

Abstract
Pancreatic cancer is characterized by very low survival rates because of high intrinsic resistance to conventional therapies. Ionizing radiation (IR)-enhanced tumor invasiveness is emerging as one mechanism responsible for the limited benefit of radiotherapy in pancreatic cancer. In this study, we establish the role of heparanase - the only known mammalian endoglycosidase that cleaves heparan sulfate - in modulating the response of pancreatic cancer to radiotherapy. We found that clinically relevant doses of IR augment the invasive capability of pancreatic carcinoma cells in vitro and in vivo by upregulating heparanase. Changes in the levels of the transcription factor Egr-1 occurred in pancreatic cancer cells following radiation, underlying the stimulatory effect of IR on heparanase expression. Importantly, the specific heparanase inhibitor SST0001 abolished IR-enhanced invasiveness of pancreatic carcinoma cells in vitro, while combined treatment with SST0001 and IR, but not IR alone, attenuated the spread of orthotopic pancreatic tumors in vivo. Taken together, our results suggest that combining radiotherapy with heparanase inhibition is an effective strategy to prevent tumor resistance and dissemination, observed in many IR-treated pancreatic cancer patients. Further, the molecular mechanism underlying heparanase upregulation in pancreatic cancer that we identified in response to IR may help identify patients in which radiotherapeutic intervention may confer increased risk of metastatic spread, where anti-heparanase therapy may be particularly beneficial.
Keywords: Invasiveness, Pancreatic cancer, Heparanase, Ionizing radiation, Extracellular matrix
Introduction
Pancreatic cancer is one of the most aggressive neoplasms with an extremely low 5-year survival rate (1–3). Currently, pancreaticoduodenectomy is the only curative form of treatment, however ~90% of pancreatic cancer patients miss the opportunity for complete surgical resection at the time of diagnosis (3, 4). Thus, radiotherapy remains a major component of treatment modalities for controlling pancreatic tumor progression (5). However, pancreatic cancer often shows resistance to radiation, and randomized trials could not demonstrate benefit from radiation, revealing rather conflicting results (6, 7). Although the high resistance of pancreatic tumors to radiation treatment is likely multifactorial, ionizing radiation (IR)-induced increase in carcinoma cell aggressiveness is emerging as one of the important mechanisms responsible for the limited benefit of radiotherapy in pancreatic cancer (8, 9). Indeed, accumulating preclinical and clinical data suggest that under some circumstances IR may stimulate tumor aggressiveness (8–14), although the identity of downstream effectors acting at the cell or tissue levels and responsible for this effect, remains poorly investigated. Here we report that IR promotes pancreatic cancer aggressiveness through up-regulation of the heparanase gene. Heparanase is a single mammalian endoglycosidase capable of degrading heparan sulfate (HS), the main polysaccharide component of the basement membrane and other types of extracellular matrix (ECM) (15–17). HS is a key element participating in the self-assembly, insolubility, and barrier properties of the ECM (18–20). In addition, HS moieties in the ECM are responsible for binding of heparin-binding growth factors (i.e., bFGF, VEGF, HGF) (20, 21), which are thereby protected, stabilized and sequestered from their site of action, but upon enzymatic degradation of HS can be readily mobilized to induce growth factor-dependent processes (i.e., neovascularization, tissue repair, tumor progression) (22–24). Thus, cleavage of HS by heparanase leads to disassembly of extracellular barriers, enables cell invasion (15, 16), releases HS–bound angiogenic and growth factors from the ECM depots, and generates bioactive HS fragments which promote growth factor-receptor binding, dimerization and signaling (18, 22, 24). Direct evidence for a causal role of heparanase in tumor progression was provided by demonstration of an accelerated primary tumor growth (25–29) and increased metastatic ability of melanoma, lymphoma, and prostate carcinoma cells (26, 30) following over-expression of the heparanase gene, as well as by a marked decrease in the tumorigenic/metastatic potential of cells following heparanase silencing (26, 31, 32). The role of heparanase in sustaining the pathology of malignant tumors was further confirmed by numerous reports, demonstrating preferential over-expression of the enzyme in numerous cancers (reviewed in (15)). Causal involvement of heparanase in pancreatic carcinoma progression is particularly well-documented. There was a 30-fold increase in heparanase mRNA in pancreatic cancer tissue samples, in comparison to normal pancreatic tissue (33). Moreover, elevated levels of the enzyme have been found in body fluids of patients with active pancreatic cancer disease (34) as compared to healthy donors. Pancreatic cancer patients whose tumors exhibit high levels of the heparanase mRNA had a significantly shorter postoperative survival time than patients whose tumors contained relatively low levels of heparanase (33, 35). Cultured pancreatic cancer cells over-expressing the heparanase enzyme displayed enhanced invasiveness in vitro (33, 35). Recent finding that heparanase is a highly significant independent variable for lymph node metastasis in pancreatic carcinoma patients further demonstrate a crucial role of the enzyme in the aggressiveness of pancreatic cancer (36).
In the present study we investigated heparanase involvement in radiation-enhanced aggressiveness of pancreatic cancer. We found that clinically relevant doses of IR augment invasive ability of pancreatic carcinoma cells in vitro and in vivo through upregulation of heparanase expression, and revealed that the molecular mechanism responsible for IR-induced heparanase transcription involves the Early growth response 1 (Egr-1) transcription factor. These observations may assist in defining a subset of patients in which radiotherapeutic intervention may confer increased risk of tumor growth and metastatic spread. Moreover, compound SST0001 (non-anticoagulant N-acetylated, glycol split heparin 100NA,RO-H) a specific heparanase inhibitor (37), abolished IR-enhanced invasiveness of pancreatic carcinoma cells in vitro, while combined treatment with SST0001and IR, but not IR alone, attenuated orthotopic pancreatic tumor spread in vivo. Taken together, our results suggest that combination of radiotherapy with a specific heparanase inhibitor as an effective strategy to prevent tumor resistance and progression, observed in many IR-treated pancreatic cancer patients.
Materials and methods
Cell culture, irradiation and immunostaining
PANC1 and AsPc-1 human pancreatic carcinoma cells (ATCC, Manassas, VA) were routinely maintained in DMEM supplemented with 1 mM glutamine, 50 µg/ml streptomycin, 50 U/ml penicillin and 10% fetal calf serum (FCS) (Biological Industries, Beit-Haemek, Israel) at 37°C and 7.5% CO2. Prior to IR treatment, cells were maintained for 16 h in serum-free medium and then irradiated with doses indicated in the Results section, using a 60Co Picker unit irradiator (1.56 Gy/min). Cells were then lysed and processed for RNA/protein extraction and assessment of heparanase enzymatic activity. In some experiments, cells were cultured on glass coverslips (12 mm; Carolina Biological Supply Company), fixed with 100% ice-cold methanol and processed for immunofluorescent staining with 1:500 diluted anti-heparanase monoclonal antibody H100 (38, 39), kindly provided by Dr. P. Kussie (ImClone Systems Inc., New York, NY).
Orthotopic injection of pancreatic carcinoma cells
Six weeks old male severe combined immunodeficient (SCID) mice were anesthetized, a small left abdominal flank incision was made and the spleen exteriorized. The luciferase expressing PANC1 (PANC1-Luc) cells (0.5×106) were resuspended in 0.02 ml PBS and injected subcapsularly to the pancreas. A successful subcapsular intrapancreatic injection of tumor cells was verified by the appearance of a fluid bleb without i.p leakage. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee.
In vivo imaging
To monitor in vivo progression of Luc-expressing tumors, the cooled-charge-coupled device (CCCD) camera model LN/CCD-1300EB equipped with ST-133 controller and a 50-mm Nikon lens (Roper Scientific, Princeton Instrument, Trenton, NJ), supported with appropriate software was used for light detection, as described (26, 41). In all experiments animals were anesthetized before light detection. The exposure conditions (including exposure time, distance of lens from the object, and time after injection of luciferin) were kept identical. Ten minutes before monitoring light emission, the animals were injected intraperitoneally with Beetle luciferin (Promega Corp., Madison, WI) in PBS at 126 mg/kg body weight. Animals were placed in a dark box supplemented with a controlled light to take pictures of the background gray-scale image and then exposed to the CCCD to generate a pseudo-color image that represents light intensity. For co-localization of the bioluminescent emission on the animal body, gray-scale and pseudo-color images were merged and quantification of bioluminescence was performed applying Meta Imaging Series software (Molecular Devices, Silicon Valley, CA).
Tumor irradiation
Mice were anesthetized and in vivo bioluminescent imaging was used to delineate the luciferase-expressing pancreatic tumors. The tumor margins were marked on the skin and the animals were restrained by adhesive tape during irradiation. Tumors were irradiated applying brachytherapy afterloader (I192 Nucletron microSelectron HDR, Veenendaal, The Netherlands) through bronchial sleeve applicator. The dose (6 Gy) was calculated to 1.0 cm isodose line and 0.5-cm width silicon bolus was placed above and below the sleeve to achieve dose homogenicity. The treatment field was designed to cover the tumors and protect the rest of the body. The prescribed dose was confirmed by film dosimetry. Variation of dose inside the tumors was estimated to be within ±10% of the prescribed dose. Tumor growth was monitored at the indicated times immediately prior to irradiation and 7 days after irradiation by whole-body bioluminescent imaging and quantification of bioluminescence, applying Meta Imaging Series software.
Statistics
The results are presented as the mean ± SD. Differences between groups were assessed by the unpaired Student’s t-test. Chi-test was performed to examine the association between IR and PANC1-Luc tumor dissemination in vivo.
Results
IR increases heparanase expression and invasiveness of pancreatic carcinoma cells in vitro and in vivo
As radiation-induced tumor aggressiveness was previously studied applying primarily in vitro model systems (8, 9, 13), we initially investigated whether similar mechanism operates in vivo. For this purpose, human pancreatic carcinoma PANC1 cells, genetically engineered to express the luciferase gene (PANC1-Luc), were implanted orthotopically into the pancreas of SCID mice, and whole-body bioluminescent imaging was applied to monitor tumor progression. Once the primary orthotopic tumors became detectable by bioluminescent imaging (2 weeks post injection, Supplementary Fig. S1A), the mice were divided into two groups (n>10 in each group); tumors growing in mice from the experimental group were treated with IR (6 Gy), as shown in Supplementary Fig. S1B and described in 'Methods', while mice of the control group remained untreated. Whole body bioluminescent imaging was repeated 7 days post IR treatment to assess tumor progression. As expected, the increase in total bioluminescent signal emitted by the IR-treated primary tumors was ~2 fold lower than that emitted by the untreated tumors, indicating inhibitory effect of IR on tumor cell proliferation rate. This finding is in agreement with the inhibitory effect exerted by IR on Panc1-LUC cell proliferation in vitro (Fig. 1A). However, whole body imaging analysis revealed Panc1-LUC tumor dissemination, evidenced by the appearance of newly formed bioluminescent foci on day 7 post IR, in 73% of IR-treated mice vs. only 23% of untreated mice (chi-square test p = 0.008). Notably, the increase in overall tumor area (which includes both primary and secondary tumor sites and reflects tumor spread rather than growth) did not differ statistically in IR-treated and untreated mice on day 7 post IR treatment (not shown). Collectively these results suggest that along with its anti-proliferative action on PANC1 cells, IR may promote dissemination of orthotopic pancreatic tumor in vivo. These findings are in full agreement with the ability of IR to augment invasiveness of PANC1 cells in vitro (Fig. 2B and ref. (9)).
Figure 1. Effect of IR on proliferation and invasiveness of PANC1 cells.
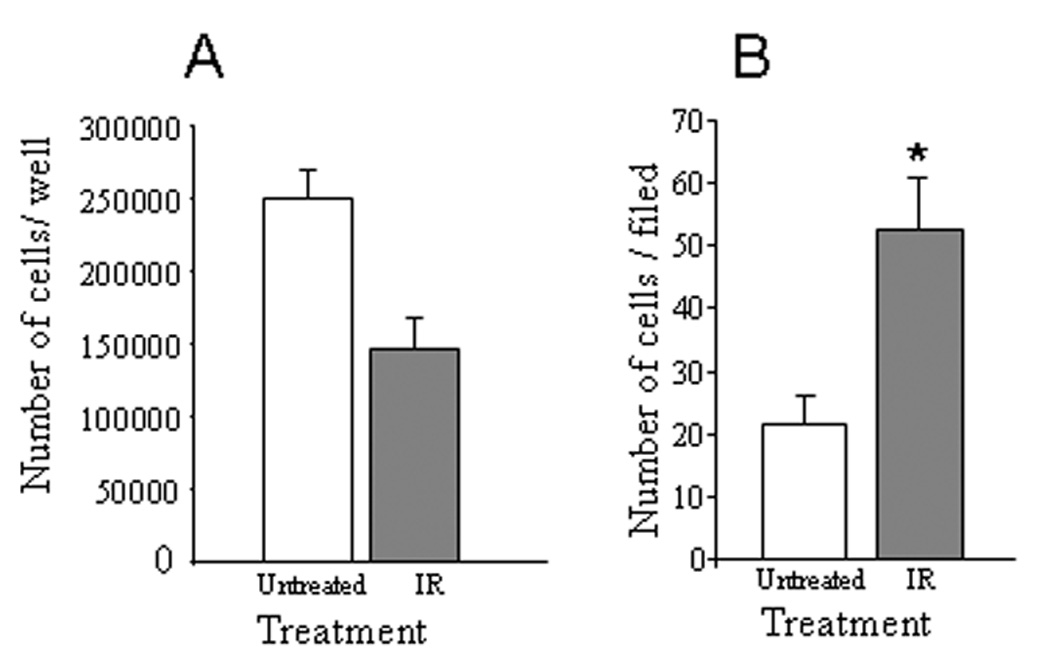
PANC1 cells, untreated (empty bars) or irradiated with 10 Gy (grey bars), were assessed for proliferation rate (A) and in vitro invasiveness through Matrigel (B). A. The cells were seeded in quadruplicates into 2 cm2 dishes (1 × 105 cells per dish) and remained untreated or treated with IR (10Gy). Twenty four hours later, the cells were dissociated with trypsin/EDTA and counted. Each point represents mean ± SD of quadruplicate dishes. B. Non-irradiated and irradiated PANC1 cells were added on top of Matrigel-coated filters, and incubated for 6 hours as described in 'Methods'. The mean number of invading cells per field was determined in five random microscopic fields per filter. Bars represent mean ± SD of triplicate filters, p<0.0001.
Figure 2. Effect of IR on heparanase expression.

A. Conformal irradiation increases heparanase mRNA levels in PANC1-LUC orthotopic tumors. SCID mice were orthotopically injected with 5×105 PANC1-LUC cells. When pancreatic tumors became detectable by bioluminescent examination (~2 weeks post injection), the mice were divided into two groups (n = 4 per group) and their tumors were either irradiated (6 Gy, grey bar) as described in ‘Methods’, or remained untreated (empty bar). Twenty four hours later, mice were sacrificed and their tumors excised. RNA was isolated from the tumor tissue and qRT-PCR was performed using human heparanase-specific primers, as described in ‘Methods’. Each bar represents mean ± SD of 4 animals, p < 0.05. B–D. IR induces heparanase expression in pancreatic carcinoma cell lines. Prior to IR treatment, PANC1 (left) and AsPc-1 (right) cells were maintained for 16 h in serum-free medium. Then, the cells were treated with the indicated doses of IR. Six hours later, heparanase levels were measured by semi-quantitative RT-PCR (B), immunofluorescent staining (C) and enzymatic activity assay (D). B. Dose-dependent changes in heparanase and Egr1 mRNA levels following IR treatment. RNA was isolated from the cells and comparative semi-quantitative RT-PCR was performed, as described in ‘Methods’. Aliquots (10 µl) of the amplification products were separated by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining. RT-PCR products obtained with GAPDH-specific primers were used as a control for RNA integrity and equal loading. The gels shown are representative of three independent experiments. C. Immunofluorecsent staining with anti-heparanase antibody, performed as described in ‘Methods’, reveals IR increased heparanase protein content in AsPc1 pancreatic carcinoma cells following IR treatment (10 Gy). D. Heparanase activity was determined in lysates obtained from PANC1 cells untreated (black squares) or irradiated with 10Gy (empty squares), as described in ‘Methods’. The graphs shown are representative of three independent experiments.
Critical involvement of heparanase in tumor cell invasiveness (and particularly in aggressive behavior of pancreatic cancer (33–36)) prompted us to examine the effect of IR on heparanase expression in this experimental setting. For this purpose, PANC1-Luc orthotopic tumors growing in SCID mice were either untreated or irradiated with a dose of 6 Gy, as described above. Twenty four hours post irradiation, the tumor-bearing mice were sacrificed, their tumors excised and heparanase mRNA expression levels in IR-treated vs. untreated tumor tissue were compared by quantitative real-time PCR (qRT-PCR). Since our experimental model consists of PANC1 human pancreatic carcinoma cells growing in a mouse host, utilization of primers specific to either human or mouse heparanase mRNA sequences enabled us to distinguish between tumor- and host- derived heparanase. As shown in figure 2A, qRT-PCR analysis with human heparanase specific primers revealed a 4-fold increase in heparanase mRNA levels in the orthotopic tumor following in vivo irradiation. When mouse heparanase specific primers were utilized, no significant difference in host-derived heparanase expression was detected between irradiated and non-irradiated tumors (not shown). To further validate a stimulatory effect of IR on heparanase expression, two pancreatic cancer cell lines, PANC1 and AsPc-1 (characterized by relatively low basal level of heparanase expression) were untreated or treated with clinically relevant doses of IR, and heparanase mRNA levels were assessed by RT-PCR. As shown in figure 2B, RT-PCR analysis demonstrated a dose dependent increase in heparanase mRNA levels following cell exposure to IR. Consistent with the above findings, increased levels of heparanase protein in IR-treated vs. untreated pancreatic cancer cell lines were detected by immunofluorecsent staining (Fig. 2C), and confirmed by increased enzymatic activity (Fig. 2D), further corroborating a stimulatory effect of IR on heparanase expression. Noteworthy, treatment with gemcitabine, the backbone of chemotherapy in pancreatic cancer (5), often given concurrently with radiation therapy, did not abolish IR induced heparanase expression in pancreatic carcinoma cells (not shown).
Effect of IR on heparanase expression is mediated by Egr1 transcription factor
We next investigated the molecular mechanism underlying IR-driven increase of heparanase expression in pancreatic cancer. The Egr1 transcription factor is an important regulator of heparanase promoter activity. Egr1 was shown to control heparanase expression acting as either activator or repressor of heparanase transcription, depending on the cell and tissue type (44, 45). Notably, ionizing radiation was reported to affect Egr1 expression in several cell types (46–48). Applying immunoblot analysis to investigate IR-induced changes in Egr1 protein levels, we found that in PANC1 cells transient increase (3-fold) in Egr1 during the first 15 minutes post IR was followed by a profound and continuous down-regulation of Egr1, as compared to its basal levels in the untreated cells (Fig. 3A, B). These results are in accordance with the RT-PCR data showing decreased levels of Egr1 mRNA in IR-treated vs. untreated PANC1 and AsPc-1 cells (Fig. 2B, middle panels). Of note, in pancreatic carcinoma cells the decrease in Egr1 levels occurred in parallel with IR-induced overexpression of heparanase (Fig. 2B).
Figure 3. Downregulation of Egr1 mediates IR-induced activation of heparanase promoter in pancreatic carcinoma cells.

A. Changes in Egr1 protein levels following IR treatment of PANC1 pancreatic carcinoma cells. PANC1 cells remained untreated or were irradiated (10 Gy) as described in ‘Methods’, lysed on the indicated time points and analyzed for Egr1 protein by immunoblotting. B Densitometric quantification of Egr1 protein levels presented in A. The immunoblot membrane was stripped and re-probed with anti- α-tubulin monoclonal antibody. Egr1 and α-tubulin band intensity was quantified using Scion image software, and Egr1/α-tubulin intensity ratio was calculated and expressed as percent of the intensity ratio observed in the untreated cells. The data shown are representative of three independent experiments. C. Egr1 suppresses heparanase promoter activity in PANC1 cells. PANC1 cells were co-transfected with plasmid encoding for Luciferase driven by heparanase promoter together with either Egr1 expressing vector, pEgr1 (filled bar) or control empty pcDNA3 plasmid (empty bar), along with the normalizing beta-galactosidase construct. Luciferase activity was measured in cell lysates 48 h post transfection and normalized with beta-galactosidase. Each transfection was carried out in triplicate and the data are representative of three independent experiments. Error bars represent mean±SD, * p value =0.0001. D. Decreased occupancy of the heparanase promoter by Egr1 following IR treatment. PANC1 cells were untreated or treated with IR (10 Gy). Four hours later chromatin was prepared as described in ‘Methods’ and immunoprecipitated (IP) with antibodies against Egr1 or an unrelated protein, Flt-1. The final DNA extractions were amplified using primer set that covers functional Egr1 binding site (44) in the heparanase promoter (three upper panels), or primer set specific to unrelated GAPDH gene sequence, used as control (lower panel). Input lanes show DNA that was PCR amplified from chromatin preparations before immunoprecipitation. The gels shown are representative of three independent experiments.
These observations led us to hypothesize that, unlike in the majority of cancer cells, in pancreatic carcinoma Egr1 acts as a repressor rather then activator of heparanase transcription. To test this assumption, PANC1 cells were co-transfected with plasmids encoding for luciferase (LUC) driven by the heparanase promoter (49) together with either Egr1 expressing vector (pEgr1) or empty pcDNA3 vector. Luciferase activity was measured in cell lysates 48 hours post transfection and normalized with β-galactosidase. Four fold decrease in heparanase promoter activity was detected in PANC1 cells co-transfected with Egr1 expressing vector, as compared to cells co-transfected with empty vector (p=0.0001; Fig. 3C), validating the inhibitory effect of Egr1on heparanase promoter in pancreatic carcinoma. Moreover, ChIP analysis demonstrated decreased occupancy of the heparanase promoter by Egr1 in IR-treated vs. untreated cells 4 hours post IR (Fig. 3D), in further support of the role of Egr1 in IR-induced heparanase expression.
Specific heparanase inhibitor abolishes IR-induced invasiveness of Panc-1 cells in vitro
We next utilized the Matrigel invasion assay (50) to test whether the stimulatory effect of IR on pancreatic carcinoma cell invasiveness is due (at least in part) to the IR-induced increase in heparanase expression. For this purpose, PANC1 cells added to the upper compartment of Boyden chambers, were untreated or irradiated with 10 Gy and allowed to migrate through Matrigel in the presence or absence of a specific inhibitor of heparanase enzymatic activity SST0001 (= 100NA,RO-H, 100% N-acetylated, 25% glycol-split heparin) (37). SST0001 inhibits the heparanase enzyme at 10 nanomolar concentrations in vitro (37), as well as melanoma metastasis (51) and myeloma tumor growth (29) in vivo. As shown in figure 4, IR markedly (4 fold) enhanced invasion of PANC1 cells through Matrigel-coated filters and this enhancement was abolished in the presence of SST0001.
Figure 4. IR-induced invasiveness of PANC1 cells in vitro is abolished in the presence of heparanase inhibitor.

PANC1 cells were added (3 × 105 cells/well, 6 h, 37°C, 5% CO2) on top of Matrigel-coated filters, and incubated in 1 mL DMEM containing 0.1% BSA. Cells were first untreated, irradiated (10 Gy), treated with 2 µg/ml SST0001, or subjected to combined treatment (IR+SST0001) followed by incubation on top of the Matrigel-coated filters. The bottom chambers were filled with medium conditioned by NIH 3T3 fibroblasts. For a negative control, the bottom chamber was filled with serum-free DMEM containing 0.1% BSA. The number of cells/field on the lower surface of the filter was determined as described in ‘Materials and Methods’. Each data point represents the mean ± SD of quadriplicate filters. The graph shown is representative of three independent experiments. *p = 0.0016, **p = 0.0036.Inset. Representative micrographs showing an increase in the number of invading PANC1 cells after IR (left vs. middle panels). Treatment with compound SST0001 blocks the IR-induced increase in invading cells (right panel).
Enhanced anti-tumor effect of combined SST0001 and IR treatment
To examine the effects of combining SST0001 and radiation in vivo, SCID mice were orthotopically injected with PANC1-LUC cells. Two weeks after cell injection, when the bioluminescent pancreatic tumors were detected (as described in 'Methods'), the mice were divided into 4 groups (n=7) and treated with either radiation alone (6Gy), SST0001 alone (600 µg/mouse, administered daily i.p.), radiation (6 Gy) followed by treatment with SST0001, or vehicle alone (saline, administered daily i.p.). Progression of the orthotopic PANC1-Luc tumors was monitored by measurements of the total tumor size (including primary and secondary sites) on day 1 and day 7 of the experiment, applying Meta Imaging Series software and the effect of the different treatments on PANC1-Luc tumor spread was estimated as the change in total tumor size between experimental day 1 and 7. Administration of both SST0001 and IR, but not each treatment alone, attenuated PANC1-Luc tumor spread, as evidenced by an unchanged total tumor size (tumor area on day seven divided by tumor area on day one = 1) measured in mice under combined treatment, vs. ~3 fold increased tumor size observed in mice that were untreated or treated with either SST0001 or IR alone (Fig. 5). In addition, newly-formed bioluminescent tumor foci were detected on day 7 post IR in only 21% of mice that received combined treatment with SST0001 and radiation, as compared to >70% of mice that received IR treatment alone (chi-square test p = 0.009).
Figure 5. Enhanced antitumor effect of combined SST0001 and IR treatment.

SCID mice were orthotopically injected with PANC1-LUC cells and either remained untreated (white bar), treated with conformal IR (6Gy) alone (grey bar), SST0001 alone (daily i.p injection of 600 µg/mouse; white dotted bar) or IR in combination with SST0001 (administered daily i.p. for 7 days) (grey dotted bar). A. Tumor progression was monitored by whole-body bioluminescence imaging of the orthotopic PANC1-LUC tumor, prior (day 1) and 7 days after (day 7) administration of IR. Representative images are shown. B. The effect of different treatments on tumor spread presented as the change of total tumor size (primary + secondary tumors), analyzed applying Meta Imaging Series software. Tumor spread was attenuated by a combined treatment with IR and SST0001 (*p=0.037), but not IR or SST0001 alone.
Discussion
Although improved surveillance and therapy have decreased the mortality of most cancer types during the last decades, the impact of these advances on pancreatic carcinoma patients remains rather modest. Pancreatic cancer is the fourth leading cause of cancer death in the United States and the 5-year survival rate is less than 5% (2). This poor outcome is often explained by a high intrinsic resistance of pancreatic tumors to conventional treatment, primarily due to overexpression of different effector molecules that prevent the normal response to drugs and radiotherapy (3). Thus, more data about the identity of downstream effectors responsible for pancreatic cancer cell resistance to therapy are needed to properly address the disease treatment. Targeting these effector molecules may represent an attractive therapeutic approach in pancreatic carcinoma. Considerable progress has been made in deciphering the role of multiple mutations that render pancreatic cancer resistance to treatment by triggering proto-oncogene expression or inactivating tumor suppressor genes (3). Much less is known about the involvement of ECM-degrading enzymes in pancreatic cancer resistance. Here we demonstrate for the first time the functional importance of heparanase in modulating response of pancreatic cancer to radiotherapy and describe a new molecular mechanism underlying radio-resistance of pancreatic tumors.
It should be noted that like nearly any known treatment modality, radiotherapy may exert an adverse effect upon metastasis under some conditions (52–54). Several researchers have observed that local irradiation increases invasiveness/aggressiveness of different tumor cell types, including pancreatic cancer (8–14). In light of these findings it is conceivable that radiation-induced aggressiveness may hamper effectiveness of radiotherapy in pancreatic and probably other types of cancer. Our present study indicates that stimulation of heparanase expression by IR may be an integral part of this phenomenon.
Heparanase plays a pivotal role in creating a supportive microenvironment for tumor development and dissemination (16, 32, 55). Potential contributions of heparanase activity to pancreatic cancer progression (via enhanced tumor growth, invasiveness, angiogenesis, metastasis) are well documented (33–35), and further supported by a significant independent association of heparanase positivity with pancreatic adenocarcinoma dedifferentiation and lymph node metastasis (36). Here we have shown that IR augments heparanase expression and thereby aggressiveness of pancreatic carcinoma both in vitro and in vivo. Importantly, we provide evidence that highly specific changes in the levels of Egr1, occurring in pancreatic cancer cells following radiation, may underlie the stimulatory effect of IR on heparanase expression in pancreatic carcinoma. As a transcriptional regulator, Egr1 can both induce and repress the expression of its target genes, including heparanase (44, 45, 56). Egr1 was previously shown to activate heparanase expression in T lymphocytes, prostate, breast, and colon carcinomas, but to inhibit its transcription in melanoma cells (44). Transactivation studies using Egr1 expression vector, co-transfected with a reporter construct encoding for LUC under the heparanase promoter, showed that in pancreatic carcinoma cells Egr1 acts to repress heparanase transcription (Fig. 3C). In fact, IR treatment of PANC1 pancreatic carcinoma cells resulted initially in a transient increase in Egr1, followed by profound and continuous decline in Egr1 levels, as compared to its basal levels in untreated PANC1 cells (Fig. 3A). At the same experimental setting, ChIP analysis revealed a marked decrease in occupancy of the heparanase promoter by Egr1 following IR treatment (Fig. 3D). In this respect it should be noted that applying quantitative real-time PCR we observed that pancreatic carcinoma cell lines (i.e., PANC1, AsPc-1) are characterized by relatively higher basal levels of Egr1, as compared to cancer cells of non-pancreatic origin (i.e., breast and colon carcinoma, melanoma, glioma) (not shown). In addition, we found that pancreatic carcinoma cells express high basal levels of NGFI-A/Egr1-binding protein NAB2 (3 to 6 fold higher than in breast and colon carcinoma, not shown). NAB2 is a transcriptional co-repressor that directly interacts with Egr1 and represses activation of its target promoters (57). It is therefore plausible that in the presence of both Egr1 and NAB2 the heparanase promoter is repressed in pancreatic cells. However, following IR-associated temporal decrease in Egr1, this repression is released, allowing for activation of the heparanase promoter. Clearly, since heparanase is not the only target gene of Egr1, additional Egr1-controlled genes may contribute to the observed effect of IR on tumor aggressiveness in vivo as well.
Combination of radiotherapy with drugs that inhibit IR-induced tumor aggressiveness may be an attractive strategy to diminish adverse pro-metastatic action while retaining the therapeutic benefit of radiation, thus reducing resistance of pancreatic cancer to treatment. Our study presents evidence that SST0001, a specific inhibitor of heparanase enzymatic activity (37) attenuates radiation-induced invasiveness in an orthotopic model of pancreatic cancer (Fig. 5). Spread of orthotopically growing pancreatic tumors was significantly reduced in mice treated with a combination of SST0001 and IR, as compared with either modality alone (Fig. 5). The ability of SST0001 to inhibit IR-induced tumor invasiveness (Fig. 4) is a most likely explanation for the enhanced antitumor effect of combined SST0001 and IR treatment in this system. It should be noted that SST0001 alone did not exert a significant inhibitory effect on PANC1-Luc orthotopic tumor growth (Fig. 5). A likely explanation for the lack of effectiveness of SST0001 monotherapy in this system is that PANC1 cells are characterized by low endogenous levels of heparanase. Interestingly, in a model of orthotopic Panc02 mouse pancreatic carcinoma (58, 59), expressing high levels of endogenous heparanase, administration of SST0001 alone resulted in 2-fold inhibition of orthotopic Panc02 tumor growth (p= 0.044, Supplementary Fig. S2).
Attenuation of IR mediated pancreatic tumor spread by SST0001 represents a proof of concept for heparanase inhibition as a relevant approach to diminish radio-resistance in IR treated pancreatic cancer patients. Our results warrant further studies aimed at identifying the most effective dose and schedule administration schemes for combined SST0001 and IR treatment, toward future translation to clinical setting. In parallel, elucidation of the molecular mechanism underlying heparanase upregulation in pancreatic cancer in response to IR will help to better define target patient populations in which future anti-heparanase therapies could be particularly beneficial.
Supplementary Material
Acknowledgments
We are grateful to Sigma-Tau Research Switzerland S.A. (Mendrisio, CH) for kindly providing compound SST0001, and to Drs. Israel Vlodavsky (Rappoport Faculty of Medicine, Technion, Israel), Sergio Penco, Annamaria Naggi and Benito Casu ('Ronzoni Institute, Milano, Italy) for their continuous help and active collaboration.
Grant Support: This work was supported by grants from the Israel Cancer Research Fund (ICRF); Israel Science Foundation (grant 593/10); Chief scientist office - Israeli ministry of health, and the National Cancer Institute, NIH (grant RO1-CA106456-06).
Footnotes
Conflict of interest statement:
CP is employed by Sigma-Tau Industrie Farmaceutiche Riunite S.p.A.
CP is listed as inventor on a patent for SST0001.
AM, EH, IL, EZ, TP and ME declare no conflict of interest.
References
- 1.Raimondi S, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an overview. Nat Rev Gastroenterol Hepatol. 2009;6:699–708. doi: 10.1038/nrgastro.2009.177. [DOI] [PubMed] [Google Scholar]
- 2.Shaib YH, Davila JA, El-Serag HB. The epidemiology of pancreatic cancer in the United States: changes below the surface. Aliment Pharmacol Ther. 2006;24:87–94. doi: 10.1111/j.1365-2036.2006.02961.x. [DOI] [PubMed] [Google Scholar]
- 3.Borja-Cacho D, Jensen EH, Saluja AK, Buchsbaum DJ, Vickers SM. Molecular targeted therapies for pancreatic cancer. Am J Surg. 2008;196:430–441. doi: 10.1016/j.amjsurg.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller MW, Friess H, Koninger J, Martin D, Wente MN, Hinz U, Ceyhan GO, Blaha P, Kleeff J, Buchler MW. Factors influencing survival after bypass procedures in patients with advanced pancreatic adenocarcinomas. Am J Surg. 2008;195:221–228. doi: 10.1016/j.amjsurg.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 5.Network N. C. C. NCCN Clinical Practice Guidelines in Oncology: Pancreatic Adenocarcinoma. 2008;v.1.2008 [Google Scholar]
- 6.Cohen SJ, Dobelbower R, Jr, Lipsitz S, Catalano PJ, Sischy B, Smith TJ, Haller DG. A randomized phase III study of radiotherapy alone or with 5-fluorouracil and mitomycin-C in patients with locally advanced adenocarcinoma of the pancreas: Eastern Cooperative Oncology Group study E8282. Int J Radiat Oncol Biol Phys. 2005;62:1345–1350. doi: 10.1016/j.ijrobp.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 7.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, Falconi M, Pederzoli P, Pap A, Spooner D, Kerr DJ, Buchler MW the European Study Group for Pancreatic, C. A Randomized Trial of Chemoradiotherapy and Chemotherapy after Resection of Pancreatic Cancer. N Engl J Med. 2004;350:1200–1210. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 8.Ohuchida K, Mizumoto K, Murakami M, Qian L-W, Sato N, Nagai E, Matsumoto K, Nakamura T, Tanaka M. Radiation to Stromal Fibroblasts Increases Invasiveness of Pancreatic Cancer Cells through Tumor-Stromal Interactions. Cancer Res. 2004;64:3215–3222. doi: 10.1158/0008-5472.can-03-2464. [DOI] [PubMed] [Google Scholar]
- 9.Qian L-W, Mizumoto K, Urashima T, Nagai E, Maehara N, Sato N, Nakajima M, Tanaka M. Radiation-induced Increase in Invasive Potential of Human Pancreatic Cancer Cells and Its Blockade by a Matrix Metalloproteinase Inhibitor, CGS27023. Clin Cancer Res. 2002;8:1223–1227. [PubMed] [Google Scholar]
- 10.Camphausen K, Moses MA, Beecken W-D, Khan MK, Folkman J, O'Reilly MS. Radiation Therapy to a Primary Tumor Accelerates Metastatic Growth in Mice. Cancer Res. 2001;61:2207–2211. [PubMed] [Google Scholar]
- 11.Kaliski A, Maggiorella L, Cengel KA, Mathe D, Rouffiac V, Opolon P, Lassau N, Bourhis J, Deutsch E. Angiogenesis and tumor growth inhibition by a matrix metalloproteinase inhibitor targeting radiation-induced invasion. Mol Cancer Ther. 2005;4:1717–1728. doi: 10.1158/1535-7163.MCT-05-0179. [DOI] [PubMed] [Google Scholar]
- 12.Madani I, De Neve W, Mareel M. Does ionizing radiation stimulate cancer invasion and metastasis? Bull Cancer. 2008;95:292–300. doi: 10.1684/bdc.2008.0598. [DOI] [PubMed] [Google Scholar]
- 13.Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, Park IC, Rhee CH, Hong SI. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006;66:8511–8519. doi: 10.1158/0008-5472.CAN-05-4340. [DOI] [PubMed] [Google Scholar]
- 14.Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001;61:2744–2750. [PubMed] [Google Scholar]
- 15.Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Parish CR, Freeman C, Hulett MD. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta. 2001;1471:M99–M108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- 17.Vlodavsky I, Goldshmidt O, Zcharia E, Metzger S, Chajek-Shaul T, Atzmon R, Guatta-Rangini Z, Friedmann Y. Molecular properties and involvement of heparanase in cancer progression and normal development. Biochimie. 2001;83:831–839. doi: 10.1016/s0300-9084(01)01318-9. [DOI] [PubMed] [Google Scholar]
- 18.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 19.Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–652. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 20.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2:521–528. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 21.Vlodavsky I, Miao HQ, Medalion B, Danagher P, Ron D. Involvement of heparan sulfate and related molecules in sequestration and growth promoting activity of fibroblast growth factor. Cancer Metastasis Rev. 1996;15:177–186. doi: 10.1007/BF00437470. [DOI] [PubMed] [Google Scholar]
- 22.Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I, Vlodavsky I. Heparanase as mediator of angiogenesis: mode of action. Faseb J. 2001;15:1661–1663. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]
- 23.Kadenhe-Chiweshe A, Papa J, McCrudden KW, Frischer J, Bae JO, Huang J, Fisher J, Lefkowitch JH, Feirt N, Rudge J, Holash J, Yancopoulos GD, Kandel JJ, Yamashiro DJ. Sustained VEGF blockade results in microenvironmental sequestration of VEGF by tumors and persistent VEGF receptor-2 activation. Mol Cancer Res. 2008;6:1–9. doi: 10.1158/1541-7786.MCR-07-0101. [DOI] [PubMed] [Google Scholar]
- 24.Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat Med. 1998;4:691–697. doi: 10.1038/nm0698-691. [DOI] [PubMed] [Google Scholar]
- 25.Cohen I, Pappo O, Elkin M, San T, Bar-Shavit R, Hazan R, Peretz T, Vlodavsky I, Abramovitch R. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int J Cancer. 2006;118:1609–1617. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 26.Lerner I, Baraz L, Pikarsky E, Meirovitz A, Edovitsky E, Peretz T, Vlodavsky I, Elkin M. Function of heparanase in prostate tumorigenesis: potential for therapy. Clin Cancer Res. 2008;14:668–676. doi: 10.1158/1078-0432.CCR-07-1866. [DOI] [PubMed] [Google Scholar]
- 27.Mahtouk K, Hose D, Raynaud P, Hundemer M, Jourdan M, Jourdan E, Pantesco V, Baudard M, De Vos J, Larroque M, Moehler T, Rossi JF, Reme T, Goldschmidt H, Klein B. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–4923. doi: 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, Macleod V, Bendre M, Huang Y, Theus AM, Miao HQ, Kussie P, Yaccoby S, Epstein J, Suva LJ, Kelly T, Sanderson RD. Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood. 2005;105:1303–1309. doi: 10.1182/blood-2004-06-2141. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, MacLeod V, Dai Y, Khotskaya-Sample Y, Shriver Z, Venkataraman G, Sasisekharan R, Naggi A, Torri G, Casu B, Vlodavsky I, Suva LJ, Epstein J, Yaccoby S, Shaughnessy JD, Jr, Barlogie B, Sanderson RD. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood. 2007;110:2041–2048. doi: 10.1182/blood-2007-04-082495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5:793–802. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- 31.Roy M, Reiland J, Murry BP, Chouljenko V, Kousoulas KG, Marchetti D. Antisense-mediated suppression of Heparanase gene inhibits melanoma cell invasion. Neoplasia. 2005;7:253–262. doi: 10.1593/neo.04493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edovitsky E, Elkin M, Zcharia E, Peretz T, Vlodavsky I. Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. J Natl Cancer Inst. 2004;96:1219–1230. doi: 10.1093/jnci/djh230. [DOI] [PubMed] [Google Scholar]
- 33.Koliopanos A, Friess H, Kleeff J, Shi X, Liao Q, Pecker I, Vlodavsky I, Zimmermann A, Buchler MW. Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 2001;61:4655–4659. [PubMed] [Google Scholar]
- 34.Quiros RM, Rao G, Plate J, Harris JE, Brunn GJ, Platt JL, Gattuso P, Prinz RA, Xu X. Elevated serum heparanase-1 levels in patients with pancreatic carcinoma are associated with poor survival. Cancer. 2006;106:532–540. doi: 10.1002/cncr.21648. [DOI] [PubMed] [Google Scholar]
- 35.Rohloff J, Zinke J, Schoppmeyer K, Tannapfel A, Witzigmann H, Mossner J, Wittekind C, Caca K. Heparanase expression is a prognostic indicator for postoperative survival in pancreatic adenocarcinoma. Br J Cancer. 2002;86:1270–1275. doi: 10.1038/sj.bjc.6600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffmann AC, Mori R, Vallbohmer D, Brabender J, Drebber U, Baldus SE, Klein E, Azuma M, Metzger R, Hoffmann C, Hoelscher AH, Danenberg KD, Prenzel KL, Danenberg PV. High expression of heparanase is significantly associated with dedifferentiation and lymph node metastasis in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA and via HIF1a to HB-EGF and bFGF. J Gastrointest Surg. 2008;12:1674–1681. doi: 10.1007/s11605-008-0628-2. discussion 1681-1672. [DOI] [PubMed] [Google Scholar]
- 37.Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, Vlodavsky I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J Biol Chem. 2005;280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 38.Abboud-Jarrous G, Atzmon R, Peretz T, Palermo C, Gadea BB, Joyce JA, Vlodavsky I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J Biol Chem. 2008 doi: 10.1074/jbc.M801327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, Sanderson RD. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–8756. [PubMed] [Google Scholar]
- 40.Vlodavsky I, Fuks Z, Bar-Ner M, Ariav Y, Schirrmacher V. Lymphoma cell-mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: relationship to tumor cell metastasis. Cancer Res. 1983;43:2704–2711. [PubMed] [Google Scholar]
- 41.Zcharia E, Philp D, Edovitsky E, Aingorn H, Metzger S, Kleinman HK, Vlodavsky I, Elkin M. Heparanase regulates murine hair growth. Am J Pathol. 2005;166:999–1008. doi: 10.1016/S0002-9440(10)62321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miao HQ, Elkin M, Aingorn E, Ishai-Michaeli R, Stein CA, Vlodavsky I. Inhibition of heparanase activity and tumor metastasis by laminarin sulfate and synthetic phosphorothioate oligodeoxynucleotides. Int J Cancer. 1999;83:424–431. doi: 10.1002/(sici)1097-0215(19991029)83:3<424::aid-ijc20>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 43.Cohen I, Maly B, Simon I, Meirovitz A, Pikarsky E, Zcharia E, Peretz T, Vlodavsky I, Elkin M. Tamoxifen induces heparanase expression in estrogen receptor-positive breast cancer. Clin Cancer Res. 2007;13:4069–4077. doi: 10.1158/1078-0432.CCR-06-2546. [DOI] [PubMed] [Google Scholar]
- 44.de Mestre AM, Rao S, Hornby JR, Soe-Htwe T, Khachigian LM, Hulett MD. Early growth response gene 1 (EGR1) regulates heparanase gene transcription in tumor cells. J Biol Chem. 2005;280:35136–35147. doi: 10.1074/jbc.M503414200. [DOI] [PubMed] [Google Scholar]
- 45.de Mestre AM, Khachigian LM, Santiago FS, Staykova MA, Hulett MD. Regulation of inducible heparanase gene transcription in activated T cells by early growth response 1. J Biol Chem. 2003;278:50377–50385. doi: 10.1074/jbc.M310154200. [DOI] [PubMed] [Google Scholar]
- 46.Ahmed MM, Venkatasubbarao K, Fruitwala SM, Muthukkumar S, Wood DP, Jr, Sells SF, Mohiuddin M, Rangnekar VM. EGR-1 induction is required for maximal radiosensitivity in A375-C6 melanoma cells. J Biol Chem. 1996;271:29231–29237. doi: 10.1074/jbc.271.46.29231. [DOI] [PubMed] [Google Scholar]
- 47.Datta R, Rubin E, Sukhatme V, Qureshi S, Hallahan D, Weichselbaum RR, Kufe DW. Ionizing radiation activates transcription of the EGR1 gene via CArG elements. Proc Natl Acad Sci U S A. 1992;89:10149–10153. doi: 10.1073/pnas.89.21.10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zagurovskaya M, Shareef MM, Das A, Reeves A, Gupta S, Sudol M, Bedford MT, Prichard J, Mohiuddin M, Ahmed MM. EGR-1 forms a complex with YAP-1 and upregulates Bax expression in irradiated prostate carcinoma cells. Oncogene. 2009;28:1121–1131. doi: 10.1038/onc.2008.461. [DOI] [PubMed] [Google Scholar]
- 49.Elkin M, Cohen I, Zcharia E, Orgel A, Guatta-Rangini Z, Peretz T, Vlodavsky I, Kleinman HK. Regulation of heparanase gene expression by estrogen in breast cancer. Cancer Res. 2003;63:8821–8826. [PubMed] [Google Scholar]
- 50.Albini A, Iwamoto Y, Kleinman HK, Martin GR, Aaronson SA, Kozlowski JM, McEwan RN. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 1987;47:3239–3245. [PubMed] [Google Scholar]
- 51.Hostettler N, Naggi A, Torri G, Ishai-Michaeli R, Casu B, Vlodavsky I, Borsig L. P-selectin- and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. Faseb J. 2007;21:3562–3572. doi: 10.1096/fj.07-8450com. [DOI] [PubMed] [Google Scholar]
- 52.Kaplan HS, Murphy ED. The effect of local roentgen irradiation on the biological behavior of a transplantable mouse carcinoma; increased frequency of pulmonary metastasis. J Natl Cancer Inst. 1949;9:407–413. [PubMed] [Google Scholar]
- 53.Suit HD, Sedlacek RS, Gillette EL. Examination for a correlation between probabilities of development of distant metastasis and of local recurrence. Radiology. 1970;95:189–194. doi: 10.1148/95.1.189. [DOI] [PubMed] [Google Scholar]
- 54.von Essen CF. Radiation enhancement of metastasis: a review. Clin Exp Metastasis. 1991;9:77–104. doi: 10.1007/BF01756381. [DOI] [PubMed] [Google Scholar]
- 55.Vlodavsky I, Elkin M, Abboud-Jarrous G, Levi-Adam F, Fuks L, Shafat I, Ilan N. Heparanase: one molecule with multiple functions in cancer progression. Connect Tissue Res. 2008;49:207–210. doi: 10.1080/03008200802143281. [DOI] [PubMed] [Google Scholar]
- 56.de Mestre AM, Staykova MA, Hornby JR, Willenborg DO, Hulett MD. Expression of the heparan sulfate-degrading enzyme heparanase is induced in infiltrating CD4+ T cells in experimental autoimmune encephalomyelitis and regulated at the level of transcription by early growth response gene 1. J Leukoc Biol. 2007;82:1289–1300. doi: 10.1189/jlb.0507315. [DOI] [PubMed] [Google Scholar]
- 57.Srinivasan R, Mager GM, Ward RM, Mayer J, Svaren J. NAB2 represses transcription by interacting with the CHD4 subunit of the nucleosome remodeling and deacetylase (NuRD) complex. J Biol Chem. 2006;281:15129–15137. doi: 10.1074/jbc.M600775200. [DOI] [PubMed] [Google Scholar]
- 58.Bauer C, Bauernfeind F, Sterzik A, Orban M, Schnurr M, Lehr HA, Endres S, Eigler A, Dauer M. Dendritic cell-based vaccination combined with gemcitabine increases survival in a murine pancreatic carcinoma model. Gut. 2007;56:1275–1282. doi: 10.1136/gut.2006.108621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, Autiero M, Wyns S, Plaisance S, Moons L, van Rooijen N, Giacca M, Stassen JM, Dewerchin M, Collen D, Carmeliet P. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.