| 2625 | MZ | TMA, WB | M | 76 | 2 | 2 | 0 | 2 | High | High | NED | 120,0 | |
| 2663 | MZ | TMA | M | 50 | 2 | 3 | 1 | 2 | High | NA | DOD | 5,0 | |
| 2752 | MZ | TMA | F | 50 | 2 | 1 | 0 | 9 | 3 | Low | NA | NA | NA |
DOD, death of disease; M, male; F, female; MT, metastasis; N, node; NA, not available; NED, no evidence of disease; MZ, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; VA, Van Andel Research Institute, Grand Rapids, Michigan; T, tumor stage; TMA, tissue microarray; WB, Western blot.
Antibodies
Monoclonal anti-HA antibody (12CA5) was obtained from Roche Molecular Biochemicals (Basel, Switzerland). Polyclonal rabbit anti-VHL antibody was obtained from Cell Signaling (Pickering, Ontario, Canada). Anti-CAIX and anti-HIF-2α antibodies were obtained from Novus Biologicals Inc. (Littleton, CO). Anti-HIF1α antibody was obtained from BD Biosciences (Mississauga, Ontario, Canada). Anti-Vinculin was obtained from Sigma-Aldrich (Oakville, Ontario, Canada). Anti-GLUT-1 was obtained from Abcam (Cambridge, MA). Anti-E2EPF was obtained from Abgent (San Diego, CA).
Tissue Microarrays
Tissue microarrays, consisting of representative 0.6 to 1.0 mm cores from 37 PRCC and 20 PRCC of various Fuhrman grades (grades 1 to 4) and stages (pT1 to pT3) from University Medical Center of the Johannes Gutenberg University and Van Andel Research Institute, respectively, were immunostained using anti-E2EPF and anti-GLUT-1 antibodies, as previously described,16 and were scored blind by two independent observers. Briefly, at least half of the tissue microarray (TMA) core was required to be present on the slide with at least 50% of the tissue present being tumor cells. Staining intensity was categorized as negative (score = 0), weak (score = 1), moderate (score = 2), or strong (score = 3). The percentage of positive cells at each stated intensity was also considered and segregated into 0% (score = 0), <10% (score = 1), 10% to 50% (score = 2), or >50% (score = 3). The final positivity rating was determined by combining the aforementioned intensity score with the percentage score in which 0 to 2 total score would indicate weak, 3 to 4 as moderate, and >4 as strong.
Cells
HEK293 embryonic kidney cells, 786-MOCK, 786-VHL, UMRC2, and UMRC2-VHL were previously described,17 and CAKI-2 cell line was obtained from American Type Culture Collection (Rockville, MD). Cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich) at 37°C in a humidified 5% CO2 atmosphere.
Hypoxia Treatment of Cells
Cells were maintained at 1% O2 for 24 Hours in a ThermoForma (Marietta, OH) hypoxia chamber (5% CO2, 10% H2, 85% N2).
Protein Extraction
Renal tumor tissue was pulverized with a mortar under liquid nitrogen and suspended on ice in lysis buffer (20 mmol/L HEPES, pH 7.7, 0.2 M NaCl, 1.5 mmol/L MgCl2, 0.4 mmol/L EDTA, 1% Triton X-100, 0.5 mmol/L DTT, 100 μg/ml leupeptin, 100 μg/ml aprotinin, 10 mmol/L benzamidine, 2 mmol/L phenylmethylsulphonyl fluoride, 20 mmol/L β-glycerophosphate, and 0.1 mmol/L sodium-orthovanadate). Protein extraction of tumor cells were then prepared in radioimmunoprecipitation assay lysis buffer (10 mmol/L Tris-HCl pH 8.0, 140 mmol/L NaCl, 1% Triton X-100, 1% Na-Deoxycholate, 0.1% SDS, and 0.025% NaN3) supplemented with protease and phosphatase inhibitors on ice.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and Western blotting were performed as described previously.17
Gene Expression Analysis
Gene expression profiles from 35 PRCC (22 type 1, and 13 type 2), 10 CCRCC tumor samples and 12 nondiseased kidney tissue samples from Van Andel Research Institute that were not included in the TMAs were analyzed using the Affymetrix (Santa Clara, CA) HG-U133 Plus 2.0 GeneChip platform as previously described,18 and deposited at the Gene Expression Omnibus (GDS1344). These kidney samples were obtained from the Cooperative Human Tissue Network and internal review board approval was obtained from the Van Andel Research Institute to study these samples.
Electrophoretic Mobility Shift Assay
E2-EPF oligos [5′-AAAACCAAGGAACGTGCGCGCTGA-3′ were labeled with 50 μCi of γ-(32P)-ATP, 10 × PNK buffer NEB, Pickering, Ontario, Canada], 1 mmol/L spermidine (Sigma), and polynucleotide kinase (NEB) followed by purification using Illustra ProbeQuant G-50 Micro column (GE Health Care, Baie d'Urfe, Quebec, Canada). The probe was annealed at 95°C for 5 minutes and cooled to room temperature. TNT T7 quick-coupled (Promega, Nepean, Ontario, Canada) in vitro translated HA-HIF1α, HA-HIF2α, pcDNA3, and ARNT were combined with 10 × binding buffer (40% glycerol, 50 mmol/L Tris pH 7.5, 100 mmol/L KCl, 10 mmol/L DTT, 2 mg/ml BSA, and 0.2% Triton x100), 300 ng of salmon testes DNA (Sigma), and 200,000 cpm of labeled probe to a total volume of 25 μl. For competition, 250 molar excess of unlabeled HRE probe or scramble probe were incubated with the reaction mixture. The reaction was incubated at room temperature for 15 minutes. Supershift was performed using 3 μl of anti-HA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) antibody and incubating the reaction for an additional 15 minutes. Complexes were resolved on a nondenaturing 5% polyacrylamide gel and visualized by autoradiography.
VHL Sequencing
All 3 exons and the promoter region of the VHL gene were sequenced, including at least 20 bases flanking each exon. Samples were amplified in three exonic fragments using the Invitrogen (Carlsbad, CA) High-Fidelity Platinum TaqPCR kit and intronic VHL PCR primers were designed to not to lie on any known intronic polymorphic position. Amplified products were sequenced using ThermoSequenase (GE Health Care), termination mixes, and Cy 5.0 labeled VHL gene-specific sequencing primers. Sequence products were electrophoresed on Siemens' Long-Read Tower sequencers and compared to normal VHL sequence (GenBank accession #AC_000046).
Methylation-Specific PCR
DNA samples were bisulfite-treated by initial denaturation in NaOH followed by incubation in sodium bisulfite, NaOH, and hydroquinone for 16 hours at 55°C. The DNA was then purified using the Wizard DNA Clean-Up kit (Promega). Bisulfite treatment converted all unmethylated cytosines to uracils, leaving only methylated cytosines in CpG dinucleotides unaltered. The bisulfite-treated DNA samples were then amplified in two separate PCR reactions, one using a primer set specific for the sequence of the bisulfite-treated methylated VHL promoter and one using a primer set for the bisulfite-treated unmethylated VHL promoter. The primers used were as previously described.19 Samples were amplified using the Qiagen (Mississauga, Ontario, Canada) Multiplex PCR Kit at an annealing temperature of 60°C to 62°C. Products were analyzed on 2% agarose gel stained with ethidium bromide. All tumor samples tested reacted with the “unmethylated” primer set due to the presence of at least some nontumor DNA. Only samples with a methylated VHL promoter yielded a band with the “methylated” primer set. Unmodified genomic DNA was not amplified under tested conditions.
siRNA-Mediated Knockdown
ON-TARGETplus SMARTpool siRNA targeted to HIF1α and UBE2S (Dharmacon, Lafayette, CO) and siGENOME RISC-Free Control siRNA (Dharmacon) were transfected into the indicated cell lines according to the manufacturer's instruction.
Statistical Analysis
Groups were compared using the unpaired test. Cancer-specific survival and progression-free survival were estimated using the Kaplan-Meier method. Log-rank test was used to compare survival between different groups. As this is an exploratory study and no adjustment for multiple testing was done, P values are only descriptive.
Results
Significant Fraction of PRCC Overexpresses HIF1α
Tumor hypoxia or overexpression of HIFα, in particular HIF2α, is a hallmark feature of CCRCC, which shows response to TKIs designed against HIF-induced signaling events, such as vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and epidermal growth factor receptor pathways.14,20 Patients with metastatic PRCC are often excluded from prospective clinical trials involving TKIs due to their general lack of response to these agents.21 However, some PRCCs do respond to small molecular inhibitors and also to Temsirolimus, an inhibitor of mTOR that regulates HIFα translation.20,22 The molecular explanation underlying the differential response of PRCC to TKIs is unknown. Thus, we investigated the protein expression levels of HIFα and a well-established HIF-target gene product, glucose transporter-1 (GLUT-1), in 15 PRCC samples and matched ipsilateral normal renal cortex. Remarkably, 6 of 15 specimens showed strong HIF1α expression and 8 of 15 showed marked elevation of overlapping GLUT-1 expression in comparison to the normal counterparts (Figure 1A). CAIX, another HIF-target protein, was likewise noticeably elevated in 6 of 8 PRCC samples (Supplementary Figure S1, see http://ajp.amjpathol.org). Intriguingly, HIF2α was not detected in PRCC samples (data not shown), and concordantly, expression array profiling revealed that HIF2α mRNA expression in patient PRCC tumors is substantially below normal level (Supplementary Figure S2, see http://ajp.amjpathol.org). Consistent with this notion, CAKI-2 cells, which were originally defined as CCRCC, but recently re-categorized morphologically and cytogenetically as the only available PRCC cell line (type 2; ATCC database),23 showed elevated levels of GLUT-1 and HIF1α with undetectable HIF2α expression (Figure 1, B and C). 786-MOCK (VHL−/−; HIF1α−/−) and VHL-reconstituted 786-VHL cells were used as internal controls for HIF2α expression. As expected, 786-MOCK cells showed an increased level of GLUT-1 and HIF2α in comparison to 786-VHL or HEK293 human embryonic kidney epithelial cell line that expresses endogenous wild-type VHL (Figure 1B). Unlike 786-O cells, CCRCC line UMRC2 (VHL−/−) expresses HIF1α. CAKI-2 cells expressed increased HIF1α levels similar to UMRC2 (VHL−/−) or UMRC2 reconstituted with wild-type VHL (UMRC2-VHL) maintained under hypoxia (Figure 1C). These results suggest that HIF1α and HIF1-mediated response are elevated in a significant fraction of PRCC.
Figure 1.
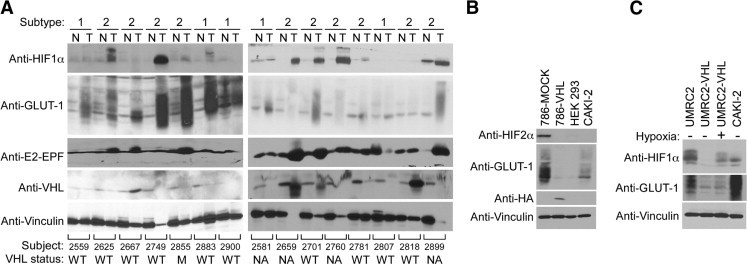
Significant fraction of papillary renal cell carcinoma (PRCC) show increased hypoxic profile. A: Equal amounts of total cell lysates generated from type 1 or type 2 tumor tissue (T) and matching normal tissue (N) from surgical specimens of 15 patients with PRCC were immunoblotted with the indicated antibodies. B: Equal amounts of the total cell lysates generated from the indicated cell lines were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the indicated antibodies. C: Equal amounts of the total cell lysates generated from CAKI-2, UMRC2, and UMRC2-VHL maintained in normoxia (21% O2: −) or hypoxia (1% O2: +) for 24 hours were resolved on SDS-PAGE and immunblotted with the indicated antibodies. VHL, von Hippel-Lindau.
VHL Mutations Are Rare in PRCC
Loss of VHL leads to the stabilization of the otherwise oxygen-labile HIFα subunits.10 VHL locus is on chromosome 3p25, and loss-of-heterozygosity analysis have demonstrated that approximately 14% to 50% of PRCC have 3p alterations.24 We asked whether the upregulation of HIF1α observed in PRCC is attributable to a loss or mutation in VHL. Direct genomic VHL sequencing from 11 PRCC samples did not detect any VHL mutation with the exception of c341G > C change in the first codon of exon 2 (GGT > GCT) resulting in Gly114Ala amino acid substitution in a single PRCC specimen (patient code 2855) (Supplementary Figure S3, see http://ajp.amjpathol.org). To our knowledge, this exact amino acid change has not been reported, but Gly114toSer, Arg, and Cys have been documented in VHL patients.25 However, the status of these remaining VHL allele was uninformative and therefore the functional activity of VHL in this PRCC sample remains unresolved and potentially normal. None of the samples tested showed VHL promoter methylation, which accounts for VHL silencing in approximately 20% to 25% of sporadic CCRCC.26 These results suggest that the increased expression of HIF1α observed in PRCC is not attributable to a loss or loss-of-function mutation of VHL.
E2-EPF Ubiquitin Carrier Protein Is Overexpressed in PRCC
We asked whether other major regulators of HIF were deregulated in PRCC, and observed that E2-EPF protein level was higher in 7 of 15 PRCC samples examined compared to the matched normal renal tissue (Figure 1A). Consistent with this finding, gene expression profiles from 12 nondiseased kidney samples and 35 PRCC tumor samples (22 type 1 and 13 type 2) using the Affymetrix HG-U133 Plus 2.0 GeneChip platform determined that E2-EPF mRNA was significantly elevated in PRCC, in particular type 2 (Figure 2A). E2-EPF has been shown to target VHL for proteasomal degradation.27 In keeping with this notion, most of the PRCC samples with high E2-EPF status were associated with lower endogenous VHL levels and increased HIF1α or GLUT-1 levels (Figure 1A). These results suggest that the overexpression of E2-EPF in PRCC promotes HIF1α induction via downregulation of VHL, the principal negative regulator of HIFα.
Figure 2.
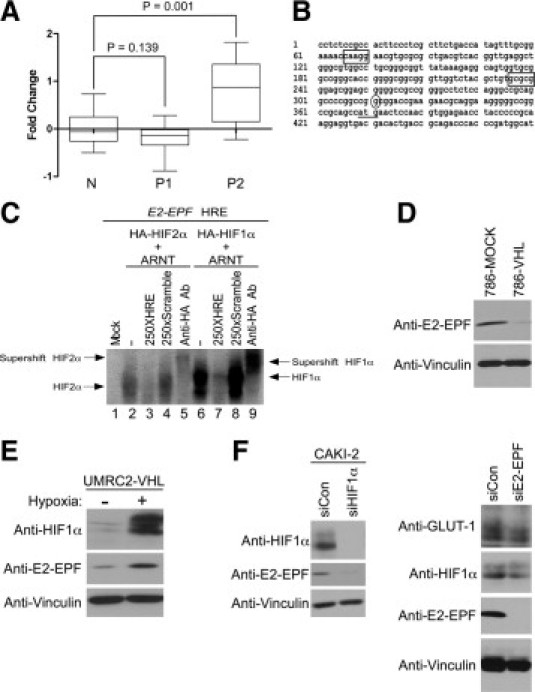
E2-EPF is a HIF-responsive gene that contains multiple HREs. A: Relative comparison of E2-EPF mRNA levels are shown in normal kidney cortex (N, n = 12), papillary renal cell carcinoma (PRCC) type 1 (P1, n = 22) and PRCC type 2 (P2, n = 13) using data from the Gene Expression Omnibus (GDS406, GDS495, and GSE2489. B: HRE (box), transcription start site (oval) and translation start site (solid line) are indicated on the human E2-EPF (UBE2S) promoter sequence. C: Binding profiles of in vitro translated HIF1 (HA-HIF1α/ARNT) and HIF2 (HA-HIF2α/ARNT) to 32P-labeled E2EPF HRE probes were determined via electrophoretic mobility shift assay (see Materials and Methods). D: Equal amounts of total cell lysates generated from 786-MOCK and 786-VHL cells were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the indicated antibodies. E: UMRC2-VHL cells were maintained in normoxia (21% O2: −) or hypoxia (1% O2: +) for 24 hours, lysed and immunoblotted with the indicated antibodies. F: Equal amounts of total cell lysates generated from CAKI-2 cells treated with HIF1α-specific siRNA (siHIF1α) or nontargeting scrambled siRNA (siCon) were resolved on SDS-PAGE and immunoblotted with the indicated antibodies.
E2-EPF Is Regulated via HIF1 and May Be a Prognostic Indicator in PRCC
We identified two conserved HREs within E2-EPF (also known as UBE2S) promoter that can bind in vitro translated HIF1 (HIF1α + ARNT) or HIF2 (HIF2α + ARNT) (Figure 2, B and C), which suggest that E2-EPF is a novel HIF-responsive gene. Consistent with this notion, 786-MOCK (high HIF2α) (Figure 1B) cells expressed higher E2-EPF levels than 786-VHL (low HIF2α) (Figure 1B) counterparts (Figure 2D). UMRC2-VHL cells maintained under hypoxia resulted in increased HIF1α level concomitant with increased E2-EPF level (Figure 2E). To directly assess the role of HIF1α in the regulation of E2-EPF, we attenuated the endogenous HIF1α level via siRNA in PRCC cell line CAKI-2 and observed marked reduction in E2-EPF expression (Figure 2F, left panel). Furthermore, siRNA-mediated knockdown of endogenous E2-EPF reduced the levels of HIF1α and GLUT1 (Figure 2F, right panel). These results demonstrate for the first time a direct HIF1-dependent regulation of E2-EPF.
We further investigated this notion using TMAs consisting of 57 PRCC tumor samples. TMAs were immunostained for E2-EPF and GLUT-1 as a reliable indicator of HIF activity, instead of direct anti-HIF1α immunostaining, which were generally of lower quality and consistency among the quadruplicate samplings. Strikingly, 26% of PRCC samples showed strong E2-EPF positivity, whereas 56% showed moderate E2-EPF staining (Figure 3A). In addition, significant fraction of the samples with either moderate or strong E2-EPF staining was associated with strong GLUT-1 positivity (Figure 3A). Similarly, a significant fraction of the samples showing moderate (42%) or strong (45%) GLUT-1 staining was associated with moderate and strong E2-EPF positivity (Figure 3A). Notably, the elevated GLUT-1 and E2EPF expression profiles were present in both type 1 and type 2 PRCC (Figure 3B).
Figure 3.
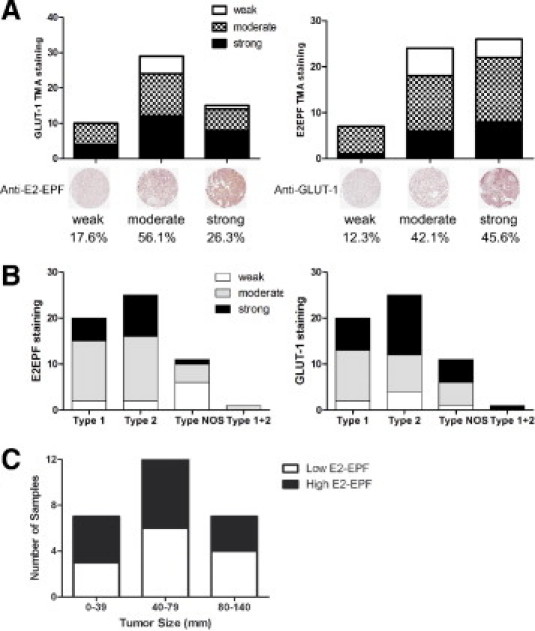
E2-EPF expression is associated with GLUT-1 expression. A: Tissue microarrays (TMAs) consisting of 57 PRCC samples in quadruplicate were immunostained with anti-E2-EPF (right) and anti-GLUT-1 (left) antibodies and scored blind by two independent observers. Tumor cores meeting the quality standard criteria (see Materials and Methods) were considered for analysis. Representative papillary renal cell carcinoma (PRCC) cores stained with anti-E2-EPF and anti-GLUT-1 antibodies showing the different staining intensities (weak, moderate, and strong) are shown below the graphs. B: Intensities in GLUT-1 (right) and E2EPF (left) expression were analyzed in type 1, type 2, and type not otherwise specified (NOS: type 1 or 2), and mixed type 1 + 2 PRCC groups. C: E2-EPF protein expression levels in 27 patient samples with available clinical follow-up information (see Table 1) as determined via TMA immunohistochemisty and/or Western blot analysis were plotted against tumor size.
Clinicopathologic features and oncological follow-up could be obtained from 27 of 57 patients studied (Table 1). From this collection of samples, E2-EPF expression was observed to not be associated with tumor size (Figure 3C). Furthermore, although a statistical significance could not be reached, mainly due to the limited sample size, patients with high tumor-specific E2-EPF expression revealed via immunohistochemistry on TMAs and/or Western blot analysis showed a trend toward increased cancer-related mortality and disease progression (Figure 4).
Figure 4.
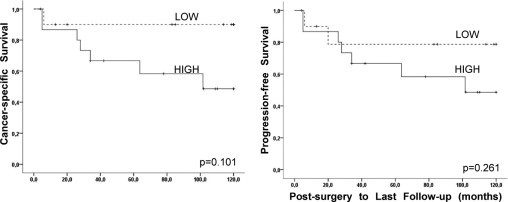
E2EPF overexpression shows a trend toward increased cancer progression. Kaplan-Meier analysis was performed in 27 patients with available clinical follow-up information (see Table 1) presenting with papillary renal cell carcinoma (PRCC) where E2-EPF protein expression was determined to be either high (n = 15) or low (n = 12) via TMA immunohistochemisty and/or Western blot analysis. Differences in cancer-specific (left) and progression-free (right) survival were calculated using the log-rank test.
Discussion
We show that a significant fraction of sporadic PRCC tumors exhibit a classic hypoxic signature with the overexpression of HIF1α and HIF-target gene products. Intriguingly, unlike CCRCC, HIF2α mRNA expression was suppressed and protein level undetectable in PRCC. The molecular mechanism underlying HIF2α suppression and the associated oncological significance in PRCC are unknown. A reduced protein level of VHL was observed in PRCC samples exhibiting strong HIF1α expression. Because VHL targets HIF1α for ubiquitin-mediated destruction, the attenuated VHL expression in PRCC likely provides a molecular explanation underlying HIF1α overexpression in PRCC. However, VHL mutation or epigenetic promoter methylation was determined to be an uncommon event in PRCC.9 Instead, we show for the first time an inordinately elevated expression of E2-EPF, an ubiquitin carrier protein that targets VHL for proteasome-dependent degradation,15 whose expression level was associated with decreased VHL and increased HIF1α levels in PRCC. In addition, we identified multiple HREs within E2-EPF promoter that were capable of recruiting HIF1, as well as HIF2, and showed that E2-EPF transcription is dependent on HIF1. These results identify E2-EPF as a novel HIF-target gene that is governed via HIF-dependent positive feedback mechanism. Consistent with tumor hypoxia being associated with increased disease aggressiveness and poor prognosis, high E2-EPF profile was associated, albeit in a small available number of PRCC patients with follow-up, with increased cancer-related mortality.
PRCC is characterized by complex cytogenetic alterations. For example, trisomies of chromosomes 7, 12, 16, 17, and 20 are the most frequently noted karyotype aberrations,27,28 whereas loss of chromosomes Y, 3p, 9p, 14q, and 19p and gain of 1q and 8q have also been reported.18,24,28 Gains of chromosome 7 and 17 are associated with type 1, whereas loss of 9p is associated with type 2 PRCC.7 Consistent with the established causal relationship between MET mutations with HPRCC (type 1 PRCC) autosomal dominant cancer predisposition syndrome,29 our regional gene expression profile analysis showed significant upregulation of MET proto-oncogene (located on chromosome 7q31) in sporadic PRCC type 1 tumors (Figure 5A; Supplementary Figure S2, see http://ajp.amjpathol.org). Remarkably, chromosomes that frequently undergo cytogenetic alterations in sporadic PRCC contained the genetic loci of several genes involved in the oxygen-sensing pathway, such as PHD2/3, VHL, and E2-EPF (Figure 5, A and B). These results suggest that the deregulation of the oxygen-sensing pathway may have pathologic roles in sporadic PRCC.
Figure 5.
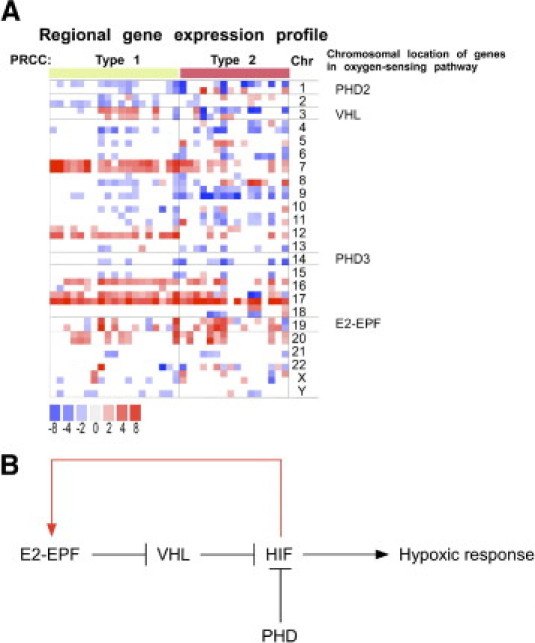
The oxygen-sensing pathway plays a role in papillary renal cell carcinoma (PRCC) pathogenesis. A: Regional gene expression profiles derived from type 1 (P1, n = 22) and type 2 (P2; n = 13) PRCC samples were compared against gene expression profiles derived from nondiseased kidney tissue samples (n = 12). The chromosomal location of the indicated HIF pathway-related genes are noted next to the chromosome number. Fold changes in comparative gene expression are represented as intensities of red (+ change) and blue (− change). Only the most significant data are displayed (P < 0.005). B: A model of oxygen-sensing pathway in PRCC. See text for details. VHL, von Hippel-Lindau.
Although it is unclear whether the upregulation of E2-EPF was due to a yet-defined (cyto)genetic alteration or/and to the increased hypoxic content of PRCC tumors that have led to HIF1-mediated transactivation of E2-EPF, the present findings implicate deregulation of the oxygen-sensing pathway in PRCC, and provide a compelling argument that a select group of PRCC patients exhibiting strong tumor-specific E2-EPF or hypoxic profile may represent responders to small molecule inhibitors designed to antagonize the HIF-signaling pathway.
Footnotes
Supported by funds from the Kidney Foundation of Canada and the Canadian Cancer Society (18460). F.C.R. is a recipient of the German Research Foundation (DFG, Ro 3750/1-1). M.O. is a Canada Research Chair in Molecular Oncology.
Supplemental material for this article can be found on http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.10.033.
Supplementary data
References
- 1.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Storkel S., Eble J.N., Adlakha K., Amin M., Blute M.L., Bostwick D.G., Darson M., Delahunt B., Iczkowski K. Classification of renal cell carcinoma: workgroup No. 1: Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC) Cancer. 1997;80:987–989. doi: 10.1002/(sici)1097-0142(19970901)80:5<987::aid-cncr24>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 3.Reuter V.E. The pathology of renal epithelial neoplasms. Semin Oncol. 2006;33:534–543. doi: 10.1053/j.seminoncol.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Delahunt B., Eble J.N. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10:537–544. [PubMed] [Google Scholar]
- 5.Klatte T., Pantuck A.J., Said J.W., Seligson D.B., Rao N.P., LaRochelle J.C., Shuch B., Zisman A., Kabbinavar F.F., Belldegrun A.S. Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma. Clin Cancer Res. 2009;15:1162–1169. doi: 10.1158/1078-0432.CCR-08-1229. [DOI] [PubMed] [Google Scholar]
- 6.Rosner I., Bratslavsky G., Pinto P.A., Linehan W.M. The clinical implications of the genetics of renal cell carcinoma. Urol Oncol. 2009;27:131–136. doi: 10.1016/j.urolonc.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng L., Zhang S., MacLennan G.T., Lopez-Beltran A., Montironi R. Molecular and cytogenetic insights into the pathogenesis, classification, differential diagnosis, and prognosis of renal epithelial neoplasms. Hum Pathol. 2009;40:10–29. doi: 10.1016/j.humpath.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Koivune P., Hirsila M., Remes A.M., Hassinen I.E., Kivirikko K.I., Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282:4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- 9.Baldewijns M.M., van Vlodrop I.J., Vermeulen P.B., Soetekouw P.M., van Engeland M., de Bruine A.P. VHL and HIF signalling in renal cell carcinogenesis. J Pathol. 2010;221:125–138. doi: 10.1002/path.2689. [DOI] [PubMed] [Google Scholar]
- 10.Ohh M., Park C.W., Ivan M., Hoffman M.A., Kim T.Y., Huang L.E., Pavletich N., Chau V., Kaelin W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 11.Jaakkola P., Mole D.R., Tian Y.M., Wilson M.I., Gielbert J., Gaskell S.J., Kriegsheim A., Hebestreit H.F., Mukherji M., Schofield C.J., Maxwell P.H., Pugh C.W., Ratcliffe P.J. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 12.Kaelin W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 13.Roberts A.M., Ohh M. Beyond the hypoxia-inducible factor-centric tumour suppressor model of von Hippel-Lindau. Curr Opin Oncol. 2008;20:83–89. doi: 10.1097/CCO.0b013e3282f310de. [DOI] [PubMed] [Google Scholar]
- 14.Rini B.I., Campbell S.C., Escudier B. Renal cell carcinoma. Lancet. 2009;373:1119–1132. doi: 10.1016/S0140-6736(09)60229-4. [DOI] [PubMed] [Google Scholar]
- 15.Jung C.R., Hwang K.S., Yoo J., Cho W.K., Kim J.M., Kim W.H., Im D.S. E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat Med. 2006;12:809–816. doi: 10.1038/nm1440. [DOI] [PubMed] [Google Scholar]
- 16.Evans A.J., Russell R.C., Roche O., Burry T.N., Fish J.E., Chow V.W., Kim W.Y., Saravanan A., Maynard M.A., Gervais M.L., Sufan R.I., Roberts A.M., Wilson L.A., Betten M., Vandewalle C., Berx G., Marsden P.A., Irwin M.S., Teh B.T., Jewett M.A., Ohh M. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–169. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stickle N.H., Cheng L.S., Watson I.R., Alon N., Malkin D., Irwin M.S., Ohh M. Expression of p53 in renal carcinoma cells is independent of pVHL. Mutat Res. 2005;578:23–32. doi: 10.1016/j.mrfmmm.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 18.Yang X.J., Tan M.H., Kim H.L., Ditlev J.A., Betten M.W., Png C.E., Kort E.J., Futami K., Furge K.A., Takahashi M., Kanayama H.O., Tan P.H., Teh B.S., Luan C., Wang K., Pins M., Tretiakova M., Anema J., Kahnoski R., Nicol T., Stadler W., Vogelzang N.G., Amato R., Seligson D., Figlin R., Belldegrun A., Rogers C.G., Teh B.T. A molecular classification of papillary renal cell carcinoma. Cancer Res. 2005;65:5628–5637. doi: 10.1158/0008-5472.CAN-05-0533. [DOI] [PubMed] [Google Scholar]
- 19.Kuroki T., Trapasso F., Yendamuri S., Matsuyama A., Alder H., Mori M., Croce C.M. Promoter hypermethylation of RASSF1A in esophageal squamous cell carcinoma. Clin Cancer Res. 2003;9:1441–1445. [PubMed] [Google Scholar]
- 20.Choueiri T.K., Plantade A., Elson P., Negrier S., Ravaud A., Oudard S., Zhou M., Rini B.I., Bukowski R.M., Escudier B. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol. 2008;26:127–131. doi: 10.1200/JCO.2007.13.3223. [DOI] [PubMed] [Google Scholar]
- 21.Motzer R.J., Bacik J., Mariani T., Russo P., Mazumdar M., Reuter V. Treatment outcome and survival associated with metastatic renal cell carcinoma of non-clear-cell histology. J Clin Oncol. 2002;20:2376–2381. doi: 10.1200/JCO.2002.11.123. [DOI] [PubMed] [Google Scholar]
- 22.Atkins M.B., Hidalgo M., Stadler W.M., Logan T.F., Dutcher J.P., Hudes G.R., Park Y., Liou S.H., Marshall B., Boni J.P., Dukart G., Sherman M.L. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22:909–918. doi: 10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 23.Furge K.A., Chen J., Koeman J., Swiatek P., Dykema K., Lucin K., Kahnoski R., Yang X.J., Teh B.T. Detection of DNA copy number changes and oncogenic signaling abnormalities from gene expression data reveals MYC activation in high-grade papillary renal cell carcinoma. Cancer Res. 2007;67:3171–3176. doi: 10.1158/0008-5472.CAN-06-4571. [DOI] [PubMed] [Google Scholar]
- 24.Sanders M.E., Mick R., Tomaszewski J.E., Barr F.G. Unique patterns of allelic imbalance distinguish type 1 from type 2 sporadic papillary renal cell carcinoma. Am J Pathol. 2002;161:997–1005. doi: 10.1016/S0002-9440(10)64260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patocs A., Gergics P., Balogh K., Toth M., Fazakas F., Liko I., Racz K. Ser80Ile mutation and a concurrent Pro25Leu variant of the VHL gene in an extended Hungarian von Hippel-Lindau family. BMC Med Genet. 2008;9:29. doi: 10.1186/1471-2350-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim W.Y., Kaelin W.G. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 27.Lim J.H., Jung C.R., Lee C.H., Im D.S. Egr-1 and serum response factor are involved in growth factors- and serum-mediated induction of E2-EPF UCP expression that regulates the VHL-HIF pathway. J Cell Biochem. 2008;105:1117–1127. doi: 10.1002/jcb.21914. [DOI] [PubMed] [Google Scholar]
- 28.Gunawan B., von Heydebreck A., Fritsch T., Huber W., Ringert R.H., Jakse G., Fuzesi L. Cytogenetic and morphologic typing of 58 papillary renal cell carcinomas: evidence for a cytogenetic evolution of type 2 from type 1 tumors. Cancer Res. 2003;63:6200–6205. [PubMed] [Google Scholar]
- 29.Linehan W.M., Vasselli J., Srinivasan R., Walther M.M., Merino M., Choyke P., Vocke C., Schmidt L., Isaacs J.S., Glenn G., Toro J., Zbar B., Bottaro D., Neckers L. Genetic basis of cancer of the kidney: disease-specific approaches to therapy. Clin Cancer Res. 2004;10:6282S–6289S. doi: 10.1158/1078-0432.CCR-050013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.