

Abstract
Target-based drug discovery for Alzheimer's disease (AD) centered on modulation of the amyloid ß peptide has met with limited success. Therefore, recent efforts have focused on targeting the microtubule-associated protein tau. Tau pathologically accumulates in more than 15 neurodegenerative diseases and is most closely linked with post-symptomatic progression in AD. We endeavored to identify compounds that decrease tau stability rather than prevent its aggregation. An extract from Myrica cerifera (bayberry/southern wax myrtle) potently reduced both endogenous and over-expressed tau protein levels in cells and murine brain slices. The bayberry flavonoids myricetin and myricitrin were confirmed to contribute to this potency but a diarylheptanoid, myricanol, was the most effective anti-tau component in the extract with potency approaching the best targeted lead therapies. (+)-aR,11S-myricanol, isolated from M. cerifera and reported here for the first time as the naturally occurring aglycone, was significantly more potent than commercially available (±)-myricanol. Myricanol may represent a novel scaffold for drug development efforts targeting tau turnover in AD.
Targeting the microtubule-associated protein tau (MAPT) that pathologically accumulates in AD has emerged as perhaps the most effective strategy for treating post-symptomatic AD.1-8 Tau normally functions to stabilize microtubules in neurons, but in disease it pathologically aggregates in AD and other tauopathies.9-14 Tau forms neurofibrillary tangles (NFTs) that are intracellular aggregates composed of paired helical filaments (PHFs) of the tau protein.15,16 While aggregation of tau into tangles is indeed the strongest post-mortem correlate of disease severity, tau intermediates may be more neurotoxic than higher order aggregates. It was long thought that the NFTs impair synaptic plasticity and cognitive function,17 however, it is now clear that soluble tau intermediates, rather than pathologically visible aggregates, are responsible for the cognitive dysfunction in AD and other tauopathies.6-8,18 Thus, while a number of efforts are underway to prevent tau aggregation,19,20 approaches to enhance tau turnover are less advanced.21 Our goal is to fill this void in the AD drug development pipeline. While reducing all tau in cells would likely lead to toxicity, we speculated that removing some tau from a symptomatic AD brain may be an effective therapeutic strategy. Moreover, normal, functional tau is less affected by clearance pathways in the cell than aberrant tau, suggesting that modulators of tau clearance may selectively target abnormal tau.21,22
Natural products chemistry has produced several effective AD therapy leads, including the amyloid aggregation inhibitor curcumin, isolated from tumeric,23 the microtubule stabilizer, paclitaxel from the Pacific yew tree,24 and the Streptomyces-derived Hsp90 inhibitor, geldanaymcin.21 By analyzing extracts from natural sources with a modern cell-based screening platform that measures tau levels25-27 we found that bayberry (Myrica cerifera) extract potently enhanced tau clearance. Fractionation of this extract revealed (+)-aR,11S-myricanol, a novel AD scaffold that could be used to develop future compounds to reduce tau burden.
Results and Discussion
Cell-based Natural Product Screen Identifies Bayberry Extract as Potent Tau Reducer
A small natural product library was screened for anti-tau efficacy. M17 neuroblastoma cells were treated for 24-hours and an In-Cell Western (ICW) screen was performed to assess tau and GAPDH levels. This cell line is derived from striatal neurons and has high levels of endogenous tau. The results from 10 random extracts, in triplicate, are shown in Figure 1A. Wells lacking a GAPDH signal suggest toxicity. The entire assay was performed in a total of 10 plates. Analysis of this screen revealed that extract from M. cerifera (bayberry extract, Herb Pharm) was the most potent tau reducer lacking cellular toxicity (Figure 1B).
Figure 1.
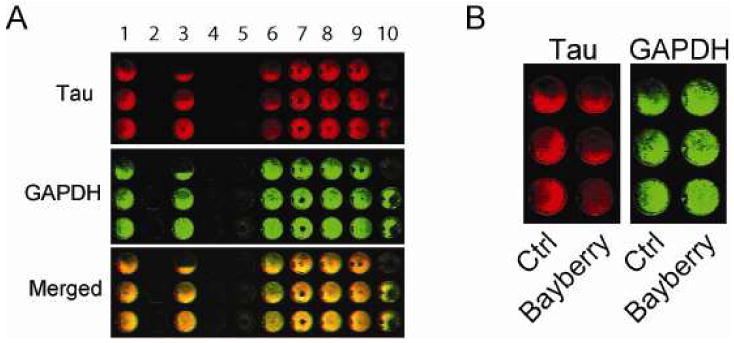
M17 cells were plated in 96-well plates and treated with 190 extracts (10%/vol, Herb Pharm) for 24 h. An ICW was used to identify extracts capable of reducing tau levels relative to GAPDH. Example of a result with 10 extracts is shown (A). Bayberry extract was identified as most potent tau reducer with no toxicity (B). Toxicity was determined by calculating GAPDH levels relative to vehicle.
Anti-tau efficacy using immunocytochemistry was then confirmed in a second neural cell line, H4 neuroglioma cells. H4 cells were chosen for secondary analysis not only because they have high levels of endogenous tau similar to M17 cells, but they are also more adherent compared to M17 cells, making them very well-suited for fluorescent immunocytochemistry (Figure 2). H4 cells were treated with indicated amounts of bayberry extract. After 24 hours, immunofluorescent staining showed a dose-dependent decrease in tau levels (Figure 2). No toxicity was observed with LDH release analyses (data not shown).
Figure 2.
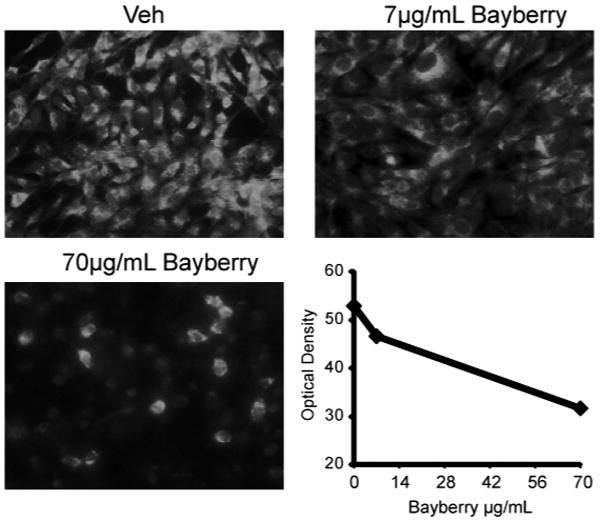
H4 neuroglioma cells in 4-well chamber slides were treated with indicated amounts of bayberry extract from Herb Pharm (mixed suspension with ethanol, glycerol and water) for 24 hours, fixed and tau immunoreactivity was assessed using standard (as opposed to near-infrared) immunofluorescent microscopy. Quantitation was derived using optical density with ImageJ software.
Bayberry Aqueous Extract Reduces Tau Levels in a Cell Model that over-expresses Tau as Well as a Cell Line Derived from Hippocampal Neurons
Raw bayberry root-bark powder was extracted in pure water resulting in a solution that contained 7 mg extract/mL solution. A HeLa cell line stably transfected with tau (HeLa-C3) and a human IMR32 cell line derived from hippocampal neurons were then treated with indicated doses (Figure 3A and 3B, respectively) for four hours to assess the effects of the aqueous extract on both over-expressed and endogenous tau levels. The hippocampus is the region first affected by tau deposition in Alzheimer's disease. Indeed, tau was reduced in both cell lines by ∼60% when treated with 70 μg/mL aqueous extract (Figure 3C). Thus bayberry extract was capable of reducing tau levels in three brain derived cell lines as well as a cell line over-expressing tau.
Figure 3.
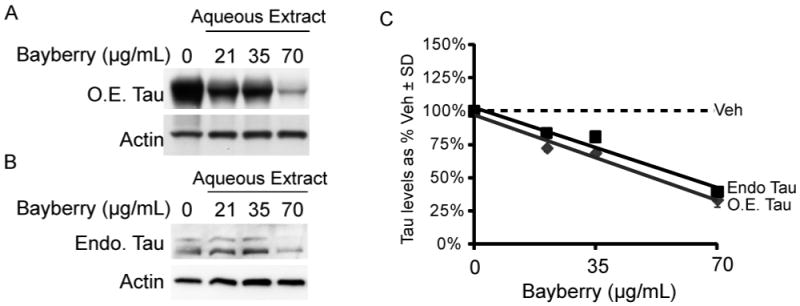
HeLa cells stably overexpressing tau (HeLa-C3; A) and IMR32 cells with endogenous tau levels (B) were treated for four hours with an aqueous bayberry extract at 0 – 70 μg/mL. Quantification of the western blots after actin normalization display a downward trend in tau levels. Tau levels are shown as a percentage of vehicle-treated cells ± SD (C).
Bayberry Aqueous Extract Significantly Reduces Tau Levels in Fresh Forebrain Tissue Slices Derived from Mice
Acute murine brain slices from wild type mice were treated with the aqueous preparation at six doses (9 – 140 μg/mL, Figure 4A) for four hours as previously described.28 Total and phospho-tau levels were reduced by as much as 40%, reaching statistical significance at doses >70μg/mL by volume (Figure 4B).
Figure 4.
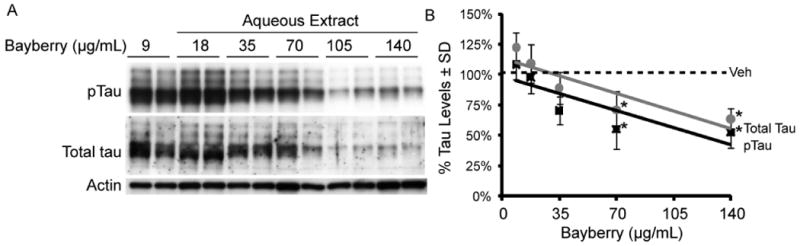
A representative western blot (A) is used to demonstrate the dose-dependent reductions in tau levels from whole brain murine slice cultures obtained from four wild type mice (2 males, 2 females) at 5 months of age. Treatments were performed in duplicate at indicated μg/ml doses for each mouse. Each point in the graph to the right (B) represents the average tau reductions (n=8 slices) for all four mice at the given doses relative to the average vehicle ± SD. *p< 0.05 by student's t test.
Fractionation of Bayberry Reveals Both Novel and Previously Described Tau-reducing Compounds
Ethanol extraction and fractionation of M. cerifera was carried out to identify the constituents29-32 of bayberry that were primary contributors to the observed effects on tau levels. HeLa-C3 cells were used to confirm that crude EtOH extraction did not reduce the potency of the bayberry extract. Cells were treated with indicated concentrations of the EtOH extract of bayberry for four hours and tau levels were assessed by Western blot. Tau levels were dose dependently reduced at all concentrations showing similar potency as the aqueous extract (Figure 5A). The EtOH extract was subjected to acid hydrolysis to enrich the aglycone component of the extract, then fractionated via MPLC. Treatment of HeLa-C3 cells with 400μg/mL of each UV active fraction from the hydrolyzed EtOH extract showed that the eighth fraction (F8) had the most anti-tau potency (Figure 5B). Even though other fractions lacked the same potency as F8, many of the fractions could reduce tau levels (Figure 5C). For example, despite modest reductions, LC/MS demonstrated that F11 contained the known tau-reducing compound myricetin, while fraction F8 contained a novel tau-modifier, the diarylheptanoid, myricanol.
Figure 5.
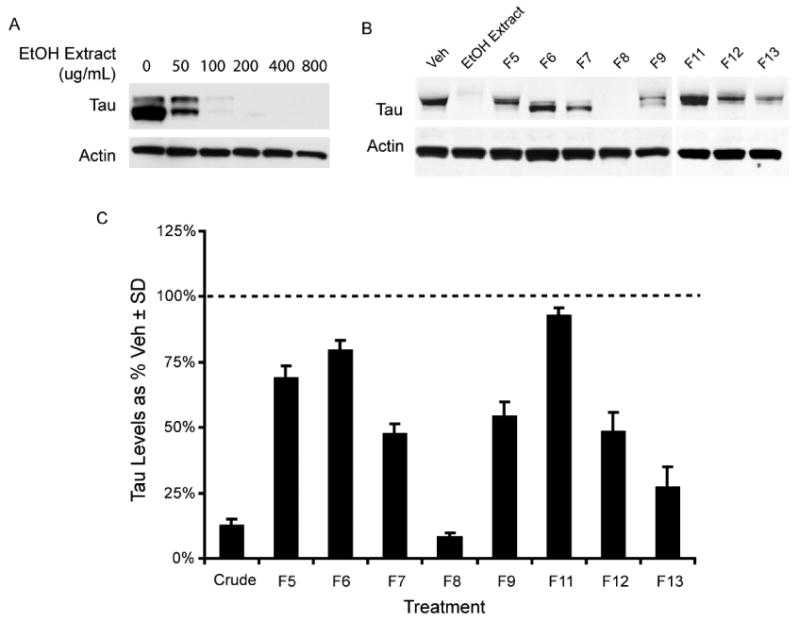
Bayberry ethanol extract displays anti-tau activity (A). Eight UV active chromatographic fractions from the hydrolyzed bayberry extract were tested on HeLa-C3 cells. The cells were treated with 400μg/ml of each UV active bayberry fraction for four hours and analyzed by western blot (B). Tau reductions are presented in the graph (C) as a percentage of vehicle treated cells (dashed line) ± SD.
Rhamnosidation of the Known Tau-reducer Myricetin does not Improve Anti-tau Efficacy
TLC analysis of crude hydrolyzed bayberry extract demonstrated that the aglycone myricetin was present when analyzed concurrently with commercially available myricetin. Myricetin was previously shown to be both a major constituent of bayberry after acid hydrolysis31 and an inhibitor of Hsp70 ATPase activity that reduced tau levels.28 Myricitrin (1), a rhamnoside of the flavonoid myricetin (2), has been found in high concentration in bayberry.33
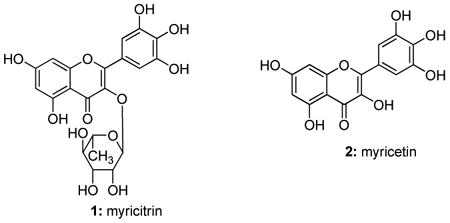
Since flavonoids typically have poor bioavailability, we speculated that myricitrin might be more effective given the addition of the carbohydrate moiety.34 A time course study was performed using commercially available myricitrin and myricetin. We tested the anti-tau efficacy of each over a 24 hr period in HeLa-C3 cells (Figure 6A & B) and found that both myricitrin and myricetrin showed similar activity (Figure 6C). Thus, myricetin efficacy could not be improved by rhamnosidation.
Figure 6.
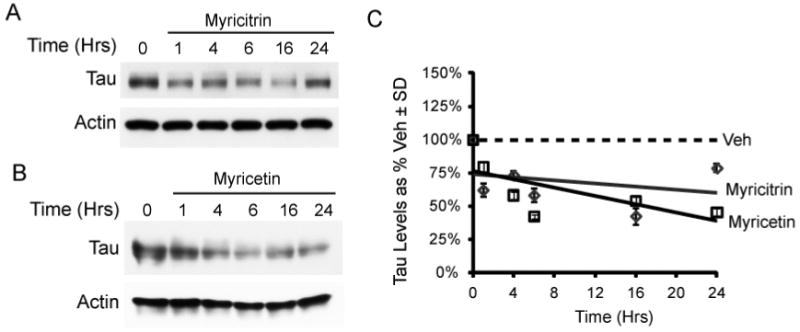
Commercial myricitrin (50 μM, DMSO) and myricetin (50 μM, DMSO) were tested over time on HeLa-C3 cells (A & B). The results are displayed as a trend line of tau levels as a percent of vehicle ± SD (C).
The Diarylheptanoid, Myricanol, Has Unique Structural Properties Compared to that Extracted from Other Species
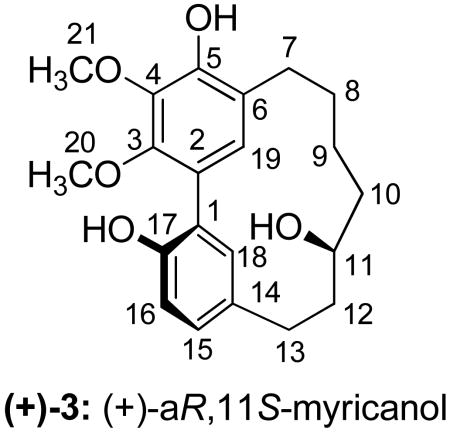
The primary constituent of F8 was found to be myricanol (3), a diarylheptanoid. Both 1H and 13C NMR data are in agreement with recently published data,35,36 but differ from those NMR data published earlier.37 Interestingly, the specific rotation for myricanol isolated in this study ([α]D20 = +48.0) differed in sign from previous reports: [α]D27.5 = -65.6 from M. nagi;38 [α]D = -62.9 from M. rubra;39 [α]D22 = -27.6 from M. rubra;40 [α]D= -64.0 from M. esculenta;37 and [α]D= 0.0 from M. cerifera.35 All previously isolated myricanol displayed negative rotations with the exception of one racemate. The negative rotation was correlated to 11R-configuration via X-ray crystallography of a brominated derivative of myricanol.38 (-)-R-myricanol from M. rubra ([α]D24= -48.3) along with (+)-S-myricanol 5-O-β-d-glucopyranoside which, upon hydrolysis yielded (+)-S-myricanol ([α]D22= +37.3), have been reported41 with the 11S configuration confirmed by Mosher's analysis. The 1H and 13C NMR data as well as the rotation reported for the hydrolyzed product matched those of the myricanol isolated in this study. The CD spectrum of our (+)-S-myricanol displayed positive Cotton effects which mirror the negative Cotton effects previously reported for (-)-R-myricanol,38,40 confirming our isolate has not only 11S configuration but also equal and opposite biphenyl twisting. Joshi et al.35 reported an X-ray structure of the enatiomeric pair of (+)-aR,11S- and (-)-aS,11R-myricanol and also noted that the crystal structure of (-)-aS,11R-myricanol closely resembled that of the previously reported brominated derivative of 11R-myricanol isolated by Begley et al.38 We surmise, from the identical NMR data shared by our compound and both the racemate isolated by Joshi et al. and the 11S-agylcone obtained by Matsuda et al.41 as well as the inverse CD spectra and specific rotations of our compound and (-)-aS,11R-myricanol reported by Begley et al., that we have in fact isolated (+)-aR,11S-myricanol. (+)-11S-Myricanol has been identified previously as a glycoside41 and part of a racemic mixture35 but the aglycone of the (+)-11S-enantiomer had not previously been found to occur naturally.
Myricanol is a Potent Tau Modulator in Several Models, Including Brain Tissue
(+)-aR,11S-Myricanol was used to treat HeLa-C3 cells (Figure 7A), IMR32 hippocampus-derived cells (Figure 7B), and murine brain slices (Figure 7C). (+)-aR,11S-Myricanol potently reduced tau levels in all models with comparable potency (Figure 7D). No toxicity was observed by a lactate dehydrogenase release assay (data not shown).
Figure 7.
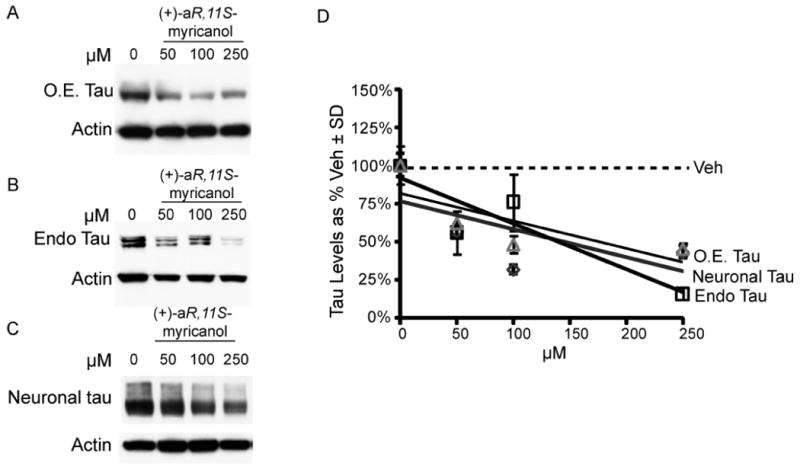
Extracted myricanol was used to treat HeLa-C3 cells that over-express tau, O.E., (A) IMR32 cells that express endogenous (endo) tau (B), and murine brain slices (C). The western blot results were quantified and plotted as a percent of vehicle ± SD (D).
Chirality Significantly Contributes to the Anti-tau Efficacy of Myricanol
Due to inconsistencies noted above in specific rotations, chiral HPLC analysis was conducted and revealed that our isolate was not enantiopure but, in fact, a scalemic mixture (86% ee, Figure 8A; racemic myricanol is shown for comparison in 8B). Investigation of tau levels in HeLa-C3 cells treated with the indicated concentrations of the isolated (+)-aR,11S-myricanol (86% ee) and commercially available “racemic” myricanol (actually 9% ee) showed that only the isolated myricanol reduced tau levels (Figure 8C): The EC50 of (+)-aR,11S-myricanol (86% ee) was ∼35μM (Figure 8D). Thus, (+)-aR,11S-myricanol isolated from M. cerifera may represent a novel scaffold from the diarylheptanoid family that drives tau reductions based on chirality.
Figure 8.
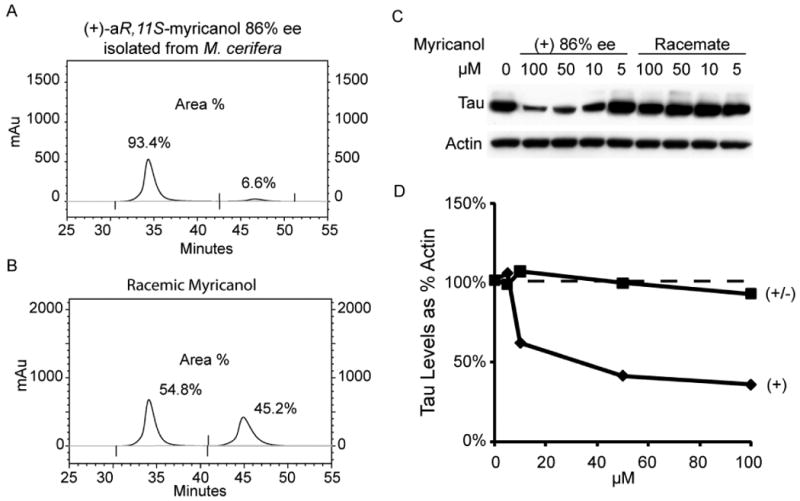
Using a chiral stationary phase, HPLC traces of (+)-aR,11S-myricanol from M. cerifera (A) and commercially available (±)-myricanol (B) reveal 86% ee and 9% ee, respectively. Tau levels in HeLa-C3 cells are reduced significantly by enantiomerically-enriched (+)-aR,11S-myricanol but the racemic mixture showed no activity at similar concentrations (C & D).
Natural product-based drug discovery has yielded a number of promising compounds that are in clinical and pre-clinical development for AD and other neurodegenerative diseases. An Extract from M. cerifera was shown to decrease tau levels in several cell models as well as murine brain slices. Three compounds capable of reducing tau levels were identified from this extract: myricetin, myricitrin, and (+)-aR,11S-myricanol. Myricetin is a flavonoid found in the rootbark of M. cerifera. In nature, it exists as the glycoside myricitrin and has been isolated in quantities of up to 2.5 g/kg.33 We have shown here that both myricetin and its “pro-drug” myricitrin reduce tau levels. Indeed, it is likely that myricitrin does function as a pro-drug in vivo due to the activity of glycoside hydrolases that are especially prevalent in the intestinal tract. Myricitrin is more stable in DMSO and more water soluble than myricetin. This could have implications for therapeutic efforts focused on flavonoid-based drug development. Flavonoids, and other antioxidants in general, have been the focus of intensive research efforts in AD and other neurodegenerative diseases. The antioxidant properties of flavonoids help to prevent oxidation of crucial proteins and biomolecules. The flavonoids quercetin and epigallocatechin gallate have demonstrated efficacy for preventing various aspects of AD pathogenesis.42-46 While flavonoids are attractive from a biological standpoint, their development into robust therapeutics has been hampered by their inherent lack of stability.27,47
Because the literature suggested that the primary component of bayberry was myricetin in the form of myricitrin, we were initially resolved to attribute the anti-tau efficacy of bayberry to these flavonoids. Surprisingly, however, the most potent anti-tau compound extracted from bayberry was not a flavonoid, but rather the diarylheptanoid, myricanol which has not previously been shown to have efficacy for AD or other tauopathies. This family of compounds also includes curcumin an extensively studied anti-inflammatory and anti-amyloidogenic agent.23 In fact, curcumin can also modulate tau phosphorylation in cultured hippocampal neurons.48 These structural similarities between curcumin and myricanol may allow for faster elucidation of the mechanism of action for myricanol. Although both compounds are diarylheptanoids, curcumin is linear while myricanol possesses a meta-meta bridged macrocyclic structure. Also, this is the first description of the dextrorotatory aglycone ((+)-aR,11S-myricanol) being found in nature or derived synthetically,49 and it is this enantiomer that seems to possess anti-tau efficacy with a potency commensurate with previously characterized targeted therapeutic lead compounds.21,28 Thus, (+)-aR,11S-myricanol is a novel and potentially highly tractable drug scaffold. The physical properties of myricanol suggest that it will abide by Lipinski's rule of five and could serve as the foundation for new and potent anti-tau therapies.
Experimental Section
Analytical Instrumentation
Specific rotations were measured on an Autopol IV automatic polarimeter using Na lamp corrected to 20°C. UV data was obtained on a Varian Cary 50-Bio UV-visible spectrophotometer using a xenon lamp. Circular Dichroism (CD) spectra were obtained with an Aviv Instruments model 215 CD spectrometer using a xenon lamp. 1H and 13C NMR spectra were recorded on a Varian 400 MHz spectrometer using residual protonated solvent as 1H internal standard or 13C absorption lines of solvents for 13C internal standard. NMR data were obtained in CDCl3 (purchased from Cambridge Isotope Labs). LC/MS data were obtained on an Agilent 1100 LC/MS TOF ESI mass spectrometer. Separations were conducted on a Teledyne Isco Combiflash Companion MPLC instrument using an appropriately sized normal phase silica gel cartridge purchased from Teledyne Isco. Enantiomeric excess was measured via HPLC (Shimadzu Prominence LC-20AT HPLC system with a SPD-N20A diode array detector using a Whelk-O 1 column, solvent system: 5% IPA:hexanes, 0.8 mL/min.).
Antibodies and Reagents
Anti-tau antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Dako (Carpinteria, CA). Anti-actin and anti-GAPDH were purchased from Sigma Aldrich and Biodesign (Birmingham, AL). All of these aforementioned antibodies were used at a dilution of 1:1,000. Anti-S396/S404 phosphorylated tau antibody (pS396/404) was used at a dilution of 1:200 and provided by P. Davies, Albert Einstein College of Medicine (New York, NY). Secondary antibodies were purchased from Southern Biotech (Birmingham, AL) and used at a 1:1,000 dilution. Fluorescent secondary antibodies were purchased from Molecular Probes and Rockland Laboratories. All antibodies were diluted into 7% nonfat dry milk (Bio-Rad Laboratories) in TBS. Myricitrin and (±)-myricanol were purchased from Indofine Chemical Company, Inc. (Hillsborough, NJ). Myricetin was purchased from Sigma-Aldrich. These compounds were all solubilized in DMSO to 50 mM.
Aqueous Extract of Bayberry
Bayberry root-bark powder was purchased from Frontier Natural Products Co-op (Norway, IA). An aqueous extract was prepared fresh for each experiment by adding 400 mg of root-bark powder to 10 mL deionized H2O. The solution was protected from light and allowed to nutate at room temperature for 1 h, after which it was centrifuged at 4,200 rpm for 15 min to remove insoluble particles. The liquid was lyophilized to determine the concentration of dissolved analytes (7 mg/mL).
Lipophilic Extract of Bayberry and Isolation of (+)-aR,11S-myricanol
50 g of raw-bayberry root-bark powder was extracted with 150 mL of toluene under agitation for 6 h in a flask protected from light. The filtrate was concentrated and the marc was then extracted under agitation with 150 mL 95% EtOH for 6 h, again protected from light. The EtOH extract was dried in vacuo yielding 5.6 g of crude extract. Acid hydrolysis was performed on 2 g of EtOH extract to facilitate the conversion of myricitrin to myricetin31 by refluxing for 2 h in 16 mL 1:1 MeOH:aqueous HCl (1.2 M) with addition of t-butylhydroquinone (TBHQ) (32 mg) as an antioxidant. The mixture was then concentrated in vacuo and fractionated on a silica gel column using MPLC (silica cartridge, Isco Combiflash Companion) with a linear gradient of 0-20% MeOH:CHCl3 (0.1% TFA). Fraction 8 (F8) was further purified on MPLC (silica, eluting with a linear gradient from 40-45% EtOAc:hexanes) to afford (+)-aR,11S-myricanol (12 mg). The previously prepared toluene extract was concentrated and found to be a mixture of compounds (∼1.4 g) with (+)-aR,11S-myricanol as the major constituent. A portion of the toluene extract was purified (430 mg) using silica gel MPLC, eluting at 40% EtOAc:hexanes to yield 180 mg (+)-S-myricanol: 86% ee; [α]D20 = +48.0 (c 1.0, CHCl3); UV λmax nm (MeOH) 215, 220, 260, 300; CD λmax nm (MeOH) (Δε): 238 (+5.6), 258 (+6.1), 301 (+4.9); 1H NMR (400 MHz, CDCl3) δ (integration, multiplicity, J (Hz), position): 1.50-1.62 (3H, m, OH11, H9a, H10a), 1.63-1.77 (2H, m, H9b, H12a), 1.89-1.99 (3H, m, H28, H10b), 2.35 (1H, m, 12b), 2.56 (1H, m, H7a), 2.81 (1H, dt, 18.2, 3.0, H7b), 2.93 (2H, m, H213), 3.89 (3H, s, H320), 4.01 (3H, s, H321), 4.10 (1H, t, 9.8, H11), 5.87 (1H, br s, OH5), 6.92 (1H, s, H19), 6.92 (1H, d, 8.1, H16), 7.10 (1H, dd, 8.1, 2.0, H15), 7.19 (1H, d, 2.0, H18), 7.68 (1H, s, OH17); 13C NMR (100 MHz, CDCl3) δ (position): 22.9 (C9), 25.4 (C7), 25.8 (C8), 26.9 (C13), 34.7 (C12), 39.4 (C10), 61.4 (2C, C20 and C21), 68.6 (C11), 116.8 (C16), 122.6 (C6), 123.4 (C2), 124.7 (C1), 129.4 (C19), 129.9 (C15), 130.7 (C14), 133.1 (C18), 138.7 (C4), 145.9 (C3), 147.7 (C5), 151.4 (C17); ESIMS m/z 359.1 [M + 1]+; HRESIMS m/z 359.1856 [M +1]+ (calc'd for C21H27O5, 359.1853).
In-cell Western (ICW) Screen
Extracts tested in the screen were supplied by Herb Pharm, (Williams, OR). ICW was performed on M17 cells as previously described.26 Briefly, M17 cells were plated in a 96-well plate with 200 μL complete media and treated with Herb Pharm extracts in triplicate (10% by volume). Protein levels were illuminated with rabbit anti-human tau (1:500; DAKO) and mouse anti-human glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:1500; BioDesign). Secondary infrared fluorescent antibodies were used for detection; one absorbing at 680 nm (AlexaFluor 680; Molecular Probes) and the other absorbing at 800 nm (IRDye 800 CW; Rockland, MD). Toxicity was assessed based on GAPDH immunoreactivity.
Cell Culture and Western Blot Analysis
HeLa cells stably expressing V5-tagged 4R0N tau were maintained under G418 selection in Opti-Mem plus 10% FBS (complete media; Invitrogen) as previously described.28 IMR32, M17 and H4 cells were maintained in Opti-mem plus 10% FBS and 2% 200 mM L-glutamine (Cellgro, Mediatech, Inc, Herndon, VA). All cells were treated as indicated and were harvested as previously described.28 Protein levels were determined by using the Peirce BCA kit. Western blotting was performed by running SDS-PAGE, followed by transfer onto Immobilon-P (Millipore).26,27
Ex-vivo Slice Cultures
Four non-transgenic 5 month-old mice were decapitated and their brains were quickly removed. 400 μm coronal sections were made using a vibratome as previously described.28,50,51 Briefly, slices were allowed to recover for 1 hour at RT in a 50/50 mixture of cutting solution (110 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 5 mM glucose, 0.6 mM ascorbate, 0.5 mM CaCl2, and 7 mM MgCl2) and artificial cerebral spinal fluid (ACSF; 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 2 mM CaCl2 and 1 mM MgCl2) and then moved into ACSF at 30-32°C for 1 h. Slices were then moved in duplicate into an oxygenated multi-well plate at 30-32°C containing ACSF and treated with aqueous bayberry extract at the following concentrations: 9, 18, 35, 70, 105, and 140 μg/mL. Sections were kept alive under the previously described conditions for 4 hrs and then snap frozen by liquid nitrogen. Sections were homogenized mechanically and analyzed as previously described.28
Supplementary Material
Acknowledgments
We thank Dr. Peter Davies (Albert Einstein COM, NY) for PHF1 (pS396/S404) antibody. CAD was supported by the Rosalinde and Arthur Gilbert Foundation/American Federation for Aging Research, CurePSP, the Alzheimer's Association and NIA grant AG031291. BJB was supported by USF's Center for Molecular Diversity in Drug Design, Discovery and Delivery.
Footnotes
Supporting Information Available: LC/MS data for F11 and commercial myricetin (2), UV, CD, 1H and 13C NMR spectra for isolated myricanol (3). This information is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH. J Am Med Assoc. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gauthier S, Aisen PS, Ferris SH, Saumier D, Duong A, Haine D, Garceau D, Suhy J, Oh J, Lau W, Sampalis J. J Nutr Health Aging. 2009;13:550–557. doi: 10.1007/s12603-009-0106-x. [DOI] [PubMed] [Google Scholar]
- 3.Lyketsos CG, Breitner JC, Green RC, Martin BK, Meinert C, Piantadosi S, Sabbagh M. Neurology. 2007;68:1800–1808. doi: 10.1212/01.wnl.0000260269.93245.d2. [DOI] [PubMed] [Google Scholar]
- 4.Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Neurology. 1988;38:1682–1687. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Acta Neuropathol (Ber) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 6.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT. J Neurosci. 2008;28:862–867. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 9.Garcia ML, Cleveland DW. Curr Opin Cell Biol. 2001;13:41–48. doi: 10.1016/s0955-0674(00)00172-1. [DOI] [PubMed] [Google Scholar]
- 10.Cassimeris L, Spittle C. Int Rev Cytol. 2001;210:163–226. doi: 10.1016/s0074-7696(01)10006-9. [DOI] [PubMed] [Google Scholar]
- 11.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. P Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Heutink P. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 13.Hardy J, Orr H. J Neurochem. 2006;97:1690–1699. doi: 10.1111/j.1471-4159.2006.03979.x. [DOI] [PubMed] [Google Scholar]
- 14.Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen RB, Gambetti P. Nat Med. 1999;5:454–457. doi: 10.1038/7454. [DOI] [PubMed] [Google Scholar]
- 15.Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. PLoS Biol. 2009;7:e34. doi: 10.1371/journal.pbio.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M. Biochemistry. 2009;48:10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 17.Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. J Neurosci. 2009;29:10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 19.Congdon EE, Necula M, Blackstone RD, Kuret J. Arch Biochem Biophys. 2007;465:127–135. doi: 10.1016/j.abb.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. P Natl Acad Sci USA. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jinwal UK, Koren J, 3rd, Borysov SI, Schmid AB, Abisambra JF, Blair LJ, Johnson AG, Jones JR, Shults CL, O'Leary JC, 3rd, Jin Y, Buchner J, Cox MB, Dickey CA. J Neurosci. 2010;30:591–599. doi: 10.1523/JNEUROSCI.4815-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ. P Natl Acad Sci USA. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickey CA, Ash P, Klosak N, Lee WC, Petrucelli L, Hutton M, Eckman CB. Mol Neurodegener. 2006;1:6. doi: 10.1186/1750-1326-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickey CA, Eriksen J, Kamal A, Burrows F, Kasibhatla S, Eckman CB, Hutton M, Petrucelli L. Curr Alzheimer Res. 2005;2:231–238. doi: 10.2174/1567205053585927. [DOI] [PubMed] [Google Scholar]
- 27.Koren J, 3rd, Jinwal UK, Jin Y, O'Leary J, Jones JR, Johnson AG, Blair LJ, Abisambra JF, Chang L, Miyata Y, Cheng AM, Guo J, Cheng JQ, Gestwicki JE, Dickey CA. J Biol Chem. 2010;285:2498–2505. doi: 10.1074/jbc.M109.057208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jinwal UK, Miyata Y, Koren J, 3rd, Jones JR, Trotter JH, Chang L, O'Leary J, Morgan D, Lee DC, Shults CL, Rousaki A, Weeber EJ, Zuiderweg ER, Gestwicki JE, Dickey CA. J Neurosci. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saito A, Sugisawa A, Umegaki K. Shokuhin Eiseigaku Zasshi. 2001;42:174–178. doi: 10.3358/shokueishi.42.174. [DOI] [PubMed] [Google Scholar]
- 30.Mehrdad M, Zebardast M, Abedi G, Koupaei MN, Rasouli H, Talebi M. J AOAC Int. 2009;92:1035–1043. [PubMed] [Google Scholar]
- 31.Hakkinen SH, Karenlampi SO, Heinonen IM, Mykkanen HM, Torronen AR. J Agric Food Chem. 1999;47:2274–2279. doi: 10.1021/jf9811065. [DOI] [PubMed] [Google Scholar]
- 32.Biesaga M, Ochnik U, Pyrzynska K. J Sep Sci. 2009;32:2835–2840. doi: 10.1002/jssc.200800730. [DOI] [PubMed] [Google Scholar]
- 33.Paul BD, Rao GS, Kapadia GJ. J Pharm Sci. 1974;63:958–959. doi: 10.1002/jps.2600630638. [DOI] [PubMed] [Google Scholar]
- 34.Hollman PC, Katan MB. Free Radical Res. 1999;31 Suppl:S75–80. doi: 10.1080/10715769900301351. [DOI] [PubMed] [Google Scholar]
- 35.Joshi BS, Pelletier SW, Newton MG, Lee D, McGaughey GB, Puar MS. J Nat Prod. 1996;59:759–764. [Google Scholar]
- 36.Kawai S, Nakata K, Ohashi M, Tomoaki N. J Wood Sci. 2008;54:256–260. [Google Scholar]
- 37.Sun DW, Zhao ZC, Wong H, Foo LY. Phytochemistry. 1988;27:579–583. [Google Scholar]
- 38.Begley MJ, Campbell MVM, Crombie L, Tuck B, Whiting DA. J Chem Soc C. 1971:3634–3642. [Google Scholar]
- 39.Inoue T, Arai Y, Nagai M. Yakugaku Zasshi. 1984;104:37–41. doi: 10.1248/yakushi1947.104.1_37. [DOI] [PubMed] [Google Scholar]
- 40.Takeda Y, Fujita T, Shingu T, Ogimi C. Chem Pharm Bull. 1987;35:2569–2573. [Google Scholar]
- 41.Matsuda H, Morikawa T, Tao J, Kazuho U, Yoshikawa M. Chem Pharm Bull. 2002;50:208–215. doi: 10.1248/cpb.50.208. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dajas F, Rivera-Megret F, Blasina F, Arredondo F, Abin-Carriquiry JA, Costa G, Echeverry C, Lafon L, Heizen H, Ferreira M, Morquio A. Braz J Med Biol Res. 2003;36:1613–1620. doi: 10.1590/s0100-879x2003001200002. [DOI] [PubMed] [Google Scholar]
- 44.Choi RC, Zhu JT, Leung KW, Chu GK, Xie HQ, Chen VP, Zheng KY, Lau DT, Dong TT, Chow PC, Han YF, Wang ZT, Tsim KW. J Alzheimers Dis. 2010;19:795–811. doi: 10.3233/JAD-2010-1293. [DOI] [PubMed] [Google Scholar]
- 45.Obregon DF, Rezai-Zadeh K, Bai Y, Sun N, Hou H, Ehrhart J, Zeng J, Mori T, Arendash GW, Shytle D, Town T, Tan J. J Biol Chem. 2006;281:16419–16427. doi: 10.1074/jbc.M600617200. [DOI] [PubMed] [Google Scholar]
- 46.Mandel S, Weinreb O, Amit T, Youdim MB. J Neurochem. 2004;88:1555–1569. doi: 10.1046/j.1471-4159.2003.02291.x. [DOI] [PubMed] [Google Scholar]
- 47.Boulton DW, Walle UK, Walle T. J Pharm Pharmacol. 1999;51:353–359. doi: 10.1211/0022357991772367. [DOI] [PubMed] [Google Scholar]
- 48.Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, Chen PP, Hudspeth B, Chen C, Zhao Y, Vinters HV, Frautschy SA, Cole GM. J Neurosci. 2009;29:9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whiting DA, Wood AF. J Chem Soc Perkin Trans I. 1980:623–628. [Google Scholar]
- 50.Qiu S, Zhao LF, Korwek KM, Weeber EJ. J Neurosci. 2006;26:12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mirnikjoo B, Brown SE, Kim HF, Marangell LB, Sweatt JD, Weeber EJ. J Biol Chem. 2001;276:10888–10896. doi: 10.1074/jbc.M008150200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.