

Abstract
The origin and role of IL-17, a T-cell derived cytokine, in cartilage and bone destruction during rheumatoid arthritis (RA) remain to be clarified. In human ex vivo models, addition of IL-17 enhanced IL-6 production and collagen destruction, and inhibited collagen synthesis by RA synovium explants. On mouse cartilage, IL-17 enhanced cartilage proteoglycan loss and inhibited its synthesis. On human RA bone explants, IL-17 also increased bone resorption and decreased formation. Addition of IL-1 in these conditions increased the effect of IL-17. Blocking of bone-derived endogenous IL-17 with specific inhibitors resulted in a protective inhibition of bone destruction. Conversely, intra-articular administration of IL-17 into a normal mouse joint induced cartilage degradation. In conclusion, the contribution of IL-17 derived from synovium and bone marrow T cells to joint destruction suggests the control of IL-17 for the treatment of RA.
Keywords: bone, cartilage, degradation, IL-17, rheumatoid arthritis
Introduction
RA is a chronic inflammatory disease characterized by articular cartilage and bone destruction. The pathogenesis of inflammation and tissue destruction in RA results from complex cell–cell interactions between, among others, T lymphocytes, monocytes and synoviocytes [1,2,3].
The contribution of the pro-inflammatory macrophage-derived cytokines IL-1 and tumor necrosis factor (TNF)-α has been well established through their capacity to modulate cartilage and bone metabolism. On the one hand, IL-1 and TNF-α stimulate osteoblasts to secrete cytokines such as granulocyte–monocyte colony stimulating factor (GM-CSF) and IL-6, which in turn regulate osteoclast formation. This leads, or not, to osteoclastic bone resorption according to the local conditions [4,5,6]. Chondrocyte activation similarly contributes to cartilage matrix destruction. On the other hand, IL-1 and TNF-α suppress matrix synthesis in cartilage and inhibit collagen synthesis in osteoblasts, leading to defective repair [7,8]. These two molecules are now, accordingly, the main targets for treatment, and the use of blocking anti-TNF-α antibodies, soluble TNF-α receptors and IL-1 receptor antagonist has shown very interesting clinical results, including protective effects on cartilage and bone [9,10,11,12].
The role of T cells and their derived cytokines on these cell interactions inside the synovium has also been the subject of a number of studies. A growing list of factors from T cells, including IL-7, IL-18, GM-CSF, TNF-α and, more importantly, RankL has similarly been shown to act on bone remodeling [6,13,14,15]. As T cells are present in bone marrow, their local contribution is probable. It has long been thought that, in cartilage, cytokines would act only by diffusion. Direct cell contact may, however, occur during the invasion of cartilage by the tumor-like synoviocytes.
Recent information regarding the contribution of T cells through their cytokines has come from results with IL-17. This cytokine is exclusively produced by activated T cells [16]. Furthermore, bioactive IL-17 is produced by RA synovium [17,18]. As IL-17 shares properties with IL-1 and TNF-α, it may affect both osteoclast and chondrocyte-mediated resorption, and thereby mediate bone and cartilage destruction [19,20]. Whether IL-17 produced by RA synovium affects bone by diffusion or whether IL-17 produced locally by juxta-articular bone is also a mediator of bone destruction remains to be clarified.
The aim of the present study was to assess the role of IL-17, used alone and in association with IL-1, in the inflammatory and destructive pattern characteristic of RA, by looking at its effects on synovium, cartilage and bone. Through the use of IL-17 inhibitors, we then looked at the respective contribution of IL-17 produced by synovium and bone. Finally, intra-articular administration of IL-17 was performed to study its direct effect on destruction.
Materials and methods
Cytokines and reagents
Dr Fossiez (Schering-Plough Research Center, Dardilly, France) kindly provided human recombinant IL-17 and anti-IL-17 monoclonal antibody (mAb) 5. One microgram per milliliter of mAb5 was able to completely inhibit the IL-6 production by synoviocytes induced by 50 ng/ml IL-17, whereas an irrelevant mAb had no effect [21]. This antibody had no effect on the action of IL-1, TNF-α, GM-CSF, and monocyte-CSF. Human recombinant IL-1β (2 × 108 U/mg) was purchased from Sigma Chemical Co (St Louis, MO, USA). Murine soluble IL-17 receptor (sIL-17R) was kindly provided by Dr Kendall Mohler (Immunex, Seattle, WA, USA), the properties of which have been previously described [22]. One microgram per milliliter of sIL-17R was able to completely inhibit the IL-6 production by synoviocytes induced by 50 ng/ml IL-17, whereas normal human IgG used as control for the fusion IgG–Fc part had no effect (data not shown).
Preparation of synovium and bone explant cultures
Synovium and bone samples were obtained from patients with RA, according to the revised criteria of the American College of Rheumatology [23], who were undergoing knee or wrist synovectomy, or joint replacement. Synovium and bone fragments were prepared as already described [24,25]. These samples show prolonged and active cytokine production and bone resorption, related to the persistence of active osteoclasts. Samples were cut into small pieces of approximately 2 mm3, and incubated in triplicate in complete medium made of MEM medium (Gibco, Grand Island, NY, USA), 2 mM L-Glutamin, 100 U/ml penicillin, 50 mg/ml gentamicin, 20 mM Hepes buffer and 1% fetal calf serum. Synovium and bone fragments were cultured in 24-well plates (Falcon, Oxnard, CA, USA) in a final volume of 2 ml. The cytokines or IL-17 inhibitors to be tested were added at the beginning of the culture. Cultures were performed at 37°C in a 5% CO2/95% humidified air. Supernatants were collected at day 7 in order to measure the accumulation of effects related to the cytokine cascade.
Measurement of type I collagen degradation in human samples
Type I collagen C-telopeptide breakdown products (CTX) were measured in synovium piece culture supernatants by a two-site enzyme-linked immunosorbent assay (ELISA) (Serum Crosslaps one step™; Osteometer Biotech®, Ballerup, Denmark) using two monoclonal antibodies raised against a synthetic peptide with an amino acid sequence specific for a part of the C-telopeptide of the a1-chain of type I collagen [26]. Intra-assay and inter-assay coefficients of variation are lower than 5 and 8%, respectively, and the sensitivity is 154 pmol/l.
Determination of human type I collagen production in human samples
The production of type I collagen was estimated in culture media by measuring the concentration of the C-propeptide of type I collagen (PICP) using a two-site ELISA, which uses a monoclonal and a polyclonal antibody raised against human PICP purified from skin fibroblast cultures (Procollagen-CTM; Metra Biosystem, Inc, Palo Alto, CA, USA) [27]. The sensitivity of the assay is 1 ng/ml, and the intra-assay and inter-assay coefficients of variation are less than 7%.
Determination of IL-6 levels by ELISA
ELISA measured IL-6 levels as previously described [28]. Briefly, supernatants or serial dilutions of IL-6 standard (Schering-Plough Research Institute, Kenilworth, NJ, USA) were incubated for 60 min at 37°C in 96-well microtiter plates (Nunc, Roskilde, Denmark) coated overnight at 4°C with the mouse 39C3 anti-IL-6 mAb (1 mg/ml), and saturated for 60 min at 20°C with phosphate buffered saline 5% bovine albumin. Biotinylated mouse MQ2 anti-IL-6 mAb (1 mg/ml) was added after washing and the mixture incubated for 60 min at 20°C. The plates were read at 492 nm after subsequent incubation with peroxidase streptavidin and revelation with orthophenylenediamine (Sigma Chemical Co). The sensitivity of the assay was 150 pg/ml.
Cartilage tissue culture
Patellae obtained from C57B/1 mice, aged 10–12 weeks, were dissected in a standardized manner from the knee joint as previously described [29]. Specimens were cultured in 200 μl RPMI 1640 (Dutch modification), with glutamic acid (2 mM) and 0.1% bovine serum albumin at 37°C in a humidified 5% CO2 atmosphere. In addition, 0.25 μg human recombinant insulin-like growth factor (IGF-1) was added to provide optimal metabolic activity of chondrocytes [30]. Explants were cultured in the presence of murine recombinant IL-1 and/or IL-17 (R&D system, Minneapolis, MN, USA). Determination of the synthesis rate and release of cartilage proteoglycan (PG) turnover was performed after culture for 48 h in the presence of cytokines.
Determination of mouse chondrocyte proteoglycan synthesis
As a measurement for PG synthetic activity of chondrocyte, the sulfate incorporation rate into PG was determined as previously described [29]. This rate was calculated from the 35SO42- incorporation and the specific activity of the medium. After culture in the presence of cytokines, specimens were pulse-labeled with 35S-sulfate (20 μCi/ml) for three additional hours. The specimens were thereafter washed three times, fixed in 4% formalin and subsequently decalcified in formic acid (5%) for 4 h. The patellar cartilage was punched out of adjacent tissue and dissolved in 0.5 ml Lumasolve (Omnilabo, Breda, The Netherlands). The 35S-sulfate incorporation label was measured by scintillation counting.
Determination of mouse cartilage proteoglycan breakdown
Patellae were pulse-labeled with 35S-sulfate for 3 h before exposure to cytokines. After three washes with medium, patella specimens were cultured for an additional 48 h in the presence of IGF-1 (0.25 μg/ml) and/or the various combinations of IL-1 and IL-17. Cultures were performed with six patella specimens per variable, and the data are expressed as mean values ± SD relative to the values found with IGF-1 alone.
Intra-articular injection of IL-17
Recombinant murine IL-17 (100 ng/ml) alone was injected into the joint space of the right knee of naïve C57Bl/6 mice at days 0 and 3. The contra-lateral joint received an equal volume (6 μl) of saline. Mice were killed by cervical dislocation 2 days after the last injection. Whole knee joints were thereafter removed and fixed for 4 days in 10% formalin. Specimens were processed for paraffin embedding after decalcification in 5% formic acid. Tissue sections (7 μm) were stained with hematoxylin and eosin, and Safranin O. Proteoglycan depletion was determined using Safranin O staining.
Statistical analysis
Results were expressed as mean ± SEM of the indicated number of experiments. Statistical evaluation was performed with the nonparametric Wilcoxon paired t test.
Results
Effect of IL-17 on IL-6 production by RA synovium explants
An ex vivo model of RA synovitis has been established using synovium pieces obtained at surgery [24]. Since RA synovium is composed of different cell types, we tested how IL-17, a T-cell product, would regulate the production of IL-6 and we compared its effect with that of IL-1. Thus synovium pieces were incubated with and without 50 ng/ml IL-17 and/or with and without 100 pg/ml IL-1. Levels of IL-6 were measured after 7 days of culture. IL-17 increased spontaneous IL-6 production (mean ± SEM, 202 ± 57 ng/ml) by 64 ± 17% (Fig. 1). IL-1 also increased spontaneous IL-6 production by 90 ± 27%. The combination of IL-17 and IL-1 induced a 189% increase of IL-6 production.
Figure 1.
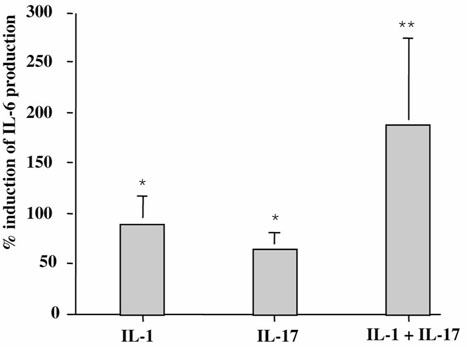
Effect of exogenous IL-17 on IL-6 production by RA synovium. Synovium samples from RA patients were incubated for 7 days in the presence of IL-17 (50 ng/ml; n = 10), IL-1 (100 pg/ml; n = 10), and IL-17 + IL-1 (n = 3). ELISA measured IL-6 levels in supernatants. Results are expressed as mean ± SEM of % induction of IL-6 production. Spontaneous production of IL-6 was 202 ± 57 ng/ml. *P < 0.01, **P < 0.05 compared with control (medium alone).
Effect of IL-17 on type I collagen metabolism in synovium explants
To investigate the consequences of addition of exogenous IL-17, we measured its effect on the destruction/formation activity of type I collagen by synovium explants. First, synthesis of type I collagen in synovium cultures was analyzed by measuring the release of PICP in supernatants by ELISA. Spontaneous production of PICP was 371 ± 36 ng/ml (range, 334–442 ng/ml). IL-17 inhibited these levels by a mean of 38% and inhibited IL-1 by a mean of 23% (n = 3; Fig. 2a).
Figure 2.

Effect of exogenous IL-17 on type I collagen metabolism in RA synovium explants. Synovium samples from RA patients were incubated for 7 days in the presence of IL-17 (50 ng/ml) or IL-1 (100 pg/ml). (a) PICP (n = 4) levels in supernatants were measured by ELISA. Results are expressed as mean ± SEM of % induction of PICP production. Spontaneous production of PICP was 371 ± 36 ng/ml. (b) CTX levels (n = 7) in supernatants were measured by ELISA. Results are expressed as mean ± SEM. Spontaneous production of CTX was 33 ± 11 ng/ml. *P < 0.05, **P < 0.01 compared with control (medium alone).
Second, to assess the degradation of type I collagen from these synovium explants, we measured levels of CTX, a C-terminal peptide released during degradation of type I collagen. Spontaneous production of CTX was 33 ± 11 nM (range, 3-77 nM). IL-17 enhanced CTX release by 211%, an effect that was as potent as that of IL-1 (274%) (Fig. 2b). These results combined indicated a dual effect of IL-17 on synovium, leading to increased destruction and reduced formation of type I collagen.
Effect of IL-17 on cartilage proteoglycan synthesis and degradation
The second important target in a joint is cartilage. There are, however, serious limitations to using the same type of ex vivo model with samples of cartilage obtained during joint surgery in RA. Only limited amounts of cartilage could obviously be obtained that way. To assess such an important target, we switch to a mouse model where patellae are cultured ex vivo in the presence of exogenous cytokines. First, we examined the capacity of IL-17 to inhibit chondrocyte PG synthesis. Whole patellae were incubated with IL-17 and/or IL-1 for 48 h under IGF-1 stimulation, which induced an optimal synthesis of PG. Addition of IL-17 and IL-1 inhibited this synthesis by 23 and 40%, respectively (Table 1). The combination of IL-17 and IL-1 was more potent, inhibiting PG synthesis by 63%.
Table 1.
IL-17 inhibits mouse chondrocyte proteoglycan (PG) synthesis
| Culture conditiona | |||
| IGF-1 | IL-1 | IL-17 | Chondrocyte PG synthesis (%)b |
| + | - | - | 100 ± 11 |
| + | + | - | 58 ± 5* |
| + | - | + | 66 ± 8* |
| + | + | + | 40 ± 5* |
a Mouse patellae incubated with insulin-like growth factor (IGF-1) (0.25 μg/ml), and with and without IL-1 (1 ng/ml), IL-17 (10 ng/ml), or IL-1 and IL-17 for 48 h. b Values represent percentages of sulfate incorporation into proteoglycans ± SD of four separate experiments with six mice per group per experiment. *P < 0.01 compared with IGF-1 alone, by Wilcoxon rank sum test.
Second, we looked at the effects of IL-17 and IL-1 on PG release. PG release during cartilage culture with cytokines was enhanced by 22% with 100 ng/ml IL-17, and by 25% with 10 ng/ml IL-1 (Fig. 3). Combination of IL-17 and IL-1 was more potent, increasing PG loss by 35%. Our results show that inhibition of PG synthesis by IL-17 was accompanied by PG breakdown, indicating that IL-17 induced cartilage degradation and suppression of its synthesis. These results combined also indicate a dual effect of IL-17 on cartilage, increasing PG breakdown and decreasing its synthesis.
Figure 3.
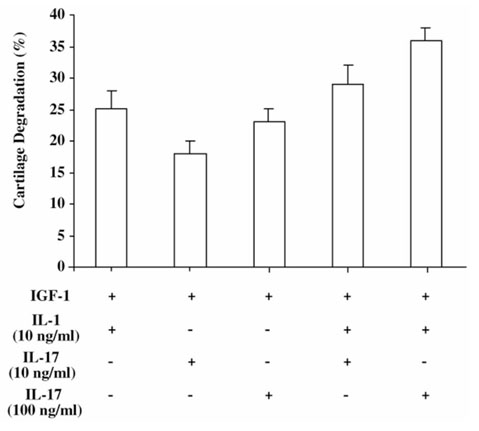
Effect of exogenous IL-17 on mouse cartilage proteoglycan breakdown. Cartilage explants of patellae were pulse-labeled with 35S-sulfate for 3 h and incubated with IGF-1 (0.25 μg/ml), with or without IL-17 (10 or 100 ng/ml) and/or IL-1 (10 ng/ml) for 48 h. Cultures were performed with six patellae per variable, and data represent percentages of sulfate incorporation into proteoglycan ± SD relative to the values found with IGF-1 alone.
Effect of IL-17 on IL-6 production by RA bone explants
Finally, the third target to consider was bone because RA leads to early juxta-articular bone loss. Regarding the origin of cytokines affecting bone, cytokines produced by synovium can reach bone by diffusion or be released by the bone microenvironment itself. IL-6 has been shown to favor osteoclastogenesis. An ex vivo model of bone resorption was established to investigate the effect of IL-17 on IL-6 release by bone using bone pieces obtained at the site of joint surgery of patients with RA as previously described [25]. Bone pieces were incubated, as for synovium pieces, with and without 50 ng/ml IL-17 and/or with and without 100 pg/ml IL-1. IL-17 increased the spontaneous production (220 ± 95 ng/ml) of IL-6 by these bone pieces by 41 ± 25% and that of IL-1 by 73 ± 21% (Fig. 4). The combination of IL-17 and IL-1 had a much larger effect on IL-6 production (171 ± 55%).
Figure 4.
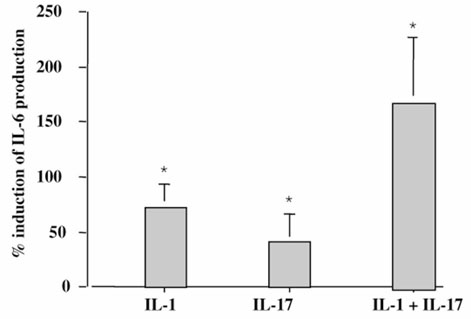
Effect of exogenous IL-17 on IL-6 production by RA bone explants. Bone samples from RA patients were incubated for 7 days in the presence of IL-17 (50 ng/ml; n = 5), IL-1 (100 pg/ml; n = 5), and IL-17 + IL-1 (n = 2). ELISA measured IL-6 levels in supernatants. Results are expressed as mean ± SEM of % induction of IL-6 production. Spontaneous production of IL-6 was 220 ± 95 ng/ml. *P < 0.05 compared with control (medium alone).
Effect of IL-17 on type I collagen metabolism in bone explants
IL-17 and/or IL-1 were, as already shown, able to induce collagen degradation and, more importantly, to decrease collagen synthesis by synovium and cartilage. It was important to extend these results to bone collagen. We thus tested how IL-17 would regulate collagen synthesis/degradation balance using this model. Samples of RA bone were incubated in the presence or absence of IL-17 and/or IL-1.
First, we investigated the consequences of IL-17 on collagen synthesis by measuring the release of PICP by bone explants. The spontaneous production of PICP was 433 ± 133 ng/ml (range, 299–567 ng/ml). IL-17 inhibited such production by 42 ± 10% and IL-1 by 21 ± 3% (Fig. 5a).
Figure 5.
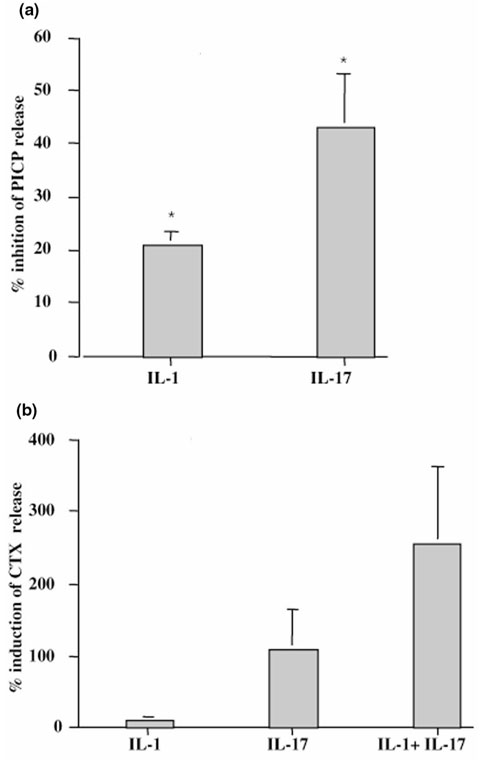
Effect of exogenous IL-17 on type I collagen metabolism by RA bone explants. Bone samples from RA patients were incubated for 7 days in the presence of IL-17 (50 ng/ml) and/or IL-1 (100 pg/ml). (a) PICP levels (n = 3) in supernatants were measured by ELISA. Results are expressed as mean ± SEM of % induction of PICP production. Spontaneous production of PICP was 433 ± 133 ng/ml. (b) CTX levels in supernatants were measured by ELISA. Bone samples were incubated with IL-17 (n = 5), IL-1 (n = 5), and IL-17 + IL-1 (n = 2). Results are expressed as mean ± SEM. Spontaneous production of CTX was 104 ± 55 ng/ml. *P < 0.05 compared with control (medium alone).
We then analyzed the effect of IL-17 on type I collagen degradation by measuring CTX release in bone culture supernatants. Spontaneous release of CTX by bone culture was variable (104 ± 55 nM; range, 8-331 nM). IL-17 increased this release by 110% and IL-1, being less potent, by 11% (Fig. 5b). The combination of IL-1 and IL-17 had a synergistic effect increasing CTX release by 257%. As shown for synovium and then cartilage, IL-17 was also able to increase bone destruction and reduce its formation.
Contribution of bone-derived endogenous IL-17 to destruction
Regarding the two possible mechanisms of bone destruction in RA (ie diffusion of cytokines derived from synovium or production by juxta-articular bone), we looked at the contribution of endogenous IL-17 produced first by RA synovium and then by RA bone explants with specific IL-17 inhibitors. When RA synovium explants were incubated with sIL-17R or with an anti-IL-17 mAb (mAb5), spontaneous CTX production was inhibited in a same fashion by 75 and 88%, respectively (n = 5; Fig. 6a).
Figure 6.
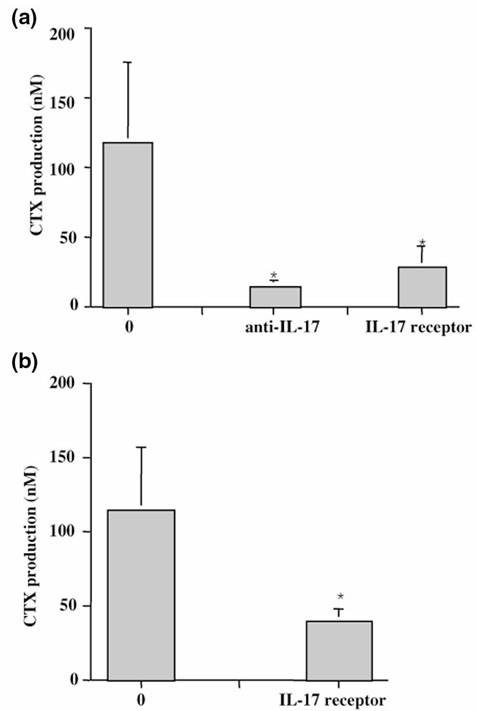
Effect of blockade of endogenous IL-17 on synovium and bone destruction. (a) Synovium (n = 5) and (b) bone explants (n = 3) from RA patients were incubated for 7 days with and without blocking anti-IL-17 mAb (10 μg/ml) or sIL-17R (1 μg/ml), which was added at the onset of the culture. CTX levels after 7 days of culture were measured by ELISA. Results are expressed as mean ± SEM of culture triplicates. Differences between IL-17 inhibitor treated groups and control groups were analyzed with the nonparametric Wilcoxon paired t test.*P < 0.05.
Previous studies regarding the in situ bone environment have demonstrated that juxta-articular bone itself can produce as many cytokines as the inflamed synovium from the same joint [25]. This was first described for cytokines produced by monocytes and mesenchymal cells such as IL-6 and leukemia inhibitory factor. To look at the potential contribution of bone-derived T-cell cytokines, namely IL-17, we performed similar blocking experiments with bone cultures. The sIL-17R was added at the onset of bone culture, and CTX release in supernatants was analyzed by ELISA. Results show that sIL-17R could reduce CTX release by a mean of 64% in bone cultures (Fig. 6b). Paired samples of bone and synovium were available from the same joint for two patients. These samples, as shown in Figure 7, indicated a much higher rate of collagen destruction in bone when compared with synovium. More importantly, sIL-17R induced a very profound reduction of CTX release by bone pieces (134.2 ± 63.7 ng/ml without versus 45.5 ± 8.8 ng/ml with sIL-17R; P < 0.05). Similar results were obtained with synovium pieces (12.6 ± 0.2 ng/ml without sIL-17R versus 4.6 ± 0.8 ng/ml with sIL-17R; P < 0.05). These final results demonstrate the direct contribution of endogenous IL-17 to synovium and, moreover, to bone destruction in RA. They suggest, in particular, that a T-cell subset secreting IL-17 inside bone can contribute significantly to bone destruction.
Figure 7.
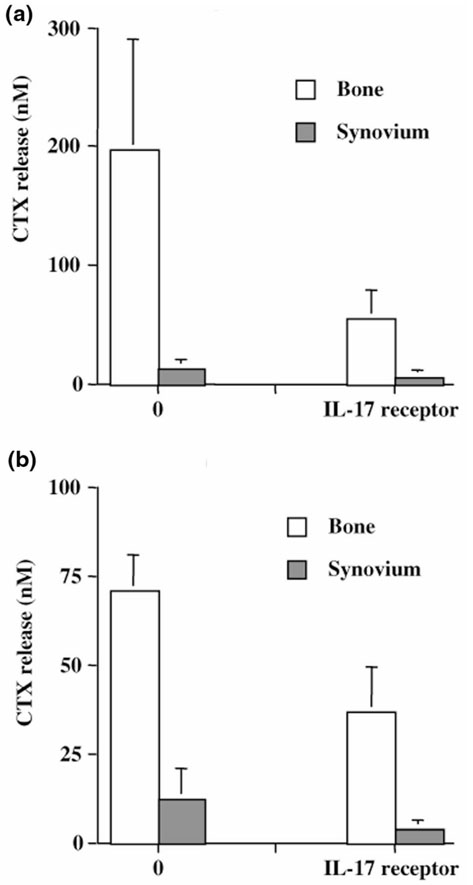
Respective contribution of endogenous IL-17 to synovium and bone destruction. Paired bone and synovium pieces from the same joint in two RA patients were cultured for 7 days in the presence of sIL-17R (1 μg/ml). CTX production in supernatants was measured by ELISA, and results are expressed in nanomoles.
Intra-articular administration of murine rIL-17 leads to joint degradation
To extend these results to an in vivo situation, murine IL-17 was injected into mouse knee joints. We used normal mice to investigate solely the direct contribution of IL-17. Whole knee sections were stained for cartilage proteoglycan content. Figure 8 shows that profound proteoglycan depletion was observed in the IL-17 treated group when compared with the control group. These findings extend the catabolic effects of IL-17 on cartilage degradation to the in vivo situation.
Figure 8.

Effect of intra-articular administration of IL-17 on cartilage destruction. Naive C57Bl/6 mice were intra-articulately injected into the knee joint with recombinant murine IL-17 (100 ng/ml) at days 0 and 3 (b and d). The contralateral joint received an equal volume (6 μl) of saline (a and c). Knee joints were taken for histology 2 days after the last injection. Proteoglycan depletion was analyzed using Safranin O staining. Cartilage depletion was visualized by diminished staining of the matrix. The effect of IL-17 can be seen as the lighter (b and d) rather than the darker staining (a and c) in cartilage. (a) and (b), Original magnification, × 50; (c) and (d), original magnification × 200. P, Patella; F, femur; JS, joint space; C, cartilage.
Discussion
The clinical problem with RA is not only chronic joint inflammation, but also moreover progressive destruction of cartilage and bone. IL-1 and TNF-α are considered the leading factors in RA that drive the enhanced production of cytokines, chemokines and degradative enzymes [9]. The definition of IL-17 as a pro-inflammatory T-cell derived cytokine produced by RA synovium has lead to the hypothesis that such a cytokine could also be involved in joint destruction. We have accordingly analyzed the role of IL-17 in the inflammation and destruction patterns associated with RA.
IL-17 shares many properties with IL-1 and TNF-α. The three cytokines activate the common transcription factor NF-κB in a variety of cell types. They all stimulate stromal cells such as dermal and synovial fibroblasts, endothelial cells and epithelial cells to secrete IL-6, IL-8 and PGE2 [16,21,31]. Interactions between these three cytokines further amplify these effects. IL-17 induces IL-1 and TNF-α production by human macrophages [32]. Monocyte activation, conversely, induces Th1 cell differentiation through IL-12 production, and IL-12 increases IL-17 production by T cells as shown with T-cell clones [33]. More importantly, combination of IL-17 with IL-1 or TNF-α often leads to synergistic or additive effects, which further increase their biological effects. The present study demonstrates the potency of IL-17, often similar to that of IL-1, to induce inhibition of type I collagen synthesis by synovium and bone, but also of PG synthesis by cartilage. As Th1 cells infiltrate RA synovium, IL-17 produced by some of these Th1 cells can increase IL-1 and TNF-α production, enhancing the rate of degradation and lack of repair, which are characteristics of RA. Conversely, in osteoarthritis, where the contribution of T cells is less pronounced, destruction occurs but collagen synthesis is not as strongly inhibited. The presence of T cells, more precisely of Th1 cells, appears to be linked to defective repair when comparing the two conditions.
Inflammation and tissue destruction in RA synovium result from complex cell-cell interactions. Interactions between antigen presenting cells and CD4+ T cells lead to macrophage activation and secretion of IL-1 and TNF-α [2,34]. These factors stimulate matrix degradation collagen in synovium, cartilage and bone. Similar results were obtained with IL-17 on induction of matrix metalloproteinase-1 production by synoviocytes and collagenase activity by synovium [35]. The present study extends these results by showing an enhanced effect when combined with IL-1.
Cartilage destruction is a major consequence of chronic synovitis. At an early stage, cytokines and other soluble factors acting on chondrocytes most probably induce such a process by diffusion. At a later stage, proliferation and activation of synovial cells lead to pannus formation that invades and destroys cartilage. Previous studies have demonstrated that cartilage degradation is largely dependent on IL-1 and TNF-α. We show in this paper that cartilage degradation can also be induced by IL-17 inhibiting PG synthesis and increasing its destruction. IL-17 has also already been shown to induce nitric oxide and stromelysin expression by isolated chondrocytes [36,37]. Administration of IL-17 into normal knee was, more importantly, able to induce such degradation [38]. It is important to notice that such effect was observed using IL-17 alone and not in an inflamed joint where IL-1, TNF-α and IL-17 would have been already present. The endogenous secretion of IL-17 from cartilage appears unlikely and was not evaluated as IL-17 is still considered a T-cell specific cytokine.
The major contribution of this work is the demonstration of the role of IL-17 and thus of T cells from bone in destruction and defective formation. This was shown using specific IL-17 inhibitors. The important inhibition observed when blocking IL-17 alone can be explained by the synergistic interaction between IL-17 and other cytokines, as first shown with fibroblasts and synoviocytes [16,21]. Th2 cell cytokines such as IL-4 have conversely been shown to inhibit osteoclastogenesis. In a previous study using the same model, we showed that exogenous IL-4 was able to decrease bone destruction through an inhibitory effect on osteoclast survival [25]. In keeping with the Th classification of cytokines, IL-17 appears to have the opposite effect in providing signals for osteoclast activity leading to increased bone destruction and reduced formation. This extends the results in the mouse showing a defective formation with IL-17 [19,20]. Previous studies have already demonstrated the direct contribution of T cells to osteoclast differentiation through cell contacts with osteoclasts [39]. T cells were shown to express RankL in particular [6,40]. Furthermore, this activator of osteoclast formation and activation is also present in RA synovium [41,42]. Administration of IL-4 in the mouse model of collagen arthritis was able to control cartilage and bone destruction through an inhibition of the expression of IL-1, TNF-α, IL-17 and RankL [43].
Osteoclasts are specialized cells with the capacity of resorbing bone, whereas osteoblastic stromal cells are responsible for matrix formation. Bone formation is regulated by an event referred to as the coupling of formation to resorption, which is mediated in part by locally produced growth factors. Because of the intimate relationship between bone marrow cells and bone-specific cells, marrow cells and their products contribute to the bone microenvironment and influence the regulation of bone cell differentiation and activity. In the context of RA, early juxta-articular bone loss appears to result from in situ bone activation. Results with paired samples have shown a much higher level of destruction in bone compared with the synovium. It appears now that juxta-articular bone can produce bone-resorbing cytokine(s) from a subset of pro-inflammatory Th1 cells. These results obtained ex vivo in man extend those obtained in the mouse, which showed a direct effect of IL-17 on bone resorption [19].
The present data have significant implication for the therapeutic control of joint damage during inflammation. We could downregulate type I collagen degradation in both synovium and bone using specific IL-17 inhibitors. As IL-17 can increase IL-1 and TNF-α production by macrophages, blocking IL-17 will also decrease IL-1 and TNF-α actions. Furthermore, combining inhibitors of IL-17 with those of IL-1 and TNF-α may lead to a better effect. IL-17 may accordingly represent a target of interest for the treatment of RA, particularly to reduce its consequences on destruction.
Abbreviations
CTX = C-terminal collagen cross-links; ELISA = enzyme-linked immunosorbent assay; GM-CSF, granulocyte–monocyte colony stimulating factor; IGF-1 = insulin-like growth factor; mAb, monoclonal antibody; PG = proteoglycan; PICP = C-propeptide of type I collagen; RA = rheumatoid arthri-tis; sIL-17R = soluble IL-17 receptor; TNF, tumor necrosis factor.
Acknowledgments
Acknowledgements
The authors thank Prof J Béjui and Prof G Herzberg for providing synovium samples, Dr P Garnero and E Thomas (Synarc, Lyon, France) for CTX/PICP determination, Dr F Fossiez for providing the anti-IL-17 antibody, and Dr K Mohler for providing the murine IL-17 receptor. These studies have been supported in part by grants from the Hospices Civils de Lyon, l'Association de Recherche sur la Polyarthrite and from the European Union (Biomed-2 program contract BMH4-CT96-1698), and by grants from the Dutch League of Rheumatism, 'Het Nationaal Reumafonds'.
References
- Arend WP. The pathophysiology and treatment of rheumatoid arthritis. Arthritis Rheum. 1997;40:595–597. doi: 10.1002/art.1780400402. [DOI] [PubMed] [Google Scholar]
- Chizzolini C, Chicheportiche R, Burger D, Dayer JM. Human Th1 cells preferentially induce interleukin (IL)-1beta while Th2 cells induce IL-1 receptor antagonist production upon cell/cell contact with monocytes. Eur J Immunol. 1997;27:171–177. doi: 10.1002/eji.1830270125. [DOI] [PubMed] [Google Scholar]
- Chomarat P, Rissoan MC, Pin JJ, Banchereau J, Miossec P. Contribution of IL-1, CD 14, CD 13 in the increased IL-6 production during monocyte synoviocyte interactions. J Immunol. 1995;155:3645–3652. [PubMed] [Google Scholar]
- Manolagas SC. Role of cytokines in bone resorption. Bone. 1995;17:63S–67S. doi: 10.1016/8756-3282(95)00180-l. [DOI] [PubMed] [Google Scholar]
- Lader CS, Flanagan AM. Prostaglandin E2, interleukin-1a, and tumor necrosis factor-α, increase human osteoblast formation and bone resorption in vitro. Endocrinology. 1998;139:3157–3164. doi: 10.1210/endo.139.7.6085. [DOI] [PubMed] [Google Scholar]
- Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT. Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun. 1999;265:144–150. doi: 10.1006/bbrc.1999.1623. [DOI] [PubMed] [Google Scholar]
- Van de Loo AAJ, Van den Berg WB. Effects of murine recombinant interleukin-1 on synovial joints in mice; measurement of patella cartilage metabolism and joint inflammation. Ann Rheum Dis. 1990;49:238–245. doi: 10.1136/ard.49.4.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghivizzani SC, Kang R, Georgescu HI, Lechman ER, Jaffurs D, Engle JM, Watkins SC, Tindal MH, Suchanek MK, McKenzie LR, Evans CH, Robbins PD. Constitutive intra-articular expression of human IL-1 beta following gene transfer to rabbit synovium produces all major pathologies of human rheumatoid arthritis. J Immunol. 1997;159:3604–3612. [PubMed] [Google Scholar]
- Arend WP, Dayer J-M. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor α in rheumatoid arthritis. Arthritis Rheum. 1995;38:151–160. doi: 10.1002/art.1780380202. [DOI] [PubMed] [Google Scholar]
- Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, Widmer MB, Blosch CM. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 1997;337:141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, Antoni C, Leeb B, Elliott MJ, Woody JN, Schaible TF, Feldmann M. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41:1548–1551. doi: 10.1002/1529-0131(199809)41:9<1552::AID-ART5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Bresnihan B, Alvaro-Garcia J-M, Cobby M, Doherty M, Domljan Z, Emery P, Nuki G, Pavelka K, Rau R, Rozman B, Watt I, Williams B, Aitchison R, McCabe D, Musikic P. Treatment of rheumatoid arthritis with recombinant interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–2104. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Horwood NJ, Udagawa N, Elliott J, Grail D, Okamura H, Kurimoto M, Dunn AR, Martin T, Gillespie MT. Interleukin 18 inhibits osteoclast formation via T cell production of granulocyte macrophage colony-stimulating factor. J Clin Invest. 1998;101:595–603. doi: 10.1172/JCI1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R. Interleukin-7 stimulates osteoclast formation by upregulating the T-cell production of soluble osteoclastogenic cytokines. Blood. 2000;96:1873–1878. [PubMed] [Google Scholar]
- Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P. Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246–1251. [PubMed] [Google Scholar]
- Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bezooijen RL, Farih-Sips HC, Papapoulos SE, Löwik CW. Interleukin-17: a new bone acting cytokine in vitro. J Bone Miner Res. 1999;14:1513–1521. doi: 10.1359/jbmr.1999.14.9.1513. [DOI] [PubMed] [Google Scholar]
- Chabaud M, Fossiez F, Taupin JL, Miossec P. IL-17 enhances the effects of IL-1 on IL-6 and LIF production by rheumatoid arthritis synoviocytes. J Immunol. 1998;161:409–414. [PubMed] [Google Scholar]
- Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Pries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA, Mitchell DM, Nenstadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GC. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Miossec P, Briolay J, Dechanet J, Wijdenes J, Martinez-Valdez H, Banchereau J. Interleukin 4 inhibits ex vivo production of proinflammatory cytokines and immunoglobulins by rheumatoid synovitis. Arthritis Rheum. 1992;35:874–883. doi: 10.1002/art.1780350805. [DOI] [PubMed] [Google Scholar]
- Miossec P, Chomarat P, Dechanet J, Moreau J-F, Roux J-P, Delmas P, Banchereau J. Interleukin 4 inhibits bone resorption through an effect on osteoclasts and proinflammatory cytokines in an ex vivo model of bone resorption in rheumatoid arthritis. Arthritis Rheum. 1994;37:1715–1722. doi: 10.1002/art.1780371202. [DOI] [PubMed] [Google Scholar]
- Bonde M, Garnero P, Fledelius C, Qvist P, Delmas PD, Christiansen C. Measurement of bone degradation products in serum using antibodies reactive with an isomerized form of an 8 amino acid sequence of the C-telopeptide of type I collagen. J Bone Miner Res. 1997;12:1028–1034. doi: 10.1359/jbmr.1997.12.7.1028. [DOI] [PubMed] [Google Scholar]
- Garnero P, Sornay-Rendu E, Chapuy MC, Delmas PD. Increased bone turnover in late postmenopausal women is a major determinant of osteoporosis. J Bone Miner Res. 1996;11:337–349. doi: 10.1002/jbmr.5650110307. [DOI] [PubMed] [Google Scholar]
- Abrams JS, Roncarolo M-G, Yssel H, Andersson U, Gleich GJ, Silver JE. Strategies of anti-cytokine monoclonal antibody development: immunoassay of IL-10 and IL-5 in clinical samples. Immunol Rev. 1992;127:5–24. doi: 10.1111/j.1600-065x.1992.tb01406.x. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LAB, Helsen MMA, Van den Berg WB. Regulatory role of interleukin-10 in joint inflammation and cartilage destruction in murine streptococcal cell wall (SCW) arthritis. Murine therapeutic benefit with IL-4/IL-10 combination therapy than with IL-10 treatment alone. Cytokine. 1998;10:361–369. doi: 10.1006/cyto.1997.0298. [DOI] [PubMed] [Google Scholar]
- van Beuningen HM, Arntz OJ, van den Berg WB. Insulin-like growth factor stimulation of articular chondrocyte proteoglycan synthesis. Availability and responses at different ages. Br J Rheumatol. 1993;32:1037–1043. doi: 10.1093/rheumatology/32.12.1037. [DOI] [PubMed] [Google Scholar]
- Awane M, Andres PG, Li DJ, Reinecker HC. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162:5337–5344. [PubMed] [Google Scholar]
- Jovanovic DV, DiBattista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- Aarvak T, Chabaud M, Kallberg E, Miossec P, Natvig JB. Change in the Th1/Th2 phenotype of memory T-cell clones from rheumatoid arthritis synovium. Scand J Immunol. 1999;50:1–9. doi: 10.1046/j.1365-3083.1999.00581.x. [DOI] [PubMed] [Google Scholar]
- Vey E, Burger D, Dayer JM. Expression and cleavage of tumor necrosis factor-alpha and tumor necrosis factor receptors by human monocytic cell lines upon direct contact with stimulated T cells. Eur J Immunol. 1996;26:2404–2409. doi: 10.1002/eji.1830261021. [DOI] [PubMed] [Google Scholar]
- Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P. Contribution of interleukin-17 to synovium matrix destruction in rheumatoid arthitis. Cytokine. 2000;12:1092–1099. doi: 10.1006/cyto.2000.0681. [DOI] [PubMed] [Google Scholar]
- Attur MG, Patel RN, Abramson SB, Amin AR. Interleukin-17 upregulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheum. 1997;40:1050–1053. doi: 10.1002/art.1780400609. [DOI] [PubMed] [Google Scholar]
- Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem. 1998;273:27467–27473. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- Dudler J, Renggli-Zulliger N, Busso N, Lotz M, So A. Effect of interleukin 17 on proteoglycan degradation in murine knee joints. Ann Rheum Dis. 2000;59:529–532. doi: 10.1136/ard.59.7.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John V, Hock JM, Short LL, Glasebrook AL, Galvin RJ. A role for CD8+ T lymphocytes in osteoclast differentiation in vitro. Endocrinology. 1996;137:2457–2463. doi: 10.1210/endo.137.6.8641199. [DOI] [PubMed] [Google Scholar]
- Kong YY, Boyle WJ, Penninger JM. Osteoprotegerin ligand: a common link between osteoclastogenesis, lymph node formation and lymphocyte development. Immunol Cell Biol. 1999;77:188–193. doi: 10.1046/j.1440-1711.1999.00815.x. [DOI] [PubMed] [Google Scholar]
- Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Romas E, Bakharevski O, Hards DK, Kartsogiannis V, Quinn JM, Ryan PF, Martin TJ, Gillespie MT. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum. 2000;43:821–826. doi: 10.1002/1529-0131(200004)43:4<821::AID-ANR12>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LA, Chabaud M, van Den Bersselaar L, Oppers B, Coenen-De Roo CJ, Richards CD, Miossec P, van Den Berg WB. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105:1697–1710. doi: 10.1172/JCI7739. [DOI] [PMC free article] [PubMed] [Google Scholar]