

Abstract
The first committed step of lipid A biosynthesis is catalyzed by UDP-(3-O-(R-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase, a metal-dependent deacetylase also known as LpxC. Because lipid A is essential for bacterial viability, the inhibition of LpxC is an appealing therapeutic strategy for the treatment of Gram-negative bacterial infections. Here we report the 1.79 Å resolution X-ray crystal structure of LpxC from Yersinia enterocolitica (YeLpxC) complexed with the potent hydroxamate inhibitor CHIR-090. This enzyme is a nearly-identical orthologue of LpxC from Yersinia pestis (99.7% sequence identity), the pathogen that causes bubonic plague. Similar to the inhibition of LpxC from Escherichia coli, CHIR-090 inhibits YeLpxC via a two-step slow, tight-binding mechanism with an apparent Ki of 0.54 ± 0.14 nM followed by conversion of the E•I to E•I* species with a rate constant of 0.11 ± 0.01 min−1. The structure of the LpxC complex with CHIR-090 shows that the inhibitor hydroxamate group chelates the active site zinc ion, and the “tail” of the inhibitor binds in the hydrophobic tunnel in the active site. This hydrophobic tunnel is framed by a βαβ subdomain that exhibits significant conformational flexibility as it accommodates inhibitor binding. CHIR-090 displays a 27 mm zone of inhibition against Y. enterocolitica in a Kirby-Bauer antibiotic assay, which is comparable to its reported activity against other Gram-negative species including E. coli and Pseudomonas aeruginosa. This study demonstrates that the inhibition of LpxC should be explored as a potential therapeutic and/or prophylatic response to infection by weaponized Yersinia species.
Septic shock is one of the leading causes of death in hospital intensive care units and results from severe hypotension and multiple organ failure associated with bacterial sepsis. Sepsis is a systemic inflammatory response to infection: specifically, macrophage activation by endotoxin, which is also known as lipopolysaccharide (LPS). Approximately 106 lipopolysaccharide molecules constitute the outer leaflet of the outer membrane of a Gram-negative bacterium (1). This “protective sheath” makes Gram-negative bacteria impermeable to many commercial antibiotics.
Each LPS molecule contains an immunodominant and highly variable repeating oligosaccharide known as the O-antigen, a core polysaccharide, and lipid A, a hydrophobic membrane anchor typically bearing six fatty acids. Lipid A is required for proper LPS assembly and cell viability, and bacteria with defective lipid A biosynthetic pathways are markedly more sensitive to antibiotics (2–5). Given that lipid A is essential for bacterial survival, and is also the toxic component of LPS, inhibition of the lipid A biosynthetic pathway will not only kill Gram-negative bacteria, but may also reduce the concentrations of immunoreactive lipid A shed from dying cells. Accordingly, inhibitors of this pathway may represent leads for the development of more potent antibiotics for the treatment of sepsis and septic shock. Additionally, such inhibitors could be developed as therapeutic and/or prophylatic drugs against weaponizable bacteria capable of causing more severe infections, such as Yersinia pestis, which causes bubonic plague.
The first committed step of lipid A biosynthesis is catalyzed by UDP-(3-O-(R-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase, a metal-dependent deacetylase also known as LpxC (Figure 1A) (6–9). X-ray crystal structures have been reported for LpxC from the Gram-negative bacteria Aquifex aeolicus (AaLpxC) (10) and Pseudomonas aeruginosa (PaLpxC) (11), as well as various ligand complexes of AaLpxC (12–16). The structure of AaLpxC has also been determined in solution by NMR methods (17, 18), and the NMR structure of its complex with the potent, slow-binding hydroxamate inhibitor CHIR-090 (Figure 1B) has been reported (19, 20). These studies have illuminated active site features important for catalysis, including a hydrophobic tunnel framed by a small, flexible βαβ domain that accommodates the 3-hydroxymyristoyl group of the substrate. No structures of LpxC enzymes from weaponizable Gram-negative pathogens have been determined to date, however, and such information is vital as we contemplate the response to potential bioterrorism threats in the twenty-first century.
Figure 1.

(A) Reaction catalyzed by LpxC and the structure of lipid A from E. coli. Reproduced from reference 10. (B) Structure of CHIR-090.
Here, we report the X-ray crystal structure of LpxC from Yersinia enterocolitica, the Gram-negative bacterium that causes yersiniosis. Although yersiniosis can resolve itself without antibiotic treatment (21), LpxC from Y. enterocolitica (YeLpxC) serves as a readily studied and nearly identical orthologue of LpxC from Y. pestis. The 306 amino acid sequence of YeLpxC shares 99.7% amino acid sequence identity (only one substitution, R174C) with LpxC from Y. pestis. The structure reported here is that of the YeLpxC-CHIR-090 complex. CHIR-090 is a slow, tight-binding inhibitor that was originally reported in a patent describing new antimicrobial agents (22), and later studied for activity against LpxC enzymes from A. aeolicus and E. coli (20, 23). In this study, CHIR-090 inhibits YeLpxC with an apparent initial Ki of 0.54 ± 0.14 nM and a rate constant for a second step (conversion of E•I to E•I*) of 0.11 ± 0.01 min−1; this inhibition is slightly more potent compared to the values reported for inhibition of AaLpxC and E. coli LpxC (EcLpxC) (20, 23). Analysis of the enzyme-inhibitor complex yields further structural insight toward the development of more potent and broad-specificity inhibitors against a wide range of Gram-negative pathogens and suggests that CHIR-090 is well suited for development as a broad-spectrum Gram-negative antibiotic.
Materials and Methods
Cloning and Protein Expression
The LpxC gene was amplified from Y. enterocolitica genomic DNA by PCR using the following primers: Forward: 5′-GCGCGGCATATGATCAAACAAAGGACTTTAAAACG-3′ and Reverse: 5′-GTCATAGGATCCTTAGTAAGCTAATACTGTAGACG-3′ with the NheI and BamHI restriction sites bolded, respectively. The amplified insert was cloned into the pET11a vector cut at the same restriction sites and transformed into BL21(DE3)pLysS cells. Transformants were grown overnight on ampicillin- and chloramphenicol-selective agar plates. The DNA from the resulting transformants was isolated and sequenced (DNA Sequencing Facility, University of Pennsylvania).
YeLpxC was recombinantly expressed in BL21(DE3)pLysS E. coli cells transformed with the pET11a-YeLpxC plasmid and purified according to procedures published for the purification of EcLpxC and AaLpxC (9, 10), with minor modifications. Briefly, cells were grown in rich medium supplemented with glucose (10 mL/L 40% (w/v)), ampicillin (50 μg/L), and chloramphenicol (34 μg/L) at 37 °C until OD600 ~ 0.5, at which point the cells were induced by addition of isopropyl β-D-thiogalactopyranoside (1 mM final concentration) and grown overnight at 25 °C. The cells were pelleted by centrifugation, resuspended in 25 mL of lysis buffer (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.0), 2 mM MgCl2, 2 mM dithiothreitol (DTT), and 2 mM phenylmethanesulfonyl fluoride), and lysed by sonication on ice. Cell debris was pelleted by centrifugation and the cell-free extract was purified by using ion exchange chromatography (HiTrap QXL, GE Healthcare) with a salt gradient (Buffer A: 25 mM HEPES (pH 7.0), 2 mM DTT; Buffer B: 25 mM HEPES (pH 7.0), 400 mM NaCl, 2 mM DTT) and size exclusion chromatography (HiLoad 26/60 Superdex, GE Healthcare) in 25 mM HEPES (pH 7.0) and 50 mM NaCl. The final yield was ~60 mg/L culture medium and the final purity was better than 95% as determined by SDS-PAGE analysis. The concentration of YeLpxC was determined from its absorbance using a calculated extinction coefficient (ε280 = 20,640 M−1cm−1) (24). The metal content of YeLpxC was determined by using inductively coupled plasma mass spectrometry (ICP-MS, University of Michigan Geological Sciences).
Structure Determination and Refinement
Diamond-shaped crystals of YeLpxC complexed with the CHIR-090 inhibitor were obtained in 1–2 weeks using the hanging drop vapor diffusion method with the following conditions: 2 μL of protein solution [10 mg/mL YeLpxC (25 mM HEPES (pH 7.0), 50 mM NaCl), 2 mM tris-(2-carboxyethyl)-phosphine (TCEP), and 1.45 mM CHIR-090] were mixed with 2 μL of precipitant solution [0.1 M Tris (pH 8.5), 0.2 M MgCl2, and 25% PEG 3350] and equilibrated against a 600 μL reservoir of precipitant solution. A stock solution of CHIR-090 (40 mM) was prepared by dissolving the compound in dimethyl sulfoxide (DMSO). This solution was added to the protein solution such that the final concentration of CHIR-090 was 1.45 mM and the final concentration of DMSO was 4%.
Larger single crystals were obtained with both micro- and macroseeding in 1–3 days. Crystals were harvested and flash-cooled using the precipitant buffer supplemented with 20% glycerol. Crystals diffracted X-rays to 1.79 Å resolution at the Advanced Photon Source, beamline NE-CAT 24-ID-E (Argonne National Lab). Diffraction data were indexed and scaled using HLK2000 (25). Crystals belonged to space group P1 with unit cell dimensions a = 46.733 Å, b = 47.511 Å, c = 68.265 Å, α = 74.478°, β = 87.883°, γ = 68.966°; with two monomers in the unit cell, the Matthews coefficient of 2.0 Å3/Da corresponds to a solvent content of 38.5%.
The structure was solved by molecular replacement (26) using the structure of PaLpxC (55% sequence identity, PDB code: 2VES, less ions and solvent) as a search probe in rotation and translation function calculations. Iterative cycles of refinement and model building were performed with CNS (27) and Phenix (28), and Coot (29), respectively, to improve the structure as monitored by Rfree. Atomic coordinates for CHIR-090 and solvent molecules were added during the last stages of refinement. Disordered segments in the final model include A299-Y306 in monomer A, and S301-Y306 in monomer B. Data collection and refinement statistics are recorded in Table 1.
Table 1.
Data collection and refinement
| Data Collection | |
| Wavelength (Å) | 0.97917 |
| Resolution (Å) | 1.79 |
| No. total reflections | 92790 |
| No. unique reflections | 47640 |
| I/σ | 15.9 (2.5)a |
| Redundancy | 2.4 (2.4)a |
| Completeness (%) | 97.1 (95.7)a |
| Rmergeb | 0.060 (0.435)a |
| Refinement | |
| R/Rfreec | 0.167/0.211 |
| No. atoms/ionsd | |
| Protein atoms | 4751 |
| Zinc ions | 2 |
| Ligand atoms | 64 |
| Water molecules | 404 |
| Root-mean-square deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.1 |
| Average B-factors | |
| Protein atoms | 24 |
| Zinc ions | 18 |
| Ligand atoms | 22 |
| Water molecules | 32 |
| Ramachandran plot (%) | |
| Most favored | 89.9 |
| Additional allowed | 9.7 |
| Generously allowed | 0.4 |
| Disallowed | 0 |
Number in parentheses refers to the outer 0.1 Å shell of data.
Rmerge = Σ|I − 〈I〉|/Σ|〈I〉|, where I is the observed intensity and 〈I〉 is the average intensity calculated for replicate data.
Crystallographic R factor, R = Σ(|F| − |F|)/Σ|F|,
Activity and Inhibition Assays
Apo-YeLpxC was incubated with ethylenediaminetetraacetic acid and dipicolinic acid to remove endogenous metal ions and reconstituted with stoichiometric ZnSO4, as previously described (15). Apo- and holo-LpxC metal content was confirmed by ICP-MS. Steady-state turnover assays were performed as described previously for E. coli LpxC (15). Briefly, assays contained 1 nM Zn2+-YeLpxC, 2 mM TCEP, 1 mg/mL bovine serum albumin (BSA), 20 mM Bis-Tris (pH 7.5), and varied concentrations of the substrate 14C-UDP-myristoyl-N-acetylglucosmine (0.05–4 μM) at 30° C. Assays were initiated by addition of YeLpxC and quenched by addition of 1.25 M sodium hydroxide. The product was separated from the substrate on PEI-cellulose TLC plates (0.1 M guanidinium HCl), and quantified by scintillation counting, as previously described (15). The kinetic parameters (kcat, KM, and kcat/KM) were determined from a fit of the Michaelis-Menten equation to the substrate-dependence of the initial velocity data. Inhibition assays were initiated by addition of enzyme (0.1 nM YeLpxC) to an assay containing 200 nM 14C-UDP-myristoyl-N-acetylglucosmine and the CHIR-090 inhibitor (0.0–2.0 nM) with the DMSO concentration maintained at 0.01% and inhibition was monitored using progress curves. The substrate concentration (0.2 μM) is 2-fold below the measured KM so the assays were carried out under kcat/KM conditions. The reaction was quenched by addition of 1.25 M sodium hydroxide and product analyzed as described above. The progress curves showing time-dependent inhibition were analyzed using an equation describing slow, tight-binding inhibition (Eq. 1, Scheme 1 in Results) (30, 31) as previously observed for CHIR-090 (19, 20, 23). The R-values for the progress curve fits were 0.96–0.99. Two parameters with standard errors are calculated from each progress curves: the initial velocity and the observed rate constant for the time-dependent inhibition. The value of Ki for the initial binding step is determined from fitting equation 2 (Results section) to the dependence of the initial velocities on the concentration of CHIR-090 and the value of the kobsmax is calculated from a fit of equation 3 (Results section) to the concentration-dependence of the observed rate constants (kobs). The kinetic parameters and standard error were determined by fitting these equations to the kinetic data using either Kaleidagraph v3.6 (Synergy Software) or Prism v4.0 (GraphPad).
Scheme 1.

Kirby-Bauer Antibiotic Assay
The antimicrobial activity of CHIR-090 against Y. enterocolitica was studied using the disk diffusion antibiotic sensitivity method (ATS Labs, Eagan, MN) (32). Briefly, Y. enterocolitica was grown on Tryptic Soy agar plates supplemented with 5% sheep blood at 37 °C for 2–3 days. Cells harvested from this plate were used to inoculate a Mueller Hinton agar plate supplemented with sheep blood. The CHIR-090 inhibitor was added (2 μL of 10 mg/mL solution) to a sterile 6 mm paper disk, and the plate was incubated at 37 °C for 2 days. The halo of killing was measured to the nearest millimeter. DMSO was used as a negative control and showed no effect against Y. enterocolitica.
Results and Discussion
CHIR-090 is a Slow, Tight-Binding Inhibitor of YeLpxC
The steady state kinetics for YeLpxC were measured as a function of substrate concentration (Figure 2A). The steady state kinetic parameters, listed in Table 2, are very similar to the mechanistically well-characterized EcLpxC, suggesting that these enzymes have similar rate-determining steps and likely share mechanistic features.
Figure 2.

Steady-state kinetics and inhibition of YeLpxC. (A) Steady-state turnover. Deacetylation of 14C-UDP-myristoyl-N-acetylglucosmine catalyzed by Zn2+-YeLpxC was assayed in 20 mM Bis-Tris (pH 7.5), 2 mM TCEP, 1 mg/mL BSA at 30° C as described in Materials and Methods. (B) Time-dependent inhibition by CHIR-090. The inhibitor CHIR-090 (0.0 nM (open circles), 0.2 nM (closed triangles), 0.4 nM (open squares), 1 nM (closed circles), and 2 nM (open triangles)) was incubated with substrate before addition of 0.1 nM Zn2+-YeLpxC and measurement of product formation. Equation 1 was fit to the progress curves (R = 0.96–0.99). (C) Measurement of inhibition constant (Ki). The value of Ki was obtained from a fit of equation 2 to the dependence of the initial velocities (obtained from the progress curves) on the concentration of CHIR-090 (R = 0.92). (D) Time-dependence of inhibition. The value of the maximal rate constant for time-dependent inhibition was calculated from a fit of equation 3 to the concentration dependence of the observed rate constants (kobs).
Table 2.
Steady-state kinetic parameters and inhibition of LpxC enzymesa
| Species | Kcat (min−1) | KM (μM) | Kcat/KM (min−1 μM−1) | Ki (nM) | Kobsmax (min−1) |
|---|---|---|---|---|---|
| E. coli | 90 ± 2b | 0.19 ± 0.01b | 460 ± 10b | 4.0 ± 1c | 1.9 ± 0.3c |
| A. aeolicus | 104e | 1.6 ± 0.2d | 65 ± 11b | 1.0 – 1.7d | 0.19d |
| Y. enterocolitica | 104 ± 3f | 0.40 ± 0.05f | 260 ± 24f | 0.54 ± 0.14f | 0.11 ± 0.01f |
All steady-state parameters and inhibition constants reported were measured at 30 °C, pH 7.5 as described in Materials and Methods or the works cited.
Adapted from reference 14.
Adapted from reference 21. In this species, k6 is significant, and the overall inhibition constant, Ki*, is 0.4 ± 0.1 nM.
Adapted from reference 19.
This work.
CHIR-090 has previously been observed to be a slow, tight-binding inhibitor of AaLpxC and EcLpxC that is competitive with substrate and does not form a covalent complex (19, 20, 23). Similarly, CHIR-090 exhibits time-dependent inhibition with YeLpxC (Figure 2B). Most commonly, time-dependent inhibition arises from relatively rapid and reversible binding of inhibitor to form an E•I complex, followed by a slower isomerization to form the E•I* complex (Scheme 1) (30, 31). This mode of time-dependent inhibition can be distinguished from a very slow binding one-step inhibitor by a decrease in the initial velocity of the reaction as the inhibitor concentration increases due to formation of the E•I complex.
The progress curves for inhibition of YeLpxC (representative curves are shown in Figure 2B) were analyzed using equation 1, where vs is the final steady state velocity, v0 is the initial velocity, and kobs is the rate constant for the time-dependent inhibition reflecting formation of the E•I* complex. Initial fits of this equation to the data indicated that the final steady-state velocity was negligible compared to the initial velocity; therefore the data were analyzed with vs set to 0. This result indicates that the conversion of E•I to E•I* is essentially irreversible under these conditions, as similarly observed for inhibition of AaLpxC by CHIR-090 (20).
| Equation 1 |
The initial velocity, v0, catalyzed by YeLpxC decreases at higher concentrations of CHIR-090, diagnostic of a two-step inhibitor binding scheme (Figure 2C). The apparent Ki for the reversible first step is calculated to be 0.54 ± 0.14 nM from a fit of equation 2 to these data. The apparent rate constant for formation of E•I*, kobs, shows a hyperbolic dependence on the inhibitor concentration. The rate constant for conversion of E•I to E•I* at saturating inhibitor concentration (kobsmax = k5) is calculated as 0.11 ± 0.01 min−1 from a fit of equation 3 to these data, assuming that formation of EI* is essentially irreversible (k6 ≪ k5 in Eq. 3) (Figure 2D). Thus, the half-time for formation of the inhibited complex is ~ 6 minutes. In summary, these data indicate that YeLpxC is inhibited by CHIR-090 in a two-step mechanism as a slow, tight-binding inhibitor.
| Equation 2 |
| Equation 3 |
Slow, tight-binding inhibition is desirable in drug development due to the longer effective half-life of the compound in biological systems (31). Unlike purely competitive inhibitors, accumulation of substrate does not result in a rapid decrease in inhibition, as the slow dissociation of inhibitor is a unimolecular process. In comparison to AaLpxC and EcLpxC, the value of Ki for inhibition of YeLpxC by CHIR-090 is 4- to 10-fold lower and the rate constant for conversion of E•I to E•I* (k5) is 2- to 20-fold slower (AaLpxC: 1.7 nM, k5 = 0.19 min−1; EcLpxC: 4 nM, k5 = 1.9 min−1) (20, 23). In contrast, LpxC from Rizobium leguminosarum is only modestly inhibited by CHIR-090 (Ki ~0.3 μM) and exhibits no time dependence, perhaps due to structural differences in the hydrophobic tunnel (23). YeLpxC is markedly more sensitive in vitro to CHIR-090 than either AaLpxC or EcLpxC, suggesting that this compound may be useful as an antibacterial agent against Y. enterocolitica and also Y. pestis.
Structure of the LpxC-CHIR-090 Complex
The two independent monomers of YeLpxC in the P1 unit cell are structurally quite similar, with r.m.s. deviations of 0.34 Å for 298 Cα atoms and 0.78 Å for all atoms (33). The structure of monomer A is shown in Figure 3. The crystal structure of YeLpxC is also similar to the structures of AaLpxC and PaLpxC. The root mean square (r.m.s.) deviation of 266 Cα atoms between YeLpxC and AaLpxC (10) is 2.27 Å; the r.m.s. deviation of 295 Cα atoms between YeLpxC and PaLpxC (11) is 1.36 Å. The catalytic zinc ion of YeLpxC is coordinated by H79, H238, and D242, which are conserved among LpxC enzymes from different species. The square pyramidal coordination geometry of zinc is completed by the hydroxamate moiety of CHIR-090.
Figure 3.

Stereoview of the YeLpxC-CHIR-090 complex. The catalytic zinc ion (red) is coordinated by H79, H238, and D242 (blue sticks). CHIR-090 is shown as a stick figure (C = green, N = blue, and O = red).
The electron density map of the YeLpxC-CHIR-090 complex clearly shows that the inhibitor binds in the hydrophobic tunnel. The inhibitor is bound with full occupancy, with thermal B factors comparable to those of surrounding residues (Figure 4A). Binding in the hydrophobic tunnel was also observed in the NMR structure of the AaLpxC-CHIR-090 complex (19). The hydroxamate moiety chelates the catalytic zinc ion, which is also bound with full occupancy, with a thermal B factor comparable to B factors of ligand residues. The hydroxamate moiety also makes polar interactions with E78, T191, and H265. Interestingly, the threonine moiety of the inhibitor adopts a different conformation from that observed in the NMR structure of the AaLpxC-CHIR-090 complex (Figure 4B) (19). In the structure of the YeLpxC-CHIR-090 complex, both the carbonyl and the backbone amide group of the inhibitor threonine moiety are positioned to make hydrogen bonds with T191 and the carbonyl of F192. The threonine methyl group also makes van der Waals interactions with the backbone carbonyl of F192 and a water molecule. The two aromatic rings and the alkyne moiety of CHIR-090 make van der Waals interactions with several residues in the hydrophobic tunnel defined by the βαβ domain, as well as residues flanking this domain, including T191, F192, I198, C207, G210, S211, A215, and V217. Finally, the morpholino ring hydrogen bonds with a water molecule and makes van der Waals interactions with M195 and F212. The morpholino ring refined well in both chair and boat conformations, as shown in Figure 4A; both conformations are included in the final structure with half occupancy. These interactions with the hydrophobic tunnel are similar to those observed in the solution NMR structure of AaLpxC (19). In that structure, the orientation of CHIR-090 is slightly different so as to allow the morpholino ring to make additional contacts with I186, K190, and Y212 (AaLpxC numbering).
Figure 4.

(A) Simulated annealing omit map contoured at 4.0 σ showing the CHIR-090 inhibitor in the hydrophobic tunnel. Both chair and boat conformations of the morpholino ring refined well into the electron density map, so each are included in the final model with half occupancy. (B) Superposition of CHIR-090 in YeLpxC (green stick figure) and the CHIR-090 binding conformation observed in the NMR structure of the AaLpxC-CHIR-090 complex (orange stick figure).
Many of the intermolecular interactions observed in the YeLpxC-CHIR-090 complex are conserved in the structure of AaLpxC complexed with palmitate (15). In the AaLpxC-palmitate complex, the metal-chelating carboxylate makes polar interactions with E78, H265, and T191, as does the hydroxamate moiety of CHIR-090. Like the aromatic rings and alkyne functionalities of CHIR-090, the hydrophobic tail of palmitate also makes van der Waals interactions with I18, I198, G210, S211, T215, and V217. Palmitate additionally makes non-polar interactions with H58 and T191.
An important feature that is evident in the comparison of the three LpxC crystal structures is the variable conformation, and hence, apparent flexibility of the βαβ domain, which forms one wall of the active site (Figure 5). Consistent with this observation, NMR measurements indicate that the βαβ domain is highly disordered in the absence of bound inhibitor (17). The binding of CHIR-090 stabilizes the βαβ domain in a well-defined conformation and the slow step that occurs after inhibitor binding may reflect reorganization of this domain. The flexibility of the βαβ domain may be a useful feature to be exploited in the design of inhibitors that will bind tightly to multiple LpxC orthologues. Despite varying degrees of overall amino acid sequence identity, ranging from 31–92%, much of the amino acid sequence of the βαβ domain is largely conserved among many species of Gram-negative bacteria, particularly those classified by the Centers for Disease Control as Class A and Class B bioterrorism agents (Figure 6) (34). The amino acid side chains that interact with the CHIR-090 inhibitor are also highly conserved among these species. Along with the catalytic metal ion, many of the residues in the active site and hydrophobic tunnel (including E78, T191, F192 and H265) are important for the catalytic activity and product affinity of EcLpxC and/or AaLpxC (15, 35), suggesting that the inhibitor makes interactions similar to those of the substrate and product. Taken together, these data strongly suggest that the hydrophobic tunnel should be a primary target for the development of more potent, broad-specificity inhibitors (10, 16, 19).
Figure 5.

(A) Superposition of YeLpxC (cyan), AaLpxC (magenta), and PaLpxC (blue). (B) Close-up view of the βαβ domain with the catalytic zinc ion shown as a red sphere and the CHIR-090 inhibitor shown as a green stick figure. This view highlights the apparent left-to-right movement of the βαβ domain, as well as the flexibility in-and-out of the plane of the paper.
Figure 6.

Partial sequence alignment of LpxC (residues 187–222, YeLpxC numbering) from selected Gram-negative bacteria. Fully conserved residues are blue and largely conserved residues are green. The residues that interact with the CHIR-090 inhibitor through van der Waals or hydrogen bond interactions are underlined. A cartoon of the secondary structure highlights the βαβ domain (arrows represent the β-strands and the cylinder represents the short α-helix).
Kirby-Bauer Antibiotic Assay
Zone of inhibition assays (32) show that CHIR-090 is a potent inhibitor of Y. enterocolitica; a 27 mm halo of killing was visible around the disk inoculated with CHIR-090 (Figure 7A). As a negative control, DMSO showed no antimicrobial activity. This result is comparable to previously reported inhibition against E. coli and P. aeruginosa; CHIR-090 displayed a 36 mm halo of killing against wild-type E. coli, 38 mm against inhibitor hypersensitive E. coli, and 34 mm against P. aeruginosa (20). The antimicrobial activity of CHIR-090 is also comparable to the activities of the commercially-available antibiotics, tobramycin and ciprofloxacin (20). These results further confirm that CHIR-090 exhibits appreciable inhibitory potency against a number of Gram-negative bacterial strains and, therefore, may serve as a model for the development of more potent, broad-specificity inhibitors.
Figure 7.
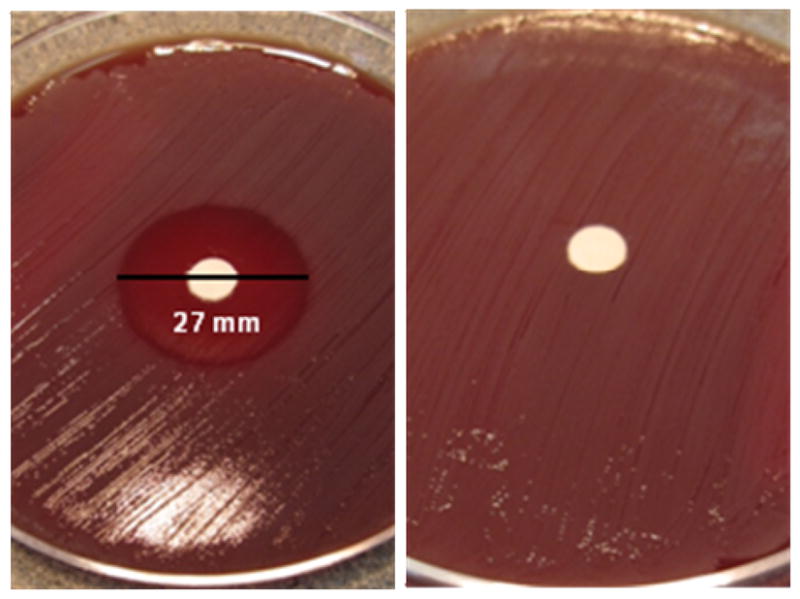
Kirby-Bauer antibiotic assay. (A) CHIR-090 displays a 27 mm zone of killing against Y. enterocolitica. (B) DMSO as the negative control.
Concluding Remarks
The structure of the YeLpxC-CHIR-090 complex is the highest resolution structure of LpxC determined to date and the first X-ray crystal structure of LpxC complexed with the potent hydroxamate inhibitor CHIR-090. Inhibition assays reveal a two-step mechanism diagnostic of a slow, tight-binding inhibitor in which the inhibition constant of the E•I complex (Ki = 0.54 ± 0.14 nM) is more potent and the rate constant for the time-dependent inhibition (kobsmax = 0.11 ± 0.01 min−1) is faster than observed with enzymes from other species (i.e. AaLpxC and EcLpxC). Consistent with this, CHIR-090 exhibits a 27 mm halo of killing in a Kirby-Bauer antibiotic assay against Y. enterocolitica. Since Y. enterocolitica is an insidious Gram-negative pathogen and LpxC from this species is a nearly identical orthologue to that of Y. pestis, the bacterium that causes bubonic plague, structure-function relationships now established for YeLpxC provide a foundation for future studies aimed at developing potent, broad-specificity inhibitors of LpxC enzymes from weaponizable Gram-negative pathogens.
Acknowledgments
This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We thank Dr. Christian Raetz and Achaogen for providing samples of CHIR-090 and Dr. Ted Huston and Caleb Joseph for assistance with ICP-MS. We also thank Dr. Daniel Dowling and Dr. Mustafa Koksal for helpful discussions.
- AaLpxC
Aquifex aeolicus LpxC
- BSA
bovine serum albumin
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- EcLpxC
Escherichia coli LpxC
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ICP-MS
inductively coupled plasma mass spectrometry
- LPS
lipopolysaccharide
- PaLpxC
Pseudomonas aeruginosa LpxC
- TCEP
tris-(2-carboxyethyl)-phosphine
- YeLpxC
Yersinia enterocolitica LpxC
Footnotes
This work was supported by the National Institutes of Health (D.W.C. GM49758 and C.A.F. GM40602). S.G.G. was supported by NIGMS training grant (T32 GM007767).
The atomic coordinates and structure factors of the YeLpxC-CHIR-090 complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession code 3NZK.
References
- 1.Raetz CRH, Whitfield C. Lipopolysaccharide Endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikaido H, Vaara M. Molecular-Basis of Bacterial Outer-Membrane Permeability. Microbiol Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaara M. Outer-Membrane Permeability Barrier to Azithromycin, Clarithromycin, and Roxithromycin in Gram-Negative Enteric Bacteria. Antimicrob Agents Chemother. 1993;37:354–356. doi: 10.1128/aac.37.2.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vuorio R, Vaara M. The Lipid-A Biosynthesis Mutation LpxA2 of Escherichia coli Results in Drastic Antibiotic Supersusceptibility. Antimicrob Agents Chemother. 1992;36:826–829. doi: 10.1128/aac.36.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A Modification Systems in Gram-Negative Bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson MS, Robertson AD, Macher I, Raetz CRH. Biosynthesis of Lipid A in Escherichia coli- Identification of UDP-3-O-(R)-3-hydroxymyristoyl-α-D glucosamine as a Precursor of UDP-N2,O3-bis (R)-3-hydroxymyristoyl-α-D glucosamine. Biochemistry. 1988;27:1908–1917. doi: 10.1021/bi00406a017. [DOI] [PubMed] [Google Scholar]
- 7.Anderson MS, Bull HG, Galloway SM, Kelly TM, Mohan S, Radika K, Raetz CRH. UDP-N-acetylglucosamine acyltransferase of Escherichia coli- the First Step of Endotoxin Biosynthesis is Thermodynamically Unfavorable. J Biol Chem. 1993;268:19858–19865. [PubMed] [Google Scholar]
- 8.Young K, Silver LL, Bramhill D, Cameron P, Eveland SS, Raetz CRH, Hyland SA, Anderson MS. The envA Permeability Cell Division Gene of Escherichia coli Encodes the Second Enzyme of Lipid A Biosynthesis-UDP-3-O-(R-3-hydroxymyristoyl)-N acetylglucosamine deacetylase. J Biol Chem. 1995;270:30384–30391. doi: 10.1074/jbc.270.51.30384. [DOI] [PubMed] [Google Scholar]
- 9.Jackman JE, Raetz CRH, Fierke CA. UDP-3-O-(R-3-hydroxymyristoyl)-N acetylglucosamine deacetylase of Escherichia coli is a Zinc Metalloenzyme. Biochemistry. 1999;38:1902–1911. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- 10.Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Crystal Structure of LpxC, a Zinc-Dependent Deacetylase Essential for Endotoxin Biosynthesis. Proc Nat Acad Sci USA. 2003;100:8146–8150. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mochalkin I, Knafels JD, Lightle S. Crystal Structure of LpxC from Pseudomonas aeruginosa Complexed with the Potent BB-78485 Inhibitor. Protein Sci. 2008;17:450–457. doi: 10.1110/ps.073324108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buetow L, Dawson A, Hunter WN. The Nucleotide-Binding Site of Aquifex aeolicus LpxC. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2006;62:1082–1086. doi: 10.1107/S1744309106041893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gennadios HA, Christianson DW. Binding of Uridine 5′-Diphosphate in the “Basic Patch” of the Zinc Deacetylase LpxC and Implications for Substrate Binding. Biochemistry. 2006;45:15216–15223. doi: 10.1021/bi0619021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gennadios HA, Whittington DA, Li XC, Fierke CA, Christianson DW. Mechanistic Inferences from the Binding of Ligands to LpxC, a Metal-Dependent Deacetylase. Biochemistry. 2006;45:7940–7948. doi: 10.1021/bi060823m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase Functions through a General Acid-Base Catalyst Pair Mechanism. J Biol Chem. 2005;280:16969–16978. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- 16.Shin H, Gennadios HA, Whittington DA, Christianson DW. Amphipathic Benzoic Acid Derivatives: Synthesis and Binding in the Hydrophobic Tunnel of the Zinc Deacetylase LpxC. Bioorg Med Chem. 2007;15:2617–2623. doi: 10.1016/j.bmc.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 17.Coggins BE, Li XC, McClerren AL, Hindsgaul O, Raetz CRH, Zhou P. Structure of the LpxC Deacetylase with a Bound Substrate-Analog Inhibitor. Nat Struct Biol. 2003;10:645–651. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coggins BE, McClerren AL, Jiang L, Li XC, Rudolph J, Hindsgaul O, Raetz CRH, Zhou P. Refined Solution Structure of the LpxC-TU-514 Complex and pK(a) Analysis of an Active Site Histidine: Insights into the Mechanism and Inhibitor Design. Biochemistry. 2005;44:1114–1126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- 19.Barb AW, Jiang L, Raetz CRH, Zhou P. Structure of the Deacetylase LpxC Bound to the Antibiotic CHIR-090: Time-Dependent Inhibition and Specificity in Ligand Binding. Proc Nat Acad Sci USA. 2007;104:18433–18438. doi: 10.1073/pnas.0709412104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan ZQ, Rudolph J, Raetz CRH. A Slow, Tight-Binding Inhibitor of the Zinc-Dependent Deacetylase LpxC of Lipid A Biosynthesis with Antibiotic Activity Comparable to Ciprofloxacin. Biochemistry. 2005;44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams EC, Barker IK. In Infections Diseases of Wild Mammals. Blackwell Publishing; Ames, Iowa: 2001. [Google Scholar]
- 22.Andersen NH, Bowman J, Erwin A, Harwood E, Kline T, Mdluli K, Pfister KB, Shawar R, Wagman A, Yabannavar A. Antibacterial Agents. World Intellectua Property Organization; 2004/062601 A2. Patent WO. 2004:324.
- 23.Barb AW, McClerren AL, Snehelatha K, Reynolds CM, Zhou P, Raetz CRH. Inhibition of Lipid A Biosynthesis as the Primary Mechanism of CHIR-090 Antibiotic Activity in Escherichia coli. Biochemistry. 2007;46:3793–3802. doi: 10.1021/bi6025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gill SC, von Hippel PH. Calculation of Protein Extinction Coefficients from Amino Acid Sequence Data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr Sect D: Biol Crystallogr. 2002;58:1948–195428. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 29.Emsely P, Cowtan K. Coot: Model Building Tools for Molecular Graphics. Acta Crystallogr Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Morrison JF. The Slow-Binding and Slow, Tight-Binding Inhibition of Enzyme Catalyzed Reactions. Trends Biochem Sci. 1982;7:102–105. [Google Scholar]
- 31.Morrison JF, Walsh CT. The Behavior and Significance of Slow-Binding Enzyme-Inhibitors. Adv Enzymol Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 32.Bauer AW, Kirby WMM, Sherris JC, Turck M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am J Clin Pathol. 1966;45:493–496. [PubMed] [Google Scholar]
- 33.Maiti R, Van Domselaas GH, Zhang H, Wishart DS. Superpose: a Simple Server for Sophisticated Structural Superposition. Nucleic Acids Res. 2004;32(Web Server Issue):W590–W594. doi: 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.http://emergency.cdc.gov/agent/agentlist-category.asp.
- 35.Hernick M, Fierke CA. Catalytic Mechanism and Molecular Recognition of E. coli UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase Probed by Mutagenesis. Biochemistry. 2006;45:15240–8. doi: 10.1021/bi061405k. [DOI] [PubMed] [Google Scholar]