

Abstract
Bromination of 3-nitro-5,10,15-triarylcorrole selectively provides two regioisomers, depending on the reaction pathway. An isocorrole species is the key intermediate to drive the reaction towards the 2-Br-17-nitro regioisomer.
It is impressive to note the attention that, over the last decade, corrole has received among porphyrinoids. While this consideration certainly arises from the intriguing chemistry shown by this macrocycle, which is unusual compared with the parent porphyrin congeners, it is obvious that these developments have been made possible by progress in synthetic routes leading to triarylcorroles.1–6 The advances achieved in the synthetic chemistry of such a macrocycle allow the preparation of a large variety of corrole derivatives, which have concomitantly opened the way for their practical applications in different fields, ranging from sensors to clinical studies.7–9 For these applications it is of paramount importance to develop methodology for diverse functionalization of corroles with peripheral substituents. It is also noteworthy that the functionalization of the preformed corrole framework is a relatively under-developed area.10,11 Different factors might explain this developmental problem in the corrole field: while from one side the instability of corrole could hamper some functionalization routes, from the other side the lower symmetry of corrole compared with porphyrin could lower the yields of the desired functionalization, because in principle a complex mixture of different regioisomers could be obtained.10 A further obstacle to the functionalization of the macrocycle has been the lack of simple protocols for the demetallation of corrole complexes. Some of these problems have been solved in the last few years, such as the admirable regioselectivity showed by corrole for substitution, and the definition of some route for the demetallation of Cu and Mn metal derivatives.12–16 The problem of corrole instability under reaction conditions for functionalization pathways usually leads to the formation of isocorrole by-products,17,18 formed because of the facile oxidation of the corrole ring, as has been observed, for example, in the case of nitration reactions.19 However, this feature should not be an unavoidable drawback but rather a tool to widen the possible pathways for corrole functionalization. To define synthetic routes for the introduction of different substituents on the peripheral positions of meso-triarylcorroles, we decided to use as starting material the 3-nitro-5,10,15-tris(4-methylphenyl)corrole, 3-NO2-ttcorrH3, which can be obtained in good yields from 5,10,15-tris-(4-methylphenyl)corrole ttcorrH320 using AgNO2 as the nitrating agent,21 followed by reductive demetallation with DBU;19 bromination of corrole was selected as the test reaction, using N-bromosuccinimide (NBS). In a first approach two equivalents of NBS were added at once to a refluxing CHCl3 solution of 3-NO2-ttcorrH3. The control of the reaction by TLC showed the complete disappearance of the starting corrole, with the formation of two spots as reaction products. Chromatographic purification on a silica gel column (CH2Cl2/hexane 1/1 as eluant) did not allow the complete separation of the two fractions, characterized by very close Rf; using the same eluant mixture, a preparative TLC allowed the tentative separation of the second green band. The characterization of this major reaction product showed in the 1H NMR spectrum two low-field resonances, while in the UV-Vis spectrum the broad Soret band of 3-NO2-ttcorrH3 was replaced by a narrow blue-shifted absorption; both of these features are typical signatures of isocorroles. However the MALDI mass spectrum indicated the formation of a disubstituted product, with a molecular peak attributable to a meso-hydroxy isocorrole bearing one bromo and one nitro group at the periphery. The X-ray characterization on a single crystal of this product allowed its unambiguous characterization as the 2-bromo-5-hydroxy-17-nitro-5,10,15-tris(4-methylphenyl)isocorrole, 2-Br-15-OH-17-NO2-ttisocorrH222(Fig. 1).
Fig. 1.
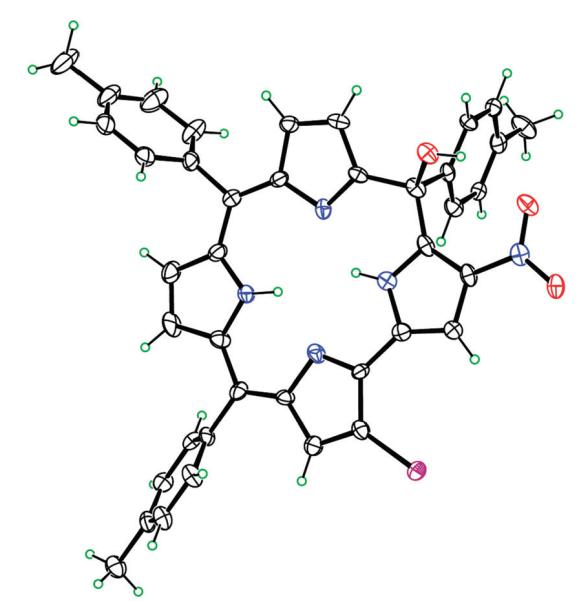
Molecular structure of 2-Br-15-OH-17-NO2-ttisocorrH2 with 50% ellipsoids.
The 23-atom ring system is only slightly nonplanar, with a mean deviation of 0.112 Å and a maximum deviation of 0.318 Å. The major distortion from coplanarity is a tipping of the unsubstituted pyrrole ring adjacent to the sp3 C atom by 15.5(3)° out of the plane of the other 18 ring atoms. The NO2 group is twisted slightly, 4.2(5)° out of the plane of the pyrrole on which it resides, and it accepts an intramolecular hydrogen bond (O⋯O 2.790(4) Å) from the OH group. The C–Br length to the fully-populated bromine is 1.849(4) Å.
It should be noted that the crystallographic characterization showed that 2-Br-15-OH-17-NO2-ttisocorrH2 was contaminated by the presence of the dibromo derivative, 2,3-dibromo-15-hydroxy-17-nitro-5,10,15-tris(4-methylphenyl)-isocorrole 2,3-Br2-15-OH-17-NO2-ttisocorrH2. The presence of this compound derived from the difficult chromatographic separation of the two reaction products. Because the separation of this isomer failed, we reacted the first chromatographic fraction with Co(II) acetate and PPh3 in methanol to prepare the corresponding triphenylphosphino cobalt complex, which allowed the spectroscopic characterization of the compound as a dibromoderivative.
While the partial formation of a dibromo derivative is not surprising, due to the difficult control of polybromination usually observed in the case of corrole derivatives, it is important to note that we were able to avoid the formation of 2,3-Br2-15-OH-17-NO2-ttisocorrH2 by a simple modification of the reaction protocol; in this case NBS was added dropwise to the 3-NO2-ttcorrH3 solution at room temperature, giving 2-Br-15-OH-17-NO2-ttisocorrH2 as the unique product in high yield (80%).
The high regioselectivity of the reaction, which afforded only one isomer in high yield, is noteworthy. While the formation of the 5-hydroxy isocorrole is not unprecedented in the case of 3-nitrocorrole precursors19 and can be attributed to the formation of an intramolecular hydrogen bond between the OH and NO2 groups, the fact that the Br substitution occurred at the 2- instead of the usually more reactive 3-position is quite unexpected.11 This different regioselectivity could be attributed to the influence of the nitro group or, even more interesting, to the isocorrole formation, if it precedes the bromination reaction. To obtain further details to examine this hypothesis, we prepared the 3-NO2 isocorrole and then ran the bromination reaction on this compound. To obtain the precursor isocorrole we attempted to follow the same route used for metal complexes, i.e. oxidation of the macrocycle in the presence of the nitrite ion as nucleophile, which led to the final functionalized isocorrole derivative. Also in this, the regioselective formation of the 5-isocorrole is due to the hydrogen bond with the 3-nitro substituent. The reaction was performed on the ttcorrH3 with NaNO2 in DMF, in the presence of chloranil as oxidant and, after work-up, the resulting isocorrole was obtained in satisfying yields (80%). The subsequent bromination, carried out under the same conditions as with the corresponding corrole, afforded again 2-Br-5-OH-17-NO2-ttisocorrH2 in similar yields. This result strongly supports the hypothesis that the isocorrole intermediate directs the subsequent bromination at the 2-position. While interesting for the regioselectivity, the reaction appears unsuccessful from the synthetic point of view because an isocorrole and not the desired corrole is obtained as product. However, we have already demonstrated that isocorrole formation is reversible and that corrole can be easily obtained from these oxidised species.23 In previous studies we exploited metal coordination to obtain the corresponding corrole complexes: with metal ions having an oxidation state higher than +2, coordination of the metal ion was followed by a metal-to-ligand charge transfer, which afforded the corrole derivative. To obtain the free base corrole, the direct reduction of the isocorrole, using NaBH4 as the reducing agent in a CH2Cl2/CH3OH solution (5 : 1) was attempted. Chromatographic separation of the reaction mixture on a silica gel (CH2Cl2/hexane 3/2 as eluant) afforded a mixture of the starting isocorrole with the desired corrole, which was then separated on basic alumina (Grade III) in a 35% yield. Spectroscopic characterization of the species confirmed the formation of the 2-bromo-17-nitro-5,10,15-tris(4-methylphenyl)corrole, 2-Br-17-NO2-ttcorrH3.
While this route confirmed the regioselective preparation of β-disubstituted corroles, we cannot completely exclude the influence of the nitro group in orienting the subsequent bromine insertion. For this reason the bromination reaction on the silver corrole complex 3-NO2-ttcorrAg19 was attempted, because metal coordination protected the corrole ring from oxidation to the isocorroles species. In this case we increased the NBS/corrole ratio to 3/1 to carry the reaction to completion. The product was obtained in 75% yield and the spectroscopic characterization confirmed the formation of the disubstituted Ag(III) corrole complex. To compare the product with the 2-Br-17-NO2-ttcorrH3 previously obtained, we demetallated the Ag(III) corrole complex using the DBU/THF route, which quantitatively afforded the corresponding free base. Spectroscopic characterization of this product confirmed the formation of a different regioisomer of 2-Br-17-NO2-ttcorrH3. While the MALDI mass spectrum indicated the presence of a Br and a nitro group, the UV-Vis spectrum was also similar to that of 2-Br-17-NO2-ttcorrH3, typical of β-nitro corrole derivative (Fig. 2). The 1H NMR spectrum, however, showed a different pattern for the proton resonances of the substituted pyrroles A and D of the corrole ring (Fig. S1, ESI†). According to these results, the corrole obtained can be definitively identified as the 3-Br-17-NO2-ttcorrH3 isomer; this identification has also been corroborated by carrying out the nitration reaction on the corresponding 3-Br-5,10,15-tris(4-methylphenyl)corrole 3-Br-ttcorrH3, recently obtained in our laboratory.24
Fig. 2.
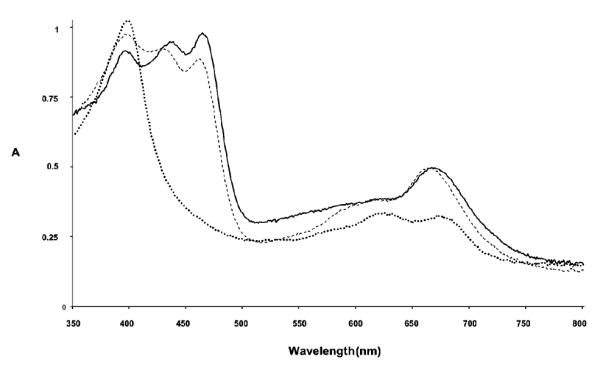
UV-Vis spectra of the 2-Br-17-NO2-ttcorrH3 (solid line), 3-Br-17-NO2-ttcorrH3 (dashed line), 2-Br-15-OH-17-NO2-ttisocorrH2 (dotted line).
The subsequent nitration afforded a single product having the same spectroscopic features of the 3-Br-17-NO2-ttcorrH3. The results confirm that it is the isocorrole intermediate that drives the subsequent bromination of the macrocyclic ring and this feature allows a definition of different pathways for the preparation of two different regioisomers in good yields, as illustrated in Scheme 1.
Scheme 1.

The different reaction pathways for the bromination reaction of 3-NO2-5,10,15-triarylcorrole.
This function for isocorroles is particularly interesting because it represents a useful and general tool for modification of the regioselectivity of corrole functionalization. A comprehensive study of the scope of these reaction pathways is currently underway in our laboratories, and the results will be presented in the due course.
Supplementary Material
Acknowledgments
This research was supported by MIUR Italy (PRIN project 2007C8RW53) and the United States National Institutes of Health (K.M.S. grant CA 132861).
Footnotes
Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data and crystallographic data in CIF format for 2-Br-5-OH-17-NO2-ttisocorrH2. CCDC 804886. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc05837h
Notes and references
- 1.Gryko DT, Fox JP, Goldberg DP. J. Porphyrins Phthalocyanines. 2004;8:1091. [Google Scholar]
- 2.Nardis S, Monti D, Paolesse R. Mini-Rev. Org. Chem. 2005;2:546. [Google Scholar]
- 3.Aviv I, Gross Z. Chem. Commun. 2007:1987. doi: 10.1039/b618482k. [DOI] [PubMed] [Google Scholar]
- 4.Gross Z, Galili N, Saltsman I. Angew. Chem., Int. Ed. 1999;38:1427. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1427::AID-ANIE1427>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 5.Paolesse R, Jaquinod L, Nurco DJ, Mini S, Sagone F, Boschi T, Smith KM. Chem. Commun. 1999:1307. [Google Scholar]
- 6.Gryko DT. J. Porphyrins Phthalocyanines. 2008;12:906. [Google Scholar]
- 7 a).Aviv-Harel I, Gross Z. Chem.–Eur. J. 2009;15:8382. doi: 10.1002/chem.200900920. [DOI] [PubMed] [Google Scholar]; (b) Kupershmidt L, Okun Z, Amit T, Mandel S, Saltsman I, Mahammed A, Bar-Am O, Gross Z, Youdim MBH. J. Neurochem. 2010;113:363. doi: 10.1111/j.1471-4159.2010.06619.x. [DOI] [PubMed] [Google Scholar]; (c) Agadjanian H, Ma J, Rentsendorj A, Valluripalli V, Hwang JY, Mahammed A, Farkas DL, Gray HB, Gross Z, Medina-Kauwe LK. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6105. doi: 10.1073/pnas.0901531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbe JM, Canard G, Brandés S, Guilard R. Chem.–Eur. J. 2007;13:2118. doi: 10.1002/chem.200601143. [DOI] [PubMed] [Google Scholar]
- 9.Lvova L, Di Natale C, D’Amico A, Paolesse R. J. Porphyrins Phthalocyanines. 2009;13:1168. [Google Scholar]
- 10.Saltsman I, Mahammed A, Goldberg I, Tkachenko E, Botoshansky M, Gross Z. J. Am. Chem. Soc. 2002;124:7411. doi: 10.1021/ja025851g. [DOI] [PubMed] [Google Scholar]
- 11.Paolesse R. Synlett. 2008:2215. [Google Scholar]
- 12.Bröring M, Hell C. Chem. Commun. 2001:2336. doi: 10.1039/b107362c. [DOI] [PubMed] [Google Scholar]
- 13.Brückner C, Barta CA, Briñas RP, Bauer J. A. Krause. Inorg. Chem. 2003;42:1673. doi: 10.1021/ic0261171. [DOI] [PubMed] [Google Scholar]
- 14.Mandoj F, Nardis S, Pomarico G, Paolesse R. J. Porphyrins Phthalocyanines. 2008;12:19. doi: 10.1142/S1088424610002513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capar C, Kolle ET, Ghosh A. J. Porphyrins Phthalocyanines. 2008;12:964. [Google Scholar]
- 16.Ngo TH, Van Rossom W, Dehaen W, Maes W. Org. Biomol. Chem. 2009;7:439. doi: 10.1039/b819185a. [DOI] [PubMed] [Google Scholar]
- 17.Nardis S, Pomarico G, Fronczek FR, Vicente MGH, Paolesse R. Tetrahedron Lett. 2007;48:8643. [Google Scholar]
- 18.Świder P, Nowak-Król A, Voloshchuk R, Lewtak JP, Gryko DT, Danikiewicz W. J. Mass Spectrom. 2010;45:1443. doi: 10.1002/jms.1860. [DOI] [PubMed] [Google Scholar]
- 19.Stefanelli M, Shen J, Zhu W, Mastroianni M, Mandoj F, Nardis S, Ou Z, Kadish KM, Fronczek FR, Smith KM, Paolesse R. Inorg. Chem. 2009;48:6879. doi: 10.1021/ic900859a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paolesse R, Marini A, Nardis S, Froiio A, Mandoj F, Nurco DJ, Prodi L, Montalti M, Smith KM. J. Porphyrins Phthalocyanines. 2003;7:25. [Google Scholar]
- 21.Stefanelli M, Mastroianni M, Nardis S, Licoccia S, Fronczek FR, Smith KM, Zhu W, Ou Z, Kadish KM, Paolesse R. Inorg. Chem. 2007;46:10791. doi: 10.1021/ic7014572. [DOI] [PubMed] [Google Scholar]
- 22.Diffraction data were collected on a Bruker Kappa Apex-II diffractometer equipped with graphite-monochromated CuKα radiation (λ = 1.54178 Å). Crystal data: C40H30BrN5O3, dark blue fragment, triclinic space group , a = 10.086(2), b = 13.041(3), c = 13.928(3) Å, α = 72.718(15)°, β = 70.046(15)°, γ = 86.335(18)°, V = 1642.8(6) Å3, Z = 2, Dcalc = 1.450 g cm−3, μ = 2.25 mm−1, T = 90.0(5) K, 27 580 reflections collected with θmax < 66.4°, 18 061 data with I > 2σ(I). The crystal was a non-merohedral twin by twofold rotation about reciprocal (101). In a substitutional disorder, the 2,3-dibromo compound is present with population 10.8(2)%. Final residuals (for 458 parameters) were R1 [I > 2σ(I)] = 0.082, wR2 (all data) = 0.242, CCDC 804886.
- 23.Pomarico G, Xiao X, Nardis S, Paolesse R, Fronczek FR, Smith KM, Fang Y, Ou Z, Kadish KM. Inorg. Chem. 2010;49:5766. doi: 10.1021/ic100730j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nardis S, Pomarico G, Mandoj F, Fronczek FR, Smith KM, Paolesse R. J. Porphyrins Phthalocyanines. 2010;14:752. doi: 10.1142/S1088424610002513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.