

Abstract
The marine-derived fungus Stachylidium sp. was isolated from the sponge Callyspongia cf. C. flammea. Four new, putatively tyrosine-derived and O-prenylated natural products, stachylines A – D (1 – 4), were obtained from the fungal extract. The structures of 1 – 4 were elucidated based on extensive spectroscopic analyses. The absolute configuration of compound 2 was established by Mosher’s method. Stachyline A (1) possesses a rare terminal oxime group and occurs as an interchangeable mixture of E/Z- isomers.
The marine environment harbours approximately half of the global biodiversity and is estimated to contain between 3 and 500 million different species, offering an almost infinite resource for novel compounds.1 Among these organisms marine-derived fungi became known as prolific producers of structurally most intriguing compounds.2 In general, tyrosine derivatives have only rarely been reported from fungi, and in most cases such compounds were obtained from strains originating from environmentally extreme habitats, e.g. tyrosol carbamate which was isolated from the deep-water fungus Arthrinium sp.3 Phytomyces sp., producing O-prenylated tyrosine derivatives, is an extremophile collected from an acid mine waste rich in toxic metals.4 Another unusual case is aspergillusol A, an α-glucosidase inhibitor obtained from the sponge-derived fungus Aspergillus aculeatus which is reported to be the only known fungal tyrosine derivative to possess an oxime group.5
Secondary metabolites with an oxime substituent are rare, and most of the reported examples have potent bioactivity, e.g. the actinomycete-derived nocardicins displayed strong antibiotic activity,6 and brevioxime from Penicillium brevicompactum inhibited the biosynthesis of insect juvenile hormones.7 P. olsonii produced 2-(4-hydroxyphenyl)-2-oxoacetaldehyde oxime (PHBA) which regenerated phosphorylated cholinesterase.8 The oxime geometrical isomers collismycins A and B were isolated from Streptomyces sp. MQ22 which inhibited dexamethasone glucocorticoid receptor binding.9
During our search for new cytotoxic natural products an extract of the marine-derived fungus Stachylidium sp. was found to be active. During chromatographic separations it became clear that this fungus produces a vast array of secondary metabolites with intriguing structural features, among them the four novel, putatively tyrosine-derived and O-prenylated natural products, stachylines A – D (1 – 4). Stachyline A (1) is distinguished by an oxime terminal group, probably derived through biosynthetic reactions similar to those known for cyanogenic glycosides and nocardicin A formation.10–14 The molecules were evaluated in a number of biological assays, to date however no considerable activity was detected.
Results and Discussion
The RP-18 HPLC chromatogram of 1 contained two peaks (ratio 1:1), which when re-injected after their individual isolation, again resulted in the same chromatogram. This result suggested that compound 1 exists as a mixture of interchangeable isomers. NMR spectra also presented two sets of data, one for each isomer (see Tables 1 and 2). For clarity in the description of the structure elucidation, only one set of data will be considered initially.
Table 1.
13C NMR Spectroscopic Data for Compounds 1 – 4.
| position |
1* |
2 |
3 |
4 |
|
|---|---|---|---|---|---|
| δC, mult.a, b, c | δC, mult.a, b, c | δC, mult.a, b,c | δC, mult.a, b, c | ||
| 1 | 130.1, qC | 128.0, qC | 132.5, qC | 127.9, qC | |
| 2 | 130.5, CH | 131.2, CH | 130.7, CH | 131.2, CH | |
| 3 | 115.5, CH | 115.3, CH | 115.2, CH | 115.2, CH | |
| 4 | (Z-), 158.5, qC | (E-), 158.7, qC | 158.8, qC | 158.3, qC | 159.0, qC |
| 5 | 115.5, CH | 115.3, CH | 115.2, CH | 115.2, CH | |
| 6 | 130.5, CH | 131.2, CH | 130.7, CH | 131.2, CH | |
| 7 | (Z-), 31.0, CH2 | (E-), 35.4, CH2 | 40.5, CH2 | 39.4, CH2 | 40.4, CH2 |
| 8 | (Z-), 150.0, CH | (E-), 149.7, CH | 173.0, qC | 64.1, CH2 | 173.2, qC |
| 1’ | 72.1, CH2 | 72.1, CH2 | 72.1, CH2 | 71.6, CH2 | |
| 2’ | 73.9, CH | 73.9, CH | 74.0, CH | 74.6, CH | |
| 3’ | 146.1, qC | 146.1, qC | 146.2, qC | 31.6, CH | |
| 4’ | 112.1, CH2 | 112.1, CH2 | 112.0, CH2 | 17.7, CH3 | |
| 5’ | 18.7, CH3 | 18.8, CH3 | 18.8, CH3 | 19.4, CH3 | |
acetone-d6,, 75.5 MHz.
Assignments are based on extensive 1D and 2D NMR experiments (HMBC, HSQC, COSY, see Supp. Inf.).
Implied multiplicities determined by DEPT.
Carbon resonances were attributed to the (Z-) or (E-) configuration, according to ACD/NMR Predictor® software.
Table 2.
1H NMR Spectroscopic Data for Compounds 1 – 4.
| position |
1* |
2 |
3 |
4 |
|
|---|---|---|---|---|---|
| δHa, b (J in Hz) | δHa, b (J in Hz) | δHa, b (J in Hz) | δHa, b (J in Hz) | ||
| 1 | |||||
| 2 | (Z-), 7.17, d (8.4) | (E-), 7.13, d (8.4) | 7.20, d (8.4) | 7.13, d (8.4) | 7.19, d (8.4) |
| 3 | 6.88, d (8.4) | 6.87, d (8.4) | 6.84, d (8.4) | 6.87, d (8.4) | |
| 4 | |||||
| 5 | 6.88, d (8.4) | 6.87, d (8.4) | 6.84, d (8.4) | 6.87, d (8.4) | |
| 6 | (Z-), 7.17, d (8.4) | (E-), 7.13, d (8.4) | 7.20, d (8.4) | 7.13, d (8.4) | 7.19, d (8.4) |
| 7 | (Z-), 3.60, d (5.5) | (E-), 3.39, d (6.2) | 3.51, br s | 2.72, t (7.1) | 3.52, br s |
| 8 | (Z-), 6.73, t (5.5) | (E-), 7.40, t (6.2) | 3.68, t (7.1) | ||
| 1’ | a: 4.01, dd (3.7, 9.5) | a: 4.01, dd (4.0, 9.8) | a: 4.00, dd (4.3, 9.8) | a: 4.00, dd (4.0, 9.8) | |
| b: 3.90, dd (7.3, 9.5) | b: 3.90, dd (7.2, 9.8) | b: 3.89, dd (7.2, 9.8) | b: 3.89, dd (6.6, 9.8) | ||
| 2’ | 4.39, m | 4.40, dd (4.0, 7.2) | 4.39, dd (4.3, 7.2) | 3.68, m | |
| 3’ | 1.86, m | ||||
| 4’ | a: 4.89, br s | a: 4.88, br s | a: 4.89, br s | 0.97, d (6.8) | |
| b: 5.08, br s | b: 5.08, br s | b: 5.09, br s | |||
| 5’ | 1.78, s | 1.79, s | 1.79, s | 0.97, d (6.8) | |
acetone-d6, 300 MHz.
Assignments are based on extensive 1D and 2D NMR experiments (HMBC, HSQC, COSY, see Supp. Inf.).
Proton resonances were attributed to the (Z-) or (E-) configuration, according to ACD/NMR Predictor® software.
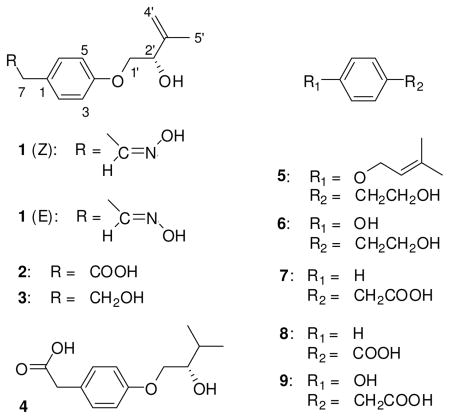
The molecular formula of 1 was deduced by accurate mass measurement (HREIMS/HRESIMS) to be C13H17NO3, requiring six sites of unsaturation. The 13C NMR and DEPT135 spectra showed the presence of thirteen resonances for one methyl, five sp2 methine, one sp3 methine, one sp2 methylene, two sp3 methylene groups and three quaternary carbons (see Tables 1 and 2). The UV maximum at 276 nm indicated the presence of an aromatic moiety, whereas an IR absorption at around 3300 cm−1 arose from a hydroxy group in the molecule.
1H NMR and 1H-13C HSQC data (see Tables 1 and 2) included two resonances for magnetically equivalent protons, i.e. H-2/H-6 and H-3/H-5 resonating at δH 7.17 and 6.88, respectively. This spectroscopic feature was interpreted as characteristic for a para-substituted aromatic ring. From the same spectra the presence of an exomethylene moiety, CH2-4’, was evident (δH 4.89, 5.08; δC 112.1). The 13C NMR spectrum showed two resonances for carbons connected to oxygen atoms, namely at δC 72.1 (C-1’) and 73.9 (C-2’) (see Table 1), and the protons attached to these carbon atoms, H2-1’ and H-2’ were coupled as evidenced by 1H-1H COSY correlations. 1H-1H COSY correlations indicated the presence of another spin system between H2-4’ and H3-5’. The partial structures deduced from these two spin systems were connected making use of 1H-13C HMBC correlations. Thus, correlations from H3-5’ to C-2’, C-3’ and C-4’ delineated an unsaturated and hydroxylated isoprene unit (see Table S1 in Supporting Information). This was confirmed by further heteronuclear long range correlations from H2-4’ to C-3’ and C-2’. 1H-13C HMBC correlations between H-1’ to qC-4 showed that the isoprene unit was connected via C-4 to the para-substituted aromatic moiety. Due to the downfield shift of C-1’ and C-4 (δC 72.1, 158.5) connection of both parts of the molecule occurred through an oxygen atom.
According to the molecular formula, the second substituent on the aromatic ring of 1 must have the composition C2H4NO. 1H-13C HMBC correlations could be detected from the aromatic protons H-2/H-6 to the methylene carbon C-7, making the connection of the second side chain via C-1 most likely. Through an 1H-1H COSY experiment, the methylene protons CH2-7 were shown to be part of a spin system including H-8 (δH 6.73), whereby the latter was bound to an sp2 hybridized carbon. The remaining presence of a nitrogen, oxygen and proton atom was assigned to an unusual oxime moiety (see Tables 1 and 2). The latter is known to be able to adopt interchangeable configurations.24 E- and Z- isomers in the case of 1 presented clearly distinguishable NMR shifts for the C-8 to C-2/C-6 part of the molecules (see Tables 1 and 2). The assignment of NMR shifts for the E- and Z- isomers was supported using the ACD NMR predictor software (ACD/Labs®). The configuration at C-2’ of compound 2 was found to be S as deduced by Mosher’s method. Based on the negative optical rotation of compound 1, which was also observed for 2, and the close biosynthetic relationship between both compounds, the S configuration is also proposed for C-2’ of 1. For compound 1 the trivial name E- and Z-stachyline A is proposed.
The molecular formula of 2 was deduced by accurate mass measurement (HREIMS) to be C13H16O4, requiring six sites of unsaturation. The 13C and 1H NMR spectra were closely related to those of compound 1 (see Table 2). One of the substituents on the aromatic ring was again an O-prenyl moiety as in 1, also connected via qC-4 and C-1’ (see Tables 1, 2 and Supporting Information). The second substituent on the aromatic ring of 2 was however, different and established by interpretation of 1H-13C HMBC long range correlations. Thus, the methylene protons H2-7 had a heteronuclear coupling with C-2 and C-6 of the aromatic ring. Additionally, H2-7 had HMBC long range correlations to the quaternary carbon C-8, which according to its characteristic 13C NMR resonance was part of a carboxylic acid group (δC 173.0). Compound 2 was thus an O-prenylated phenyl acetic acid.
The absolute configuration at C-2’ was determined by modified Mosher’s method. The evaluation of the corresponding 1H-13C HSQC NMR spectra revealed differences of the proton shifts between the (S)- and (R)-MTPA esters (see Figure 1 and Supporting Information). The calculated differences in chemical shifts [δ of protons in the (S)-MTPA-ester minus δ of the corresponding protons in the (R)-MTPA-ester] led to the assignment of the (S)-absolute configuration for C-2′. Since compounds 1 – 4 all showed a negative specific optical rotation, it was assumed that they all share the (S-) configuration at C-2’. Compound 2 is an O-prenylated phenyl acetic acid, for which we propose the trivial name stachyline B.
Figure 1.

Deduction of the absolute configuration at C-2′ using modified Mosher’s method with ΔS-R values of MTPA-esters of compound 2.
The molecular formula of 3 was deduced by accurate mass measurement (HREIMS) to be C13H18O3, requiring five sites of unsaturation. 1D and 2D NMR data of 3 were closely similar to those of 2 (see Tables 1, 2 and Supporting Information), except for the absence of resonances for the carboxylic acid group in the 13C NMR spectrum and the presence of an additional resonance for a sp3 methylene group. The latter had a downfield shifted 13C NMR resonance (δC 64.1), which evidenced connection to an oxygen atom. Thus, 3 can be regarded as the C-8 reduction product of 2, for which the trivial name stachyline C is suggested.
The molecular formula of 4 was deduced by accurate mass measurement (HRESIMS) to be C13H18O4, requiring five sites of unsaturation. 1D and 2D NMR spectroscopic data of compound 4 exhibited many identical features when compared with those of 2 (see Tables 1, 2 and Supporting Information). A difference, however concerned the exo-methylene proton signals which were absent in the 1H NMR spectrum of 4, whilst signals for a methyl (H3-4’, δH 0.97) and a methine group were present (H-3’, δH 1.86). 1H-1H COSY correlations confirmed the structure, since a spin system connected all atoms within the isoprene unit from H2-1’ to H3-5’ and H3-4’ (see Table 2 and Supporting Information). For compound 4 the trivial name stachyline D is proposed.
Further compounds isolated were established to be known molecules, including etrogol (5),4 tyrosol (6),15 phenyl acetic acid (7),16 benzoic acid (8)17 and para-hydroxyphenyl acetic acid (9).18,19
Stachyline A, B and C (1–3) were evaluated for a large number of pharmacological activities, including tumor cell cytotoxic activity, affinity to receptors from the central nervous system, anti-diabetic activity, protein kinase inhibition, NF-κB protein complex inhibition, antibacterial and antifungal activity, antiplasmodial activity, antiviral activity (HIV and influenza viruses). No activity in any of the test systems was found (see Supporting Information).
Stachylines A-D are presumably tyrosine-derived metabolites, a group of compounds rarely encountered in fungal metabolism. The oxime group in compound 1 is even more intriguing. Phenylpyruvic acid oxime derivatives,11 brominated tyrosine-derived compounds like the bastadins and psammaplins,20 as well as biosynthetically possibly related nitrile containing metabolites21 occur in marine sponges. The Stachylidium sp. used for this investigation was isolated from a marine sponge.
It was observed that the configuration of the oxime moiety in compound 1 was labile, and upon chromatographic isolation of the E- and Z-isomers, respective isomerisation took place instantaneously. None of the isomers seemed to be thermodynamically more stable under the prevailing conditions, since a 1:1 ratio of isomers was obtained (see Supporting Information). In contrast to that, the geometrical isomers collismycin A and B from Streptomyces sp. MQ22 were reported to be not interchangeable in many organic solvents and at room temperature. Only by heating in ortho-dichlorobenzene at 120 °C they were gradually interchangeable.9 For 2-(4-hydroxyphenyl)-2-oxoacetaldehyde oxime (PHBA) isolated from Penicillium olsonii, the E-configuration was described without mentioning any isomerisation reactions.7
Although the oxime functional groups are rare in natural products they occur in a variety of phyla, e.g., sponges, bacteria, fungi, and plants. Tyrosine-derivatives with an oxime moiety were found in actinomycetes, such as the β-lactam antibiotic nocardicin A from Nocardia uniformis.6 Higher plants are also capable of producing oxime groups, since the biosynthesis of glucosinolates and cyanogenic glycosides, e.g. dhurrin from Sorghum bicolor proceeds via aldoximes.12,21–23 From these studies, oximes can be regarded as biosynthetic precursors of nitriles. Aspergillusol A was reported to be the first fungal tyrosine-derivative with an oxime moiety,5 however a previous publication had described the isolation of 2-(4-hydroxyphenyl)-2-oxoacetaldehyde oxime (PHBA) from Penicillium olsonii.7 Hence, the current report is the third one of a fungal tyrosine-derived oxime derivative.
Concerning the biosynthesis of naturally occurring oximes it is not clear which isomer is produced initially. The aldoxime intermediate in the biosynthesis of the cyanogenic glycoside dhurrin has been reported to initially have the E-configuration, subsequently undergoing non-enzymatic isomerisation.12,23 Nocardicin A with Z-oxime configuration is thought to be preferentially produced over the E-configurated nocardicin B, due to favourable intramolecular hydrogen-bonding in biosynthetic intermediates.13,14 In the case of bastadins and psammaplins, it was assumed that the Z-oxime configuration is initially produced during biosynthesis, followed by isomerisation to the thermodynamically more favored E-isomers during extraction and isolation of the compounds.20
Stachylines A–D are proposed to be biosynthetically derived from tyrosine (Figure 2). For stachyline A the biosynthetic process may be similar to that of cyanogenic glycosides, e.g. dhurrin, in that the precursor tyrosine is N-hydroxylated by CYP 450 enzymes to N,N-dihydroxytyrosine and subsequently decarboxylated and dehydrated to form an aldoxime.12 A similar pathway was described for the norcardicin biosynthesis.13,14
Figure 2.

Proposed biosynthesis of stachylines A – D
Experimental section
General Experimental Procedures
Optical rotations were measured on a Jasco DIP 140 polarimeter. UV and IR spectra were obtained employing a Perkin-Elmer Spectrum BX instrument. CD spectra were recorded in MeOH at room temperature using a JASCO J-810-150S spectropolarimeter. All NMR spectra were recorded in CD3OD or (CD3)2CO employing a Bruker Avance 300 DPX spectrometer. Spectra were referenced to residual solvent signals with resonances at δH/C 3.35/49.0 for CD3OD and δH/C 2.04/29.8 for (CD3)2CO. HRESIMS were recorded on a Bruker Daltonik micrOTOF-Q Time-of-Flight mass spectrometer with ESI source. HPLC was carried out using a system composed of a Waters 515 pump together with a Knauer K-2300 differential refractometer. HPLC columns were from Knauer (250 × 8 mm, 5 μm, Eurospher-100 Si and 250 × 8 mm Eurospher-100, 5 μm, C18), flow rate 2 mL/min. Merck silica gel 60 (0.040–0.063 mm, 70–230 mesh) was used for vacuum liquid chromatography (VLC). Columns were wet-packed under vacuum using petroleum ether (PE). Before applying the sample solution, the columns were equilibrated with the first designated eluent. Standard columns for crude extract fractionation had dimensions of 13 × 4 cm.
Fungal material
The marine-derived fungus Stachylidium sp. was isolated from the sponge Callyspongia sp. cf. C. flammea (collected at Bear Island, Sydney, Australia) and identified by P. Massart and C. Decock, BCCM/MUCL, Catholic University of Louvain, Belgium. A specimen is deposited at the Institute for Pharmaceutical Biology, University of Bonn, isolation number “293K04”, strain number 220.
Culture, extraction and isolation
Compounds 1 to 4 were isolated from three different cultures of Stachylidium sp. The first and second culture comprised 12 L and 10 L of agar-biomalt medium (biomalt 20 g/L, 15 g/L agar) supplemented with sea salt incubated for 2 months (12 L) and 40 days (10 L) in Fernbach flasks at room temperature. The third culture was performed with 9.6 L liquid YPM medium (yeast extract 5 g/L, peptone from YPM 3 g/L, mannitol 25 g/L) incubated for 10 days. An extraction with 5 L EtOAc yielded 5.9 g, 2.1 g and 1.0 g of extract, respectively which were subjected to a VLC fractionation on silica gel, using a step gradient solvent system with petroleum ether - acetone from 10:1, 5:1, 2:1, 1:1 to 100 % acetone and, subsequently 100 % MeOH, resulting in 6 VLC fractions for each culture. Compounds 2 and 3 were isolated from the first culture (12 L), whilst compound 4 was isolated from the second culture (10 L) and compound 1 was isolated from the third culture (YPM medium). The known compounds 6 and 9 were isolated from the first culture and the know compounds 5, 7 and 8 from the first and third culture (isolation described for the YPM culture).
Compound 1 was isolated from VLC fraction 3 by NP-HPLC separation using petroleum ether - acetone (7:1) to yield 6 fractions. Sub-fraction 3 and 4 corresponded to a mixture of isomers of compound 1 (E- and Z-, 4.4 mg), which was again separated by RP-HPLC using 50 % MeOH (tR 11 min and 15 min), but immediately formed the initial mixture of isomers in a ratio of 1:1. Compounds 2 and 3 were isolated from VLC fraction 3 by NP-HPLC using petroleum ether - acetone (11:2) to yield 10 fractions. Further RP-HPLC fractionation with sub-fraction 5 using 50 % MeOH yielded 5 more sub-fractions, in which sub-fraction 4 was a mixture of compounds 2 and 3. Finally, we performed NP-HPLC fractionation using petroleum ether - acetone (7:1) to afford compound 2 (fraction 1 of 2, 4.1 mg, tR 19 min) and compound 3 (fraction 2 of 2, 2.2 mg, tR 26 min). Compound 4 was isolated from VLC fraction 4, followed by HPLC fractionation using petroleum ether - acetone (9:2; sub-fraction 2 of 7) which was further purified using RP-HPLC with 60 % MeOH (fraction 2 of 4, 1.8 mg, tR 10 min).
Etrogol (5) was isolated from VLC fraction 3, followed by HPLC fractionation using petroleum ether - acetone (7:1) to yield 5 sub-fractions. Sub fraction 1 was further purified using petroleum ether - acetone (7:1) to yield pure etrogol (fraction 3 of 3, 3.1 mg, tR 26 min). Tyrosol (6) was isolated from VLC fraction 3 followed by HPLC fractionation using petroleum ether - acetone (5:1) to yield 10 fractions. Further RP-HPLC fractionation with sub-fraction 6 using 30 % MeOH yielded the pure compound (fraction 3 of 7, 2.5 mg, tR 19 min). Phenyl acetic acid (7) and benzoic acid (8) were isolated from VLC fraction 3, followed by HPLC fractionation using petroleum ether - acetone (11:1) to yield 5 fractions. Benzoic acid was present in fraction 2 (7.9 mg, tR 16 min) and phenylacetic acid in fraction 3 (11.2 mg, tR 25 min). Para-hydroxyphenyl acetic acid (9) was isolated from VLC fraction 3 followed by HPLC fractionation using petroleum ether - acetone (5:1) to yield 10 fractions. Further RP-HPLC fractionation with sub-fraction 6 using 30 % MeOH yielded the pure compound (fraction 4 of 7, 1.8 mg, tR 27 min).
Stachyline A, mixture of E-/Z-isomers, 1:1 (1): white amorphous solid (458 μg/L, 0.43 %); [α] D23 -15 (c 0.29, MeOH); UV (MeOH) λmax (log ε) 225 nm (3.60), 222 nm (2.84), 276 nm (2.76) ; IR (ATR) νmax 3300 (br), 2922, 1610, 1510 cm−1; 1H NMR and 13C NMR (Tables 1 and 2 and Supporting Information); ESIMS m/z 236.1 [M+H]+; HREIMS m/z 235.1208 [M]+ (calcd. for C13H17NO3, m/z 235.1208); HRESIMS m/z 258.1125 [M+Na]+ (calcd. for C13H17NNaO3, m/z 258.1101).
Stachyline B (2): white amorphous solid (340 μg/L, 0.07 %); [α] D23 - 12 (c 0.33, MeOH); UV (MeOH) λmax (log ε) 204 nm (3.35), 227 nm (3.75), 276 nm (3.05), 283 nm (2.75); CD (c 1.06 × 10−6 mol/L, MeOH),λ (Δε) = 197 (+ 1.78); IR (ATR) νmax 3364 (br), 2929, 1610, 1511 cm−1; 1H NMR and 13C NMR (Tables 1 and 2 and Supporting Information); EIMS m/z 236.1 [M]+; HREIMS m/z 236.1051 [M]+ (calcd. for C13H16O4, m/z 236.1049).
Stachyline C (3): white amorphous solid (183 μg/L, 0.04 %); [α] D23 - 16 (c 0.33, MeOH); UV (MeOH) λmax (log ε) 205 nm (3.43), 225 nm (3.85), 277 nm (2.95), 283 nm (2.95); CD (c 1.25 × 10−6 mol/L, MeOH) λ (Δε) = 199 (+ 1.94); IR (ATR) νmax 3235 (br), 2937, 2551, 1685, 1509 cm−1; 1H NMR and 13C NMR (Tables 1 and 2 and Supporting Information); EIMS m/z 222.1 [M]+; HRESIMS m/z 245.1162 [M+Na]+ (calcd. for C13H18NaO3, m/z 245.1148).
Stachyline D (4): white amorphous solid (180 μg/L 0.09 %); [α] D23- 6 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 205 nm (3.86), 226 nm (3.86), 276 nm (3.08), 283 nm (2.90); IR (ATR) νmax 3265 (br), 1687, 1512 cm−1; 1H NMR and 13C NMR (Tables 1 and 2 and Supporting Information); ESIMS m/z 237.1 [M-H]−; HRESIMS m/z 261.1096 [M+Na]+ (calcd. for C13H18NaO4, m/z 261.1097).
Preparation of the (R)- and (S)-MTPA esters of compound 2
The secondary alcohol (1 eq, 1 mg, 4.23 × 10−6 μmol) was dissolved with the corresponding MTPA-Cl (10 eq) in an adequate quantity of deuterated pyridine and DMAP (1 eq) in an NMR tube. The reaction was followed by 1H-NMR to observe the downfield proton shift of H-2’. After 1 hour reaction time, the final 1H-NMR was recorded, the sample dried and re-dissolved in D-acetone for 1H-13C HSQC measurements. The Δ(S-R) values between (S)- and (R)- MTPA esters were recorded with the help of both 1H-NMR and 1H-13C HSQC (see Figure 1 and Supporting Information).
Bioassays
The referred compounds were tested in antibacterial (Escherichia coli, Bacillus megaterium), antifungal (Mycotypha microspora, Eurotium rubrum, and Microbotryum violaceum), and antialgal (Chlorella fusca) assays,25,26 protein kinases DYRK1A and CDK5 inhibition assays,27 HIV-1 and HIV-2 virus assays,28,29 and in the cytotoxic activity assay against a panel of 5 tumor cell lines, NCI-H460/lung, A549/lung, MCF7/breast and SF268/CNS and CAKI/renal.30,31 Compounds were further tested for antiplasmodial activity against Plasmodium berghei,32 Influenza B virus (Flu B) assay,33 in binding assays against a panel of 44 psychoactive receptors,34 for the inhibition of the NF-κB protein complex assay35 and in a panel of assays towards anti-diabetic activity.36–38
Supplementary Material
Acknowledgments
We thank the kind help of H. Greve, E. Eguereva and A. Kralj and for lab support; we also kindly thank the bioactivity assay efforts of Dr. K. Dimas (Biomedical Research Foundation of Academy of Athens, Greece), Dr. L. Meijer (CNRS, Roscoff, France), Dr. C. Pannecouque (Rega Institute for Medical Research, Leuven, Belgium), Dr. M. Prudêncio (Institute for Molecular Medicine, University of Lisbon, Portugal) and the U.S. National Institute of Health drug discovery program, NIAID; we kindly thank Indra Bergval (Royal Tropical Institute, Amsterdam, Netherlands), Dr. Marc Diederich (Laboratoire de Biologie Moleculaire et Cellulaire du Cancer (LBMCC), Luxembourg) and Dr. Steinar Paulsen (University of Tromsø, MabCent, Norway); we kindly thank Dr Brian Roth and Dr Jamie Driscol from the National Institute of Mental Health's Psychoactive Drug Screening Program (NIMH/PDSP; Inf.); finally we kindly thank the financial support from FCT (Science and Technology Foundation, Portugal); for detailed acknowledgment please see Supporting Information.
Footnotes
Supporting Information Available. Spectroscopic data and other relevant information are included for the new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Tziveleka LA, Constantinos V, Vassilios R. Curr Top Med Chem. 2003;3:1512–1535. doi: 10.2174/1568026033451790. [DOI] [PubMed] [Google Scholar]
- 2.Bhadury P, Mohammad BT, Wright PC. J Ind Microbiol Biotechnol. 2006;33:325–337. doi: 10.1007/s10295-005-0070-3. [DOI] [PubMed] [Google Scholar]
- 3.Sashidhara KV, White KN, Crews P. J Nat Prod. 2009;72:588–603. doi: 10.1021/np800817y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stierle AA, Stierle DB, Goldstein E, Parker K, Bugni T, Baarson C, Gress J, Blake D. J Nat Prod. 2003;66:1097–1100. doi: 10.1021/np030044w. [DOI] [PubMed] [Google Scholar]
- 5.Ingavat N, Dobereiner J, Wiyakrutta S, Mahidol C, Ruchirawat S, Kittakoop P. J Nat Prod. 2009;72:2049–2052. doi: 10.1021/np9003883. [DOI] [PubMed] [Google Scholar]
- 6.Hashimoto M, Komori T, Kamiya T. J Antibiot. 1976;29:890–901. doi: 10.7164/antibiotics.29.890. [DOI] [PubMed] [Google Scholar]
- 7.Amade P, Mallea M, Bouaïcha N. J Antibiot. 1993;47:201–207. doi: 10.7164/antibiotics.47.201. [DOI] [PubMed] [Google Scholar]
- 8.Moya P, Castillo M, Primo-Yúfera E, Couillaud F, Martínez-Máñez R, Garcerá MD, Miranda MA, Primo J, Martínez-Pardo R. J Org Chem. 1997;62:8544–8545. doi: 10.1021/jo970397y. [DOI] [PubMed] [Google Scholar]
- 9.Shindo K, Yiuji Y, Yukiko O, Kawai H. J Antibiot. 1994;48:1072–1074. doi: 10.7164/antibiotics.47.1072. [DOI] [PubMed] [Google Scholar]
- 10.Cimino G, de Stefano S, Minale L. Experientia. 1975;31:756–757. [Google Scholar]
- 11.Isaaki Y, Matsunaga S, Fusetani N. Tetrahedron. 1993;49:3749–3754. [Google Scholar]
- 12.Bjarnholt N, MØller BL. Phytochemistry. 2008;69:1947–1961. doi: 10.1016/j.phytochem.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 13.Kelly WL, Townsend CA. J Am Chem Soc. 2002;124:8186–8187. doi: 10.1021/ja025926g. [DOI] [PubMed] [Google Scholar]
- 14.Kelly WL, Townsend CA. J Bacteriol. 2005;187:739–746. doi: 10.1128/JB.187.2.739-746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider G, Anke H, Sterner O. Z Naturforsch C. 1996;51:802–806. doi: 10.1515/znc-1996-11-1206. [DOI] [PubMed] [Google Scholar]
- 16.Nair MG, Burke BA. Phytochemistry. 1988;27:3169–3173. [Google Scholar]
- 17.Barrero AF, Oltra JE, Poyatos JA. Phytochemistry. 1996;42:427–1433. [Google Scholar]
- 18.Chen YS. Bull Agr Chem Soc Japan. 1958;22:136–142. [Google Scholar]
- 19.Crowden RK, Ralph BJ. Aust J Chem. 1961;14:475. [Google Scholar]
- 20.Calcul L, Inman WD, Morris AA, Tenney K, Ratnam J, McKerrow JH, Valeriote FA, Crews P. J Nat Prod. 2010;73:365–72. doi: 10.1021/np9005986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flemning FF. Nat Prod Rep. 1999;16:597–606. [Google Scholar]
- 22.Akazawa T, Miljanich P, Conn EE. Plant Physiol. 1960;35:535–538. doi: 10.1104/pp.35.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sibbesen O, Koch B, Halkier BA, Moller BL. J Biol Chem. 1995;270:3506–3511. doi: 10.1074/jbc.270.8.3506. [DOI] [PubMed] [Google Scholar]
- 24.Tennant G. In: Comprehensive Organic Chemistry, The Synthesis and Reaction of Organic Compounds. Barton D, Ollis WD, editors. 2: Chapter 8. Pergamon Press; Oxford: 1979. pp. 383–590. [Google Scholar]
- 25.Schulz B, Boyle C, Draeger S, Rommert AK, Krohn K. Mycol Res. 2002;106:996–1004. [Google Scholar]
- 26.Schulz B, Sucker J, Aust HJ, Krohn K, Ludewig K, Jones PG, Döring D. Mycol Res. 1995;99:1007–1015. [Google Scholar]
- 27.Bettayeb K, Oumata N, Echalier A, Ferandin Y, Endicott JA, Galons H, Meijer L. Oncogene. 2008;27:5797–5807. doi: 10.1038/onc.2008.191. [DOI] [PubMed] [Google Scholar]
- 28.Pannecouque C, Daelemans D, De Clercq E. Nat Protoc. 2008;3:427–434. doi: 10.1038/nprot.2007.517. [DOI] [PubMed] [Google Scholar]
- 29.Zhan P, Liu X, Fang Z, Pannecouque C, De Clercq E. Bioorg Med Chem. 2009;17:6374–6379. doi: 10.1016/j.bmc.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 30.Saroglou V, Karioti A, Demetzos C, Dimas K, Skaltsa H. J Nat Prod. 2005;68:1404–1407. doi: 10.1021/np058042u. [DOI] [PubMed] [Google Scholar]
- 31.Monks A, Scudiero D, Skehan P, Shomaker R, Paull K, Vistica D, Hose C. J Natl Cancer Inst. 1991;83:661–757. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 32.Prudêncio M, Rodrigues CD, Ataide R, Mota MM. Cell Microbiol. 2008;10:218–224. doi: 10.1111/j.1462-5822.2007.01032.x. [DOI] [PubMed] [Google Scholar]
- 33.Sidwell RW, Smee DF. Antiviral Res. 2000;48:1–16. doi: 10.1016/s0166-3542(00)00125-x. [DOI] [PubMed] [Google Scholar]
- 34.Psychoactive receptors: For experimental details. see http://pdsp.med.unc.edu/UNC-CH%20Protocol%20Book.pdf.
- 35.Schumacher M, Cerella C, Eifes C, Chateauvieux S, Morceau F, Jaspars M, Dicato M, Diederich M. Biochem Pharmacol. 2010;79:610–622. doi: 10.1016/j.bcp.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 36.Marrapodi M, Chiang JYL. J Lipid Res. 2000;41:514–520. [PubMed] [Google Scholar]
- 37.Dey D, Pall BC, Biswas T, Roy SS, Bandyopadhyay A, Mandal SK, Giri BB, Bhattacharya S. Mol Cell Biochem. 2007;300:149–157. doi: 10.1007/s11010-006-9378-1. [DOI] [PubMed] [Google Scholar]
- 38.Seale AP, de Jesus LA, Kim SY, Choi YH, Lim HB, Hwang CS, Kim YS. Biotechnol Lett. 2005;27:221–225. doi: 10.1007/s10529-004-7855-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.