

Abstract
Background
The EGFR mutation T790M is reported in approximately 50% of lung cancers with acquired resistance to EGFR inhibitors and is a potential prognostic and predictive biomarker. Its assessment can be challenging due to limited tissue availability and underdetection at low mutant allele levels. Here, we sought to determine the feasibility of tumor rebiopsy and to more accurately assess the prevalence of the T790M using a highly sensitive locked nucleic acid (LNA) PCR/sequencing assay. MET amplification is also analyzed.
Methods
Patients with acquired resistance were rebiopsied and samples were studied for sensitizing EGFR mutations. Positive cases were evaluated for T790M using standard PCR-based methods and a subset were re-evaluated with an LNA-PCR/sequencing method with an analytical sensitivity of approximately 0.1%. MET amplification was assessed by FISH.
Results
Of 121 patients undergoing tissue sampling, 104 (86%) were successfully analyzed for sensitizing EGFR mutations. Most failures were related to low tumor content. All patients (61/61) with matched pretreatment and resistance specimens showed concordance for the original sensitizing EGFR mutation. Standard T790M mutation analysis on 99 patients detected 51(51%) mutants. Retesting of 30 negative patients by the LNA-based method detected 11 additional mutants for an estimated prevalence of 68%. MET was amplified in 11% of cases (4/37).
Conclusions
The re-biopsy of lung cancer patients with acquired resistance is feasible and provides sufficient material for mutation analysis in most patients. Using high sensitivity methods, the T790M is detected in up to 68% of these patients.
Introduction
Somatic mutations within the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) are the most reliable predictors of response to EGFR tyrosine kinase inhibitors in patients with non-small cell lung carcinoma (NSCLC)(1–4). In-frame deletions in exon 19, encompassing the hotspot LREA at positions 747 to 750, and a point mutation substituting leucine with arginine at position 858 (L858R) in exon 21, account for over 90% of all sensitizing mutations(4). These changes cause the constitutive activation of the kinase to promote cell proliferation and survival through multiple associated downstream pathways. Over 75% of patients harboring these mutations have dramatic or significant clinical and radiographic responses within days of treatment with EGFR TKI’s and show improved progression-free and overall survival compared to patients with WT EGFR(5, 6). Although responses may range from several months to even years, patients usually progress while on treatment after a median of 10–16 months (7–10).
Two secondary resistance mechanisms have been identified, providing a molecular explanation for clinical resistance in many cases. The more common involves the emergence of a second EGFR tyrosine kinase mutation, T790M, caused by a single base substitution, C to T, at nucleotide 2369(7, 11). The resulting methionine at codon 790 has been hypothesized to confer resistance by increasing the affinity for ATP rather than drug at the ATP binding pocket of the kinase (12). While this mutation has been reported in approximately 50% of tumors at the time of treatment failure, it is only rarely detected by conventional mutation analysis in pretreatment samples (5). It has also been suggested that the incidence may be higher but may go undetected based on most commonly used detection methods (13).
Other rare second-site mutations in the EGFR tyrosine kinase domain have been described, including L747S (14), D761Y (15) and T854A (16), but due to their relatively low prevalence, their role in conferring resistance may be limited.
A less common mechanism of TKI resistance is the amplification of the gene encoding the MET receptor tyrosine kinase (13, 17). In this case, the mechanism of resistance is due to the increased coupling of MET to ErbB3 leading to the activation of downstream signals mediated by AKT that bypass the inhibited EGFR. In the initial reports, MET amplification was reported in up to 20% of cases with acquired resistance, with a portion of these concurrently harboring the T790M mutation, but these numbers have yet to be confirmed in larger independent studies.
Several clinical trials aimed at overcoming these known mechanisms of acquired resistance are underway. The use of second-generation irreversible EGFR TKI’s (18–21), combination EGFR TKI’s with MET kinase inhibitors (22) or with anti-EGFR monoclonal antibodies (23) and Hsp90 inhibitors represent some of the therapeutic modalities under investigation. However, the successful establishment of these new therapies as effective patient specific strategies faces major challenges, many arising from limitations in the assessment of tumor tissue at the time of treatment failure. As the vast majority of patients in this setting do not undergo rebiopsy, the common lack of available resistant tumor tissue limits the molecular guided stratification of patients into separate arms of treatment and hampers the further investigation of acquired resistance. Additional challenges are specifically related to testing, such as problems with the detection of mutations in very small samples with low tumor content, the accurate identification of the T790M mutation in samples with low mutant allele burden, and the lack of a precise definition of clinically significant MET amplification.
We undertook this study with the following aims: (1) determine the feasibility of rebiopsy in the clinical setting of acquired resistance to EGFR TKI; (2) establish what constitutes adequate tissue sampling for mutation testing and MET gene assessment by Fluorescent in-situ hybridization (FISH); (3) evaluate the spectrum of EGFR mutations and MET gene copy alterations present in tumors at the time of resistance and (4) assess the value of a highly sensitive locked nucleic acid (LNA) PCR/sequencing assay developed to detect low levels of mutant EGFR T790M.
Methods
Patient recruitment
Patients with recurrent or metastatic non–small-cell lung cancer with acquired resistance to EGFR TKI’s were recruited for the study under protocols approved by the Institutional Review Board of Memorial Sloan-Kettering Cancer Center. Patients were eligible for study if they had molecular evidence of TKI sensitivity (known EGFR sensitizing mutation) or fit clinical criteria for sensitivity to TKI. Our clinical criteria for TKI sensitivity were based on published consensus guidelines (24), requiring that a patient have a partial response or durable stable disease (> 6 months) on single agent TKI therapy. To fit criteria for resistance, patients must have developed progression of disease while on continuous TKI therapy.
Rebiopsy of patients with acquired resistance
Rebiopsy procedures, including interventional radiology (IR) guided needle core biopsies, fine needle aspirates, and excisional biopsies were performed following standard hospital procedures after appropriate informed consent. For IR guided biopsies and fine needle aspirates (FNA’s), the number of passes and adequacy of the sample were subject to individual operator discretion guided by on-site immediate cytologic confirmation of tumor presence within the aspirated material. Additionally, patients with tissue available from a clinically indicated procedure (including metastectomy, diagnostic tumor tissue sampling or therapeutic thoracentesis) were consented for molecular analysis of these tissue specimens. A summary of all tissue sampling procedures is presented in Table 1.
Table 1.
Summary of Tissue Sampling Procedures
| Tissue Samples (N=153)* 121 patients | |
|---|---|
| Clinically required procedures (N=84) | Dedicated for Protocol Only (N=69) |
| Surgical procedures (33) | IR guided core biopsies/FNAs (60) |
| 11 VATS biopsies | 45 intrapulmonary: 44 cores, 1 FNA |
| 6 lung resections: 5 lobectomies, 1 pneumonectomy | 15 extrapulmonary: 13 cores, 2 FNAs |
| 6 brain metastectomies | – 1 pleural |
| 3 laminectomies | – 1 chest wall |
| 2 large bone resections | – 2 lymph nodes |
| 2 lymph node excisions | – 7 liver |
| 1 adrenalectomy | – 2 adrenal |
| 2 mediastinoscopies | – 1 omentum |
| – 1 kidney | |
| Minimally invasive biopsies (17) | Other |
| 9 IR guided biopsies (4 bone, 3 lung, 1 liver, 1 kidney) | 1 subcarinal FNA – endoscopic, US guided |
| 2 IR guided lung FNAs | 1 lymph node FNA |
| 3 kyphoplasties | 1 bone biopsy |
| 2 bedside FNA’s (1 skin, 1 lymph node) | 1 lymph node excisional biopsy |
| 1 cervical biopsy | |
| Fluid samples (34) | Bedside FNA’s |
| 26 pleural fluid | 3 lymph node |
| 5 lumbar punctures | 1 chest wall |
| 1 peritoneal fluid | 1 skin |
| 2 pericardial fluid | |
27 patients had multiple specimens studied (range 2–3)
Genomic DNA was extracted from tumor samples, including fresh, frozen, formalin fixed paraffin embedded (FFPE) or alcohol fixed (thin preparation cytology slides) specimens. Fresh fluids were spun down and DNA was extracted directly from the fresh cell block or submitted to histology to be formalin fixed and paraffin embedded for further testing. Because the vast majority of testing was based on small biopsies, aspirates or effusions, no microdissection was performed prior to extraction.
EGFR sensitizing mutation analyses (Exons 19 and 21)
Analysis for EGFR sensitizing mutations in exons 19 and 21 was carried out by standard sequencing and/or fragment analysis with primers and protocols as previously published (4, 7, 25). Results were confirmed by duplicate runs in both the forward and reverse directions. Negative results were correlated with the concurrent cytologic or histologic slide for confirmation of adequate tumor content relative to associated non-neoplastic tissue (above 10% for fragment analysis and above 25% for standard sequencing) to prevent false negative results. Samples with tumor content below this level were considered inadequate for analysis.
EGFR T790M mutation analyses by standard methods (Fragment analysis and standard sequencing)
Samples with a confirmed sensitizing mutation in exon 19 or 21 of EGFR were then tested for T790M EGFR mutation by fragment analysis as previously described (7) and/or by standard sequencing as follows. The entire coding region of exon 20 of EGFR was amplified using the following forward and reverse intronic primers (custom oligos, Proligo, Boulder, CO): EGFR-Ex20-F-seq: 5′-cat tca tgc gtc ttc acc tg-3′ and EGFR-Ex20-R-seq: 5′-cat atc ccc atg gca aac tc-3′. Each PCR reaction was performed in a 50μL volume mixture containing approximately 100ng of genomic DNA, forward and reverse primers (20 pmol each), 50μM each of dATP, dCTP, dGTP, 400μM of dUTP, 1.5mM MgCl2, 1X Qiagen PCR buffer containing 1.5mM MgCl2 and 2.5 units of HotStarTaq DNA polymerase (Qiagen). AmpErase UNG (0.5 unit) was added to each reaction mixture for carryover prevention and incubated at room temperature for 10 minutes. The PCR amplification was carried out under the following conditions: 95°C (15 min) for one cycle; 42 cycles of 95°C (30 s), 58°C (30 s), and 72°C (30 s), and a final extension step of 72°C (10 min). Amplified products were purified using Spin Columns (Qiagen) and sequenced in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol on an ABI3730 running ABI Prism DNA Sequence Analysis Software. Results were confirmed by duplicate runs in both the forward and reverse directions.
EGFR T790M mutation analysis using LNA-PCR/Sequencing assay
To increase the sensitivity of standard DNA sequencing, we modified the PCR amplification step of the assay by the addition of a 10 mer LNA probe complementary to the wild type (WT) sequence of EGFR exon 20 in the area encompassing the I789 to Q791 region (Figure 1). LNAs are nucleic acid analogs with a 2′O to 4′C methylene bridge that locks the ribose group into a C3′ endo conformation (26, 27). When incorporated into a probe, their alternate conformation allows for the hybridization to the complementary WT sequence with unprecedented affinity, effectively suppressing its amplification and leading to preferential amplification of the mutant allele (28). Identical PCR mixes and cycling conditions as in the standard assay were used except for the addition of 20pmol of the LNA probe to the PCR mixture. The sequence of the probe was as follows: +C+A+T+C+A+C+G+C+A+Gctcatg ccc t/3InvdT, where the capital letters followed by the plus (+) sign designate the locked nucleotides. The unlocked nucleotides at the end (lower case) are added to stabilize the probe to a predicted Tm of 82°C. Results were confirmed by duplicate runs in both the forward and reverse directions. Two-tailed Fisher’s exact test was used to calculate P values.
Figure 1.
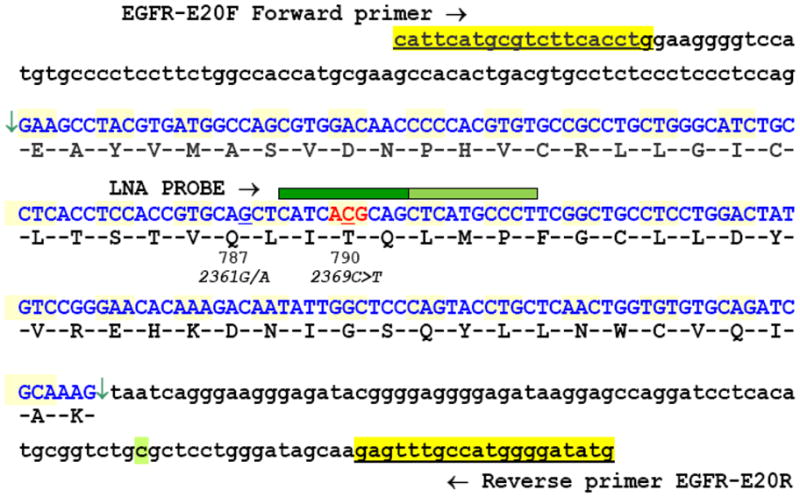
Genomic sequence of EGFR exon 20 (highlighted in blue, between arrows) with flanking intronic sequences. The sequences and location of the PCR primers are highlighted in yellow. The green bar, above positions 789–791, specifies the target area of the LNA probe with the site of the T790M (EGFR*2369C>T) located at the center of the locked portion (dark green = locked residues, light green = unmodified). The nearby common germline SNP 2361G/A (Q787Q) is also indicated.
Comparison of analytical sensitivities of standard sequencing and the T790M LNA/PCR sequencing assay
To assess the analytical sensitivity of standard sequencing and the LNA-PCR sequencing, DNA mixing studies were carried out using a mixture of mutant DNA obtained from a research patient sample containing an even proportion of mutant and germline alleles for T790M into normal DNA (In-vivoscribe, IVS-0000) in dilutions of 50, 25, 12.5, 6.25, 3, 1.5, 0.75, 0.38, 0.19 and 0.1% mutant DNA. All DNA mixtures were amplified, purified and sequenced following the standard and LNA PCR/sequencing protocols described above. The analytical sensitivity of standard sequencing was assessed in a similar fashion but dilutions of mutant to WT-DNA were carried out at 50, 25, 12.5, 6, 3 and 1.5%.
Assessment of MET gene copy number alterations by FISH
Unstained, 4 μm sections of FFPE tumor tissue (surgical biopsies or cytology cell blocks) were analyzed by a dual-color FISH assay using a MET/CEP7 probe cocktail following previously described protocols (17). The cocktail was prepared using either an in-house developed (RP 11–163C9 BAC clone derived) or a commercially available (Kreatech, Netherlands) MET probe labeled with Spectrum Orange in combination with a Spectrum Green labeled CEP7 probe (Abbott Molecular). Signals were enumerated in at least 50 tumor nuclei per specimen; samples with fewer analyzable nuclei were considered insufficient for a complete study. For each specimen, MET signals were counted and cells were stratified into groups based on the number of signals per nucleus as follows: 2 signals, 3–4 signals, 5–6 signals, 7–15 signals, >15 signals). The percentage of cells within each range was annotated and the corresponding CEP7 signal count within each group was concurrently annotated. MET to CEP7 ratios were then calculated using the mean MET and CEP7 copy numbers for each cell group. Samples with a MET:CEP7 ratio greater than 2 were considered to have MET amplification (Low amplification ≤3, high amplification >3).
Results
Patient profile
Between August 2004 and February 2010, 144 patients were consented. Of these, 14 patients were deemed ineligible, leaving 130 patients who met the study criteria. Tissue sampling procedures were performed on 121 patients while 9 did not undergo re-biopsy (Figure 2). Of the 121 sampled patients, 80 (66%) were female and 41 (34%) male with a median age of 62 years (range 27–86). Eighty one patients (67%) were never smokers while 40 were former or current smokers. All patients had a documented history of adenocarcinoma except for one who had squamous cell carcinoma. The majority (70%) had stage IV disease. The median time on EGFR-TKI before biopsy was 12 months (7–28 months).
Figure 2.

Flow chart of patients consented to rebiopsy: of 144 patients consented, 130 met strict criteria for rebiopsy; 121 underwent sampling procedures; 104 were molecularly profiled for sensitizing mutations and 99 for the T790M mutation.
Tissue samples and adequacy for mutation analyses
A total of 153 tissue samples were obtained from these 121 patients; 27 patients had multiple samples (range 2–3). Eighty four samples (55%) were acquired from clinically required procedures while 69 (45%) were protocol dedicated (Table 1). Samples for molecular testing were received as follows: 63 frozen samples, 28 fresh fluids (20 pleural, 2 pericardial, 1 peritoneal, 5 CSF’s, 4 FNA’s in RPMI culture medium), 57 FFPE tissue blocks (41 biopsies, 10 FNA cell blocks, 6 pleural fluid cell blocks) and 1 alcohol-fixed cytologic thin preparation from a FNA.
DNA extracted from whole frozen core biopsies generated an average of 3.29 μg of DNA (range 0.25–14 μg) while DNA extracted from FFPE cores and small biopsies of similar size (10 unstained sections, 4μm thick) yielded an average of 1.69μg of DNA (range 0.25–3.6μg). Pleural fluids (15 sections, 4 μm thick, prepared from 50cc of fluid into FFPE cell blocks) generated an average of 7.2 μg of DNA (range 0.3–19μg). Fine needle aspirates consistently generated low DNA amounts (average: 0.23μg; range 0.12–0.4μg).
The adequacy of tissue for successful profiling was based on the initial assessment and testing for EGFR sensitizing mutations with results summarized in Table 2. Of the total 153 samples, 126 (82%) were successfully analyzed, 9 by standard sequencing, 117 by fragment analysis (104 patients). Excluding decalcified bone tissue, biopsies provided the highest success rate with 89% (85/95) of samples fully profiled. This was followed by fine needle aspirates (79%, 11/14) and pleural fluids (77%, 20/26). The lowest yield was generated by bone samples with an overall success rate of 54% (7/13) and 40% (4/10) for the decalcified material. Two of the 5 CSF samples (40%) did not yield sufficient material for testing. Twenty eight samples were unsuitable due to insufficient or low quality DNA, very low tumor content (<10% tumor for samples analyzed by fragment analysis and <25% for standard sequencing) or complete absence of tumor compared to a corresponding H&E stained section or slides of the concurrently performed aspirate and or/biopsy for pathologic assessment. This count includes one sample partially profiled, with successful analysis for EGFR sensitizing mutations but failing T790M analysis. Large bone tissue samples undergoing extensive decalcification (several days) were found unsuitable for analysis.
Table 2.
Summary of Samples Analyzed for EGFR Mutations Based on Specimen Type
| Sample adequacy based on specimen type (N=153) | |||
|---|---|---|---|
| Sample Type | Total Samples | Successfully Profiled (%) | Failures |
| Biopsies (including non-decalcified bone) | 95 | 85 (89%) | 10 (11%) |
| Bone biopsies (decalcified) | 10 | 4 (40%) | 6* (60%) |
| FNA’s | 14 | 11 (79%) | 3 (21%) |
| Fluid samples | 34 | 24 (71%) | 10(29%) |
| Pleural | 26 | 20 (77%) | 6(23%) |
| Peritoneal | 1 | 0 | 1(100%) |
| Pericardial | 2 | 1 (50%) | 1(50%) |
| CSF | 5 | 3 (60%) | 2(40%) |
Includes 1 biopsy partially analyzed (successful EGFR exon 19 & 21, failed T790M analysis)
Tumor morphology
Of the 121 patients sampled, 106 patients had pathologic confirmation of tumor and histologic classification. The remaining 15 patients failed morphologic confirmation - 8 showed no tumor in their concurrently performed histologic or cytologic samples, 2 had suspicious cells favoring adenocarcinoma in cytologic samples but confirmation was precluded by degeneration and 5 samples had very limited material which was insufficient for both morphologic analysis and molecular testing.
Patients with histologic classification of their resistance tumors were stratified as follows: 102 adenocarcinomas, 1 squamous cell carcinoma, 2 small cell carcinomas and 1 with a combined large cell carcinoma/adenocarcinoma in one sample and a high grade neuroendocrine carcinoma in a second sample. All patients maintained a similar morphology compared to the baseline tumor except for the latter 3 patients. Despite the distinct morphology of these 3 cases, the same mutation profile was confirmed in both baseline and resistance samples in 2 patients, the one with small cell carcinoma and in all resistance samples of the patient with large cell and neuroendocrine carcinoma. Emergence of small cell histology in the setting of acquired resistance is rare but previously described (29). The resistance mutation status of the third patient could not be confirmed due to insufficient tissue.
Mutation analysis
Sensitizing EGFR mutations in resistance samples
One hundred and four patients were successfully profiled for EGFR mutations in exons 19 and 21. One hundred patients (96%) had a sensitizing EGFR mutation with the following profile: 71% (71/99) had EGFR exon 19 deletions (46 15bp, 17 18bp, 2 12bp, 5 9bp, 1 24bp), 1 patient had an 18 bp insertion and 28% (28/99) had an exon 21 point mutation (25 L858R, 1 L833V + H835L, 1 L858R + R776H, 1 L861Q). Four patients lacked any EGFR mutation in their resistance specimen. Of these, 3 were known to be EGFR WT at baseline and one was found to have two molecularly unrelated pretreatment primaries (1 EGFR mutant, 1 WT) and recurred as WT only.
Of the 27 patients undergoing multiple tissue sampling procedures, 17 had successful mutation analysis of 2 or more samples. All patients had the same sensitizing mutation at all tumor sites.
Comparison of sensitizing mutations status at baseline and rebiopsy specimens
We compared baseline and rebiopsy samples to determine whether patients known to harbor a sensitizing EGFR mutation at pretreatment maintained their mutation status at the time of resistance. Of 62 patients with a known sensitizing mutation at baseline and successfully profiled at the time of resistance, 61 (98%) had identical EGFR mutations in their resistance samples; one patient was EGFR WT in 2 separate high quality rebiopsy specimens. Review of this patient’s baseline surgical specimens demonstrated multiple bilateral pulmonary nodules, some of which were found to be EGFR WT and some sharing the same exon 19 deletion, therefore two genetically distinct lung primaries were suspected. Excluding this sample from our final analysis, 100% (61/61) of patients maintained the baseline sensitizing mutation.
Resistance mutation analysis based on standard sequencing and fragment analysis
Of 104 patients profiled for sensitizing mutations, 100 were EGFR mutant and therefore tested for the T790M resistance mutation. Ninety nine were successfully profiled (54 by standard sequencing only, 30 by fragment analysis and 15 by both methods); one sample (bone metastasis) failed due to insufficient/low quality DNA. The T790M mutation was detected in 49% (34/69) and 53% (24/45) of patients tested by standard sequencing and fragment analysis, respectively. The combined mutation rate was 52% (95% CI 42–62%) (Table 3).
Table 3.
Comparison of T790M Mutation Rate by Type of Testing Method
| STANDARD SEQUENCING | FRAGMENT ANALYSIS | COMBINED STANDARD & LNA PCR/SEQUENCING | |
|---|---|---|---|
| Total cases tested | 69 | 45 | 64 |
| Positive cases (rate) | 34 (49%) | 24 (53%)** | 45 (70%)* |
| Negative cases (rate) | 35 (51%) | 21 (47%) | 19 (30%) |
p= 0.02,
p=n.s. (Fisher’s Exact test for comparison to standard sequencing)
Resistance mutation analysis by the LNA-PCR/Sequencing method
Of 69 patients successfully profiled for the T790M mutation by standard sequencing, 64 had DNA available for further analysis by the LNA-PCR/sequencing method (30 negative and 34 positive). The remaining 5 patients had insufficient material for further assessment (all negative by standard sequencing). Retesting of all available negative cases (30) identified 11 additional mutants (37%, 11/30) raising the mutation rate to 70% (45/64) in this subset (p = 0.02 Fisher’s Exact test for comparison to standard sequencing). This figure, however may be somewhat skewed by the fact that the 5 unavailable cases were all negative by standard sequencing. Based on our direct comparison of the 2 methods, we estimate that the true mutation positive rate with the use of the LNA method for the group of 69 patients is 68% [49% (percent positive by standard sequencing) + (51% (percent negative by standard sequencing) × 0.37 (proportion positive by the LNA method)) = 68%]. Samples previously tested by fragment analysis were unavailable.
Mixing studies to assess the analytical sensitivity of both standard and LNA-PCR/sequencing showed that the detection sensitivity of sequencing after standard PCR amplification is 12.5% in both the forward and reverse directions. At dilutions below these concentrations, the mutant peak is barely seen above background, not allowing a confident call, or can no longer be detected (Figure 3). The introduction of the LNA probe improved the sensitivity of the test to 0.1% in both the forward and reverse directions. Concentrations of mutant DNA of 0.75% and above yielded sequencing graphs with only the mutant peak. At lower concentrations, the WT peak was visible, becoming progressively larger to a maximum mutant to WT peak ratio of 3:1 at the lowest mutant DNA concentration tested (0.1%).
Figure 3.

Representative electropherograms of the sensitivity study for EGFR T790M. A. Sequencing after standard PCR of undiluted mutant DNA (100%) shows both a mutant peak (T) and a WT peak (C) at position 2369 (arrow) at roughly the same proportions. Serial dilutions of mutant DNA with normal control DNA show a sequential decrease in size of the mutant peak. The mutation is clearly seen at dilutions of 12.5% or higher in both the forward and reverse directions. Below this level, the mutant peak is at the level of the background or completely absent. % M denotes percent of mutant DNA; ( ) denotes SNP at position 2361. B. LNA-PCR/sequencing. With the introduction of the LNA probe, only the mutant peak (T) is seen at position 2369 (arrow) at all dilutions of 0.8% and above. At progressively lower concentrations, the WT peak becomes visible but the mutant peak remains predominant down to 0.1% (percentages have been rounded). At the SNP position () only G is seen with complete suppression of the WT allele.
) denotes SNP at position 2361. B. LNA-PCR/sequencing. With the introduction of the LNA probe, only the mutant peak (T) is seen at position 2369 (arrow) at all dilutions of 0.8% and above. At progressively lower concentrations, the WT peak becomes visible but the mutant peak remains predominant down to 0.1% (percentages have been rounded). At the SNP position () only G is seen with complete suppression of the WT allele.
To determine whether the T790M mutation could be detected at low levels in pretreatment tumors using our sensitive LNA-PCR/sequencing assay, we tested all available baseline samples with matching successfully tested resistance samples using both the standard and the LNA-based methods. A total of 24 samples were tested – 15 from patients harboring the T790M at the time of resistance (12 by standard methods, 3 by the LNA-based method only) and 9 mutation negative. All samples (24/24) were negative for the mutation by both methods. In addition, 16 non-pulmonary malignancies were also tested during our validation procedures, all yielding negative results by LNA-PCR/sequencing. These data also support an extremely low or nil false positive rate for the T790M LNA-PCR/sequencing assay.
A notable pitfall of the LNA-PCR sequencing method is that sequencing traces sometimes show multiple, small (generally more than 2), poorly reproducible mutant peaks in the area bound by the LNA probe (Supplemental Figure 1). We found that these peaks were often generated when a mutant allele was absent or extremely low (below 0.1%), likely representing low level errors due to Taq polymerase misincorporation or formalin fixation which are unmasked by the LNA-induced suppression of the WT allele, a pattern that becomes readily recognizable and distinct from true positive results. Repeat runs in these cases do not reproduce the same pattern of mutant peaks, confirming them as artifactual. Occasionally, sequencing tracings may also be accompanied by a high background throughout. Cases showing a mutant peak in association with this background sometimes yield negative results in repeat runs. These cases should therefore be managed conservatively by repeating the testing to confirm the mutation status. It should be noted that in the majority of non-mutant cases, the LNA probe completely suppresses the amplification of wild type DNA, generating no PCR product which is indistinguishable from a PCR failure for any other reason. This method therefore requires the concurrent performance of standard PCR for quality control.
Stratification of the T790M mutation by body site and comparison of T790M mutation profile in patients with multiple resistance samples
Using a combination of all 3 methods, we examined the presence of the T790M mutation by body site. From a total of 112 cases (99 patients) the mutation was found at all biopsied sites except skin, chest wall, kidney, adrenal gland, uterine cervix and CSF. Detailed results are summarized in Supplemental Table 1. It should be noted that 4 of the 12 cases from sites where the T790 was not found, were not tested by the most sensitive LNA-based method. Of note also, the mutation was detected in 2 out of the 4 brain metastasis tested and was confirmed by both standard sequencing and the LNA/PCR sequencing method.
Of the 99 patients successfully analyzed for EGFR mutations, 14 had multiple samples for comparison (range 2–3). Eight of these had concordant T790M results in all samples while 6 (43%) showed variable positivity not related to the use of a higher sensitivity method. Four patients had initial positive results with a subsequent negative result using the same or a higher sensitivity method. Conversely, two patients, initially mutation negative by fragment analysis, had subsequent positive results by standard sequencing (lowest sensitivity method). The time frame between sampling procedures ranged from 15 days to 14 months.
Assessment of MET gene copy number alterations by FISH
Tissue for MET FISH analysis was available in 50 patients; of these, 13 patients (26%) had specimens that failed testing, specifically related to poor tissue quality and/or low tumor cell counts. The majority of the remaining 37 patients with adequate results (68%, 25/37) harbored several copies (range 3–7) of both MET and chromosome 7 (CEP7), indicating simple polysomy of chromosome 7. Specific amplification of MET relative to the CEP7 signals was found in only 4 patients (11%, 95% CI 1–20%). Amplification was confined to a subset of tumor cells in all samples (range 9%–51% of cells) with the remaining cells showing multiple copies of both MET and CEP7. Only 1 patient had high level amplification (MET:CEP ratio >10:1) while the other 3 had low levels (MET:CEP ratio ranging from 2–3:1). Three of these patients (including the one with the high level amplification) also harbored the EGFR T790M mutation.
Discussion
The emergence of resistance in patients previously sensitive to EGFR inhibitors has spurred the development of many new targeted agents designed to overcome some of the well characterized resistance mechanisms. The successful establishment of these agents as effective patient-specific therapies and the further understanding of acquired resistance depend on the careful examination of tumor samples at the time of treatment failure. Since most patients do not undergo rebiopsy in this setting, there is little published experience with the feasibility and practicality of this approach. Ideally, sampling procedures should be minimal or non-invasive but, at the same time, should yield sufficient tissue for molecular profiling based on widely accessible molecular techniques. Testing strategies must then be optimized to provide accurate and reliable mutation analysis across a broad range of tumor samples.
In the present study, we show that the rebiopsy of patients in the setting of acquired resistance to EGFR TKI is feasible. Over a 5.5 year period, 153 tissue specimens were obtained for resistance analysis; 69 of these were obtained with protocol-driven biopsies, while 84 specimens were obtained using clinically indicated procedures such as diagnostic biopsies and therapeutic procedures. It is important to note that during the period of this re-biopsy protocol, multiple therapeutic protocols in the acquired resistance setting were accruing at our institution. Thus, our data indicate that lung cancer patients who have already received benefit from an oral targeted therapy (EGFR TKI’s) are amenable to further biopsies that may uncover additional targetable tumor characteristics even if those procedures may not have direct implications for their immediate care.
Small tumor samples obtained through minimally invasive procedures (IR guided biopsies, fine needle aspirates and non-guided biopsies) provide sufficient tissue for profiling in the majority of the cases, 89% of biopsies and 79% of FNA’s. In concordance with our previous study (30), we find that dedicated frozen needle core biopsies provide ample material for molecular profiling (average of 3.29 μg of DNA). However, the assessment of suitability relies on the review of a concurrent cytology or a separate core biopsy which may not be fully representative of the tissue tested and may lead to false negative results. A preferable approach is to perform all testing on the same pathologic specimen to optimize the prospective assessment of suitability. Compared to larger samples which can be enriched for tumor content through micro-dissection, the mutation analysis of small biopsies in the resistance setting is often more challenging. Due to their small size and intimate association with non-neoplastic cells, microdissection of recurrent, resistant tumors is seldom feasible. Adequate assessment of tumor content to match the sensitivity of the testing method is therefore critical to prevent false negative results. It is important to mention that, in cases with a background of fibrosis or inflammation, the quantification of tumor content on H&E stained sections may be more challenging. Estimations based solely on the area encompassed by the tumor can lead to overestimation as non-neoplastic cells are comparatively much smaller than malignant cells but contribute a similar amount of DNA per cell. In such cases, a rough cell count in a representative field may aid in the adequate assessment of tumor content to avoid false negative results. We find that a single FFPE, 18–20 gauge, needle core biopsy can provide sufficient material for morphologic evaluation, basic immunohistochemical assessment and molecular studies when appropriately handled. Ten unstained sections, 4μm each, are generally sufficient for multiple molecular tests.
In comparison to the small biopsies and FNA’s of large, untreated tumors, needle aspirates in the resistance setting consistently generated more limited DNA, likely due to the presence of fibrosis. However, based on correlation with cytology and tissue block sections, these generally provide a purer tumor population potentially decreasing false negative results. Aspirates are particularly valuable in the mutational assessment of metastatic lesions to bone. This strategy bypasses the need for decalcification of the specimen, thereby preserving DNA integrity for molecular testing as well as other emerging pertinent studies such as FISH for MET amplification. Alternately, fresh or formalin-fixed biopsies of bone metastases may be submitted for DNA extraction without prior decalcification.
Malignant pleural effusions, which are routinely discarded after cytologic evaluation, offer another opportunity for mutation screening with applications to EGFR testing already documented (31–34). Since a significant number of lung cancer patients have a pleural effusion at the time of initial diagnosis, and many more develop a pleural effusion later in the course of disease, a large proportion of patients could potentially be evaluated in this manner. Because pleural effusions often contain significant proportions of inflammatory cells in addition to tumor, correlation with the cytologic findings is necessary to adequately interpret a negative result. The use of higher sensitivity methods such as the one we describe for T790M, would have a significant impact on the profiling success rate of these samples. Of note, samples prepared from 50cc of a pleural fluid (fresh or paraffin embedded) with cytologic confirmation of tumor cells, generated DNA amounts averaging 7.2μg of DNA, higher than many small biopsies. Higher fluid volumes can be used to prepare cell blocks in samples with low tumor content. Unstained sections from FFPE cell blocks can also be used for MET amplification by FISH, although in these cases, multiple sections might need to be analyzed to reach appropriate tumor nuclei counts.
Based on all cytologic samples analyzed, we find that suitability for testing relies heavily on both tumor content and the final DNA yield. Samples yielding as little as 2.5ng/μL (fresh) and 4.3ng/μL (FFPE) but with a tumor content (%) above the sensitivity level for the testing method were successfully tested. Cell counts were not performed in the present study but a systematic analysis of the adequacy of various cytologic lung carcinoma specimens for molecular testing over a 1 yr period has recently been completed at our institution (35).The comparison of baseline and rebiopsy specimens demonstrated that EGFR mutations are maintained in subsequent biopsies, and that “reversion” to EGFR wild-type is not a mechanism of acquired resistance. In our group of 62 patients with available baseline and resistance profiles, all retained the same sensitizing mutation except for 1 who was then proven to have 2 independent primaries at baseline. Our findings therefore support that all EGFR dependent tumors maintain their EGFR dependence and that heterogeneity for sensitizing EGFR mutations within a clonal tumor (with selection of pre-existing EGFR wild type subclones) is not a path to acquired EGFR TKI resistance. This conclusion is consistent with clinical observations demonstrating the continued sensitivity of tumors to EGFR TKIs even after disease progression has occurred (8, 36).
Through the use of our LNA-PCR/sequencing assay for the detection of the EGFR T790M mutation, we found a significantly higher proportion of T790M mutant tumors than previously reported. Based on 64 patients tested by the combination of standard sequencing and the LNA modified method, we were able to detect 11 additional mutant patients, for an estimated prevalence of 68%. Testing of 24 available paired baseline samples from this group by both methods did not detect any T790M mutants. The absence of the T790M mutation in 16 non-pulmonary malignancies further validates that positive results represent true positives. Highly sensitive techniques, such as this one, show a clear advantage over direct sequencing allowing the detection of the T790M mutation when present in low concentration, even as low as 0.1% of the total DNA. It is important to emphasize, however, that all resistance samples tested exhibited a widely variable mixture of tumor and non-neoplastic cells. Therefore, the detection of a mutant peak at a level similar to the 0.1% seen in our mixing studies cannot be equated with the presence of 0.1% mutant population in a pure tumor cell population. Instead, the combined impact of low tumor content and likely intra-tumoral heterogeneity for the secondary T790M mutation may result in very low apparent mutant allele frequencies for this mutation. Compared to other highly sensitive methods such as Scorpion Amplification Refractory Mutation System and the WAVE/Surveyor, the LNA PCR/Sequencing method preserves the broad informative genotyping capabilities of standard sequencing over a wide range of specimens and can be easily incorporated into already existing PCR-based protocols without the need to change existing cycling conditions. Furthermore, the information provided by a full sequencing tracing is unavailable in mutant-specific assays.
In contrast to previous findings, we detected the presence of the T790M mutation in 2 out of 4 brain metastases. Positive results were found by both standard sequencing and the LNA method in both patients suggesting mutant DNA levels well above 25%. At the time of testing, both patients had progression of disease systemically as well as in the CNS. These findings suggest that, in this setting, EGFR inhibitors penetrate the central nervous system in sufficient quantities to exert a selection pressure leading to the emergence of secondary resistance mechanisms in brain parenchymal lesions. All CSF samples, however, have been negative perhaps related to the protective effects of the blood brain barrier to EGFR inhibitory drugs. Patients with leptomeningeal metastases or with CNS-only progression could therefore represent a subgroup of patients with advanced disease who might still benefit from the continued use of first generation targeted therapy, however, a larger number of samples is required to confirm this preliminary observation.
In contrast to EGFR sensitizing mutations which remained 100% stable at all recurrent and metastatic sites, the T790M mutation exhibits significant heterogeneity. Forty three percent (6/14) of patients with multiple (2–3) samples tested did not show the mutation at a second site using the same method or a more sensitive method (fragment analysis assay in 5 cases and the LNA based method in 1 case. Since the mutation status could not be confirmed by the highest sensitivity method in 5 of these patients, the possibility that these represent false negative results due to the limited sensitivity of the assays remains to be explored. Whether the level of T790M allele load in tumors is also dynamic in relation to the withdrawal of EGFR therapy remains to be established by profiling of serial samples in these patients.
Lastly, compared to initial studies (17, 37), our assessment of MET gene copy alterations by FISH showed that amplification, particularly at high levels, is an uncommon event. In our cohort, the prevalence was 11% (compared to 21–22%) with only 1 case of high level amplification. Of the 4 patients identified, 3 had a concurrent T790M mutation. It must be mentioned, however, that the initial studies reporting MET amplification are based on combined data using various methodologies including aCGH, FISH and/or qPCR, all of which have yet to be standardized. When considering only post-treatment samples tested by FISH, the study by Engelman et al found only 14% (1/7) of cases with MET amplification. This is similar to our findings despite our use of stricter criteria (ratio vs difference of MET:CEP7 signals) for MET amplification. Our findings suggest that MET amplification may not explain as many cases of non-T790M acquired resistance as originally thought. Alternate pathways of TKI resistance, of which several have been hypothesized (38), are actively being investigated by multiple groups using high throughput mutation testing, siRNA screens, and gene expression microarray analysis.
The experience with imatinib in chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST) provides some precedents for the genomic assessment of neoplastic tissue at the time of clinical resistance in the management of patients with EGFR mutant adenocarcinoma. In comparison to CML, however, where testing is based on readily available blood samples, the assessment of solid tumors often requires the use of invasive techniques for acquisition of sufficient tissue. To date, the only other solid tumor with systematic assessment of rebiopsy tissue in the setting of acquired drug resistance is GIST. Closely paralleling our findings with EGFR, rebiopsy specimens in GISTs have shown secondary KIT mutations involving the ATP binding pocket of the kinase domain (exons 13 and 14)(39–41). Unlike EGFR however, where virtually all secondary mutations are T790M, several secondary KIT mutations have been identified in the ATP binding pocket as well as the kinase activation loop. In addition, imatinib-resistant GISTs have been shown to harbor significant mutational heterogeneity with distinct mutations among separate metastases and even within the same metastatic nodule (42). Our observations parallel those in GIST where tumor masses continue to show sensitivity to the TKI imatinib even after RECIST progression (43). In the GIST setting, withdrawing imatinib leads to a disease “flare” that can be abolished by the reinstitution of imatinib, suggesting that the resistance mutation is not present in every cell.
In conclusion, the re-biopsy of patients with acquired resistance is feasible and provides sufficient material for mutation analysis in most patients. Using higher sensitivity methods, the T790M may be detected in up to 68% of patients with EGFR-mutant lung cancer and acquired resistance, a higher prevalence than previously described. Detecting the T790M in this clinical setting has significant clinical implications because, among patients with acquired resistance, the presence of T790M appears to define a subset of patients with a distinct clinical course (44) and different agents and therapeutic strategies may be needed to overcome T790M-mediated acquired resistance.
Statement of Translational Relevance.
The EGFR second site mutation T790M has been reported in about half of lung cancers with acquired resistance to EGFRinhibitors but its detection is challenging due to lack of re-biopsy and underdetection at low mutant allele levels. This study sought to determine the feasibility of tumor rebiopsy and to more accurately assess the prevalence of the T790M mutation using a sensitive locked nucleic acid (LNA) PCR/sequencing assay. We show that rebiopsy is feasible and provides sufficient material for T790M mutation analysis in most patients. The higher sensitivity LNA PCR/sequencing testing method detects a significantly higher proportion of mutant tumors compared to Sanger sequencing (68% vs 49%). Detecting the T790M in this setting has significant clinical implications as this mutation appears to define a subset of patients with a distinct clinical course and different strategies may be needed to overcome T790M-mediated acquired resistance.
Supplementary Material
Sequencing tracings after LNA-PCR demonstrating multiple artifactual mutant peaks in the region bound by the LNA probe (*). In general, these cases show >2 additional peaks that are poorly reproducible in subsequent runs and should not prompt a positive mutation call. These peaks are usually generated when a genuine mutant allele is absent or extremely low (below 0.1%). In this case, an independent repeat run with excess LNA probe shows a failure pattern caused by complete suppression of the WT allele in the absence of a mutation. () denotes position of SNP. The arrow indicates where the location of the T790M, (X) indicate positions where artifactual peaks are no longer seen in the duplicate run.
Acknowledgments
Supported by NIH P01 grant CA129243 (MGK, ML), R21-CA115051 (VAM), R01-CA121210 (WP) and the Carmel Hill Fund.
References
- 1.Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol. 2008;21 (Suppl 2):S16–22. doi: 10.1038/modpathol.3801018. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 6.Jackman DM, Yeap BY, Sequist LV, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:3908–14. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 7.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riely GJ, Pao W, Pham D, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–44. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 9.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–67. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 10.Janne PA, Wang XF, Socinski MA, et al. Randomized phase II trial of erlotinib (E) alone or in combination with carboplatin/paclitaxel (CP) in never or light former smokers with advanced lung adenocarcinoma: CALGB 30406. J Clin Oncol. 2010;28:15s. doi: 10.1200/JCO.2011.40.1315. 2010 (suppl; abstr 7503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 12.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costa DB, Halmos B, Kumar A, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–79. doi: 10.1371/journal.pmed.0040315. discussion 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 16.Bean J, Riely GJ, Balak M, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–25. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–7. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–32. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 19.Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–70. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–11. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor: Results of a Phase II Trial in Patients With Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2010 doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 22.Schiller JH, Akerley WL, Brugger W, et al. Results from ARQ 197–209: A global randomized placebo-controlled phase II clinical trial of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated EGFR inhibitor-naive patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) J Clin Oncol. 2010;28:18s. 2010 (suppl; abstr LBA7502) [Google Scholar]
- 23.Riely GJ, Janjigian YY, Azzoli CG, et al. Phase II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. J Clin Oncol. 2010;28(suppl) doi: 10.1158/1078-0432.CCR-10-2662. abstr 7557. [DOI] [PubMed] [Google Scholar]
- 24.Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–60. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan Q, Pao W, Ladanyi M. Rapid polymerase chain reaction-based detection of epidermal growth factor receptor gene mutations in lung adenocarcinomas. J Mol Diagn. 2005;7:396–403. doi: 10.1016/S1525-1578(10)60569-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfundheller HM, Sorensen AM, Lomholt C, et al. Locked nucleic acid synthesis. Methods Mol Biol. 2005;288:127–46. doi: 10.1385/1-59259-823-4:127. [DOI] [PubMed] [Google Scholar]
- 27.Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–41. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 28.Arcila ME, Lau C, Nafa K, Ladanyi M. Detection of KRAS and BRAF Mutations in Colorectal Carcinoma: Roles for High Sensitivity LNA-PCR Sequencing and Broad Spectrum Mass Spectrometry (Sequenom) Genotyping. J Mol Diagn. 2010 doi: 10.1016/j.jmoldx.2010.11.005. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zakowski MF, Ladanyi M, Kris MG. EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med. 2006;355:213–5. doi: 10.1056/NEJMc053610. [DOI] [PubMed] [Google Scholar]
- 30.Solomon SB, Zakowski MF, Pao W, et al. Core needle lung biopsy specimens: adequacy for EGFR and KRAS mutational analysis. AJR Am J Roentgenol. 2010;194:266–9. doi: 10.2214/AJR.09.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shih JY, Gow CH, Yu CJ, et al. Epidermal growth factor receptor mutations in needle biopsy/aspiration samples predict response to gefitinib therapy and survival of patients with advanced nonsmall cell lung cancer. Int J Cancer. 2006;118:963–9. doi: 10.1002/ijc.21458. [DOI] [PubMed] [Google Scholar]
- 32.Soh J, Toyooka S, Aoe K, et al. Usefulness of EGFR mutation screening in pleural fluid to predict the clinical outcome of gefitinib treated patients with lung cancer. Int J Cancer. 2006;119:2353–8. doi: 10.1002/ijc.22190. [DOI] [PubMed] [Google Scholar]
- 33.Huang MJ, Lim KH, Tzen CY, et al. EGFR mutations in malignant pleural effusion of non-small cell lung cancer: a case report. Lung Cancer. 2005;49:413–5. doi: 10.1016/j.lungcan.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Soh J, Toyooka S, Ichihara S, et al. EGFR mutation status in pleural fluid predicts tumor responsiveness and resistance to gefitinib. Lung Cancer. 2007;56:445–8. doi: 10.1016/j.lungcan.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Rekhtman N, Brandt SM, Sigel CS, et al. Suitability of Thoracic Cytology for New Therapeutic Paradigms in Non-Small Cell Lung Carcinoma: High Accuracy of Tumor Subtyping and Feasibility of EGFR and KRAS Molecular Testing. J Thorac Oncol. 2010 doi: 10.1097/JTO.0b013e31820517a3. In press. [DOI] [PubMed] [Google Scholar]
- 36.Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res. 2006;12:7232–41. doi: 10.1158/1078-0432.CCR-06-0658. [DOI] [PubMed] [Google Scholar]
- 37.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 38.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–9. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 39.Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–90. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 40.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–9. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 41.Gramza AW, Corless CL, Heinrich MC. Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin Cancer Res. 2009;15:7510–8. doi: 10.1158/1078-0432.CCR-09-0190. [DOI] [PubMed] [Google Scholar]
- 42.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blay JY, Le Cesne A, Ray-Coquard I, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol. 2007;25:1107–13. doi: 10.1200/JCO.2006.09.0183. [DOI] [PubMed] [Google Scholar]
- 44.Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR mutant lung cancer: Distinct natural history of patients with tumors harboring the T790M mutation. doi: 10.1158/1078-0432.CCR-10-2692. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequencing tracings after LNA-PCR demonstrating multiple artifactual mutant peaks in the region bound by the LNA probe (*). In general, these cases show >2 additional peaks that are poorly reproducible in subsequent runs and should not prompt a positive mutation call. These peaks are usually generated when a genuine mutant allele is absent or extremely low (below 0.1%). In this case, an independent repeat run with excess LNA probe shows a failure pattern caused by complete suppression of the WT allele in the absence of a mutation. () denotes position of SNP. The arrow indicates where the location of the T790M, (X) indicate positions where artifactual peaks are no longer seen in the duplicate run.