

Abstract
Homeostatic synaptic scaling calibrates neuronal excitability by adjusting synaptic strengths during prolonged changes in synaptic activity. The molecular mechanisms that regulate the trafficking of AMPA receptors (AMPARs) during synaptic scaling are largely unknown. Here, we show that chronic activity blockade reduces PICK1 protein level on a time scale that coincides with the accumulation of surface AMPARs. PICK1 loss of function alters the subunit composition and the abundance of GluA2-containing AMPARs. Due to aberrant trafficking of these receptors, the increase in synaptic strength in response to synaptic inactivity is occluded in neurons generated from PICK1 knock-out mouse. In agreement with electrophysiological recordings, no defect of AMPAR trafficking is observed in PICK1 knock-out neurons in response to elevated neuronal activity. Overall, our data reveal an important role of PICK1 in inactivity-induced synaptic scaling by regulating the subunit composition, abundance, and trafficking of GluA2-containing AMPARs.
Introduction
The AMPA-type glutamate receptor mediates the majority of fast excitatory synaptic transmission and its trafficking into and out of the synapse plays a critical role in synaptic plasticity (Shepherd and Huganir, 2007). Hebbian forms of synaptic plasticity, such as long-term potentiation (LTP) and long-term depression (LTD), have been proposed as a physiological correlate of learning and memory. However, this type of plasticity exerts a powerful long-term destabilizing influence on neural circuits. Homeostatic plasticity maintains a stable neuronal circuit by adjusting synaptic properties to keep activity close to the internal target firing range (Turrigiano and Nelson, 2004; Davis, 2006). One such adaptation involves a cell-wide adjustment of postsynaptic AMPA receptors (AMPARs), a process termed synaptic scaling (Turrigiano and Nelson, 2004; Turrigiano, 2008). Synaptic scaling has been observed in a variety of central synapses upon chronic decrease or increase in activity in dissociated neuronal cultures (O'Brien et al., 1998; Turrigiano et al., 1998), as well as in intact animals in response to developmental or activity-dependent changes in sensory inputs (Desai et al., 2002; Maffei et al., 2006; Goel and Lee, 2007). Recently, several molecules such as TNF-α (Stellwagen and Malenka, 2006), Arc (Shepherd et al., 2006), β3-integrin (Cingolani et al., 2008), Plk2 (Seeburg et al., 2008), and retinoic acid (Aoto et al., 2008) have been shown to influence synaptic scaling. However, the molecular mechanisms underlying the trafficking of AMPARs during synaptic scaling remain elusive.
The BAR (Bin/amphiphysin/Rvs) and PDZ (PSD-95/Dlg/ZO1) domain-containing protein PICK1 (protein interacting with C-kinase 1) directly binds to the GluA2 subunit of AMPARs (Dev et al., 1999; Xia et al., 1999). PICK1 plays important roles in AMPAR surface expression, trafficking, and synaptic targeting (Xu and Xia, 2006; Hanley, 2008). Several forms of Hebbian plasticity, such as hippocampal and cerebellar LTD, are abolished in PICK1 knock-out animals, most likely due to impaired internalization, recycling, or intracellular retention of GluA2-containing AMPARs (Steinberg et al., 2006; Terashima et al., 2008; Volk et al., 2010). In contrast to the established role of PICK1 in Hebbian plasticity, the role of PICK1 in homeostatic plasticity is currently unknown.
Here, we tested whether PICK1 participates in homeostatic plasticity by investigating how loss of PICK1 function affects bidirectional synaptic scaling induced by chronic tetrodotoxin (TTX) or bicuculline treatment. We found that TTX treatment reduces PICK1 protein levels on a time scale that coincides with the accumulation of surface AMPARs. Electrophysiological recordings of AMPAR-mediated miniature EPSC (mEPSC) reveal that TTX-induced synaptic scaling is occluded in PICK1 knock-out or PICK1 knockdown neurons due to altered subunit composition and aberrant trafficking of GluA2-containing AMPARs. Interestingly, bicuculline-induced synaptic scaling is normal in PICK1 knock-out neurons. Overall, our data reveal an important role of PICK1 in inactivity-induced synaptic scaling by regulating the subunit composition, abundanc,e and trafficking of GluA2-containing AMPARs.
Materials and Methods
DNA constructs.
PICK1 shRNAs were cloned into bicistronic pSuper-Venus construct (Hayashi-Takagi et al., 2010), where Venus and shRNA expression were driven by the CMV and the H1 RNA polymerase III promoters, respectively. The PICK1 shRNA targeting sequences were as follows: sh#1, 5′-GCCTCACCATCAAGAAGTACC-3′; and sh#2, 5′-AAAGTACTATAATGACTGCTAT-3′. The PICK1 shRNA#2 sequence was reported previously (Rocca et al., 2008). The efficiency of these shRNA constructs was tested in HEK cells overexpressing Myc-PICK1. The best sequence (sh#1) was subse quently cloned into FuGW vector for lentiviral production, as described previously (Takamiya et al., 2008), and was the one used throughout the study.
Neuronal cultures.
Homozygote PICK1 knock-out mice (Gardner et al., 2005) were obtained by mating heterozygote parents. High-density cortical neurons were prepared from PICK1 knock-out pups and wild-type (WT) littermates (both males and females) at postnatal day 0 and plated onto poly-l-lysine-coated culture dish. Neurons were maintained in glial-conditioned Neurobasal medium supplemented with 1% horse serum, 2% B-27, and 2 mm Glutamax, and fed twice a week. Young neurons between days in vitro (DIV) 11–13 were used throughout experiments to minimize the contribution of presynaptic homeostatic plasticity (Wierenga et al., 2006; Han and Stevens, 2009). To induce synaptic scaling, neurons at DIV 9–11 were treated with 2 μm TTX or 40 μm bicuculline for 48 h, unless otherwise noted. To determine the pathway by which PICK1 is degraded, neurons were treated with 2 μm TTX in the presence or absence of the proteosome inhibitor lactacystin (0.5 μm) or the lysosomal inhibitor leupeptin (50 μg/ml) for 48 h. For PICK1 knockdown experiments, cortical neurons were prepared from E18 C57BL/6 mouse embryos, transfected at DIV 8, and treated with 2 μm TTX or 40 μm bicuculline at DIV 9 for 48 h. All neuronal culture reagents and drugs were obtained from Invitrogen and Tocris Bioscience, respectively.
Immunocytochemistry.
Neurons were transfected at DIV 8 with either pSuper-Venus or pSuper-Venus-PICK1 sh#1 using Lipofectamine 2000 (Invitrogen) for 72 h. Neurons were fixed with 4% paraformaldehyde/4% sucrose in PBS for 15 min, permeabilized with 0.25% Triton X-100 in PBS and incubated with 10% BSA for 1 h. Neurons were then incubated with anti-PICK1 primary antibody (JH2906) followed by Alexa-568-conjugated goat-anti-rabbit secondary antibody (Invitrogen). Images were collected with a 63× oil-immersion objective on a Zeiss LSM510 confocal microscope for both green (Venus) and red (PICK1) channels. Series of optical sections were collected at 0.38 μm intervals, and maximal intensity projection was shown.
Surface biotinylation assays.
Neurons were washed twice with artificial CSF (ACSF; in mm: 25 HEPES, 120 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 30 d-glucose, pH 7.4) and incubated with 1 mg/ml Sulfo-NHS-SS-Biotin (Pierce) for 30 min on ice. Free biotin was quenched by washing cells twice with ice-cold 50 mm glycine (pH 7.4 in ACSF). Cultures were lysed and sonicated in RIPA buffer and incubated with Neutravidin beads (Pierce) for 3 h at 4°C. Beads were washed three times, eluted with 2× SDS sample buffer followed by Western blotting analyses. Specific antibodies against GluA1 (4.9D), GluA2 (JH4297 or 6A), GluA3 (JH4300), and PICK1 (JH2906) were generated in-house. Monoclonal antibodies against α-tubulin and β-actin were purchased from Sigma.
Determination of surface GluA1 homomeric and GluA2/3 heteromeric AMPAR complexes.
Neurons were incubated with 1 mg/ml Sulfo-NHS-SS-Biotin (Pierce) for 30 min on ice and free biotin was quenched by washing neurons with ice-cold TBS (in mm: 50 Tris-HCl, 150 NaCl, pH 7.4). Neurons were lysed in RIPA buffer and the total protein concentration was determined by BCA assay. Two hundred micrograms of neuronal lysate were incubated overnight with 10 μg of anti-GluA2/3 (JH3724) or anti-GluA1 (JH4297) antibodies. The unbound fraction was subjected to another round of immunoprecipitation with the same antibodies for another 6 h at 4°C. After the second immunoprecipitation, the unbound fraction was incubated with Neutravidin beads (Pierce) at 4°C for 6 h. Beads were washed three times in RIPA buffer, eluted with 2× SDS sample buffer followed by SDS gel electrophoresis and Western blotting analyses. Quantitative analyses of Western blots were performed by determining the intensity of each band on the developed films with ImageJ software. The percentage of total and surface AMPAR subunits remaining in the unbound fraction was calculated by normalizing band intensities to the total input values.
Electrophysiology.
Pyramidal neurons were targeted for whole-cell recording with borosilicate electrodes (3–6 MΩ) at room temperature in normal ACSF (in mm: 150 NaCl, 10 HEPES, 3 KCl, 10 d-glucose, 2 CaCl2, 2 MgSO4, pH 7.4) containing 100 μm picrotoxin, 100 μm DL-APV, and 1 μm TTX. Electrode internal solution contained the following (in mm): 130 cesium methanesulfonate, 10 HEPES, 0.5 EGTA, 8 CsCl, 5 TEA-Cl, 1 QX-314, 10 Na phosphocreatine, 0.5 Na-GTP, 4 Mg-ATP, and Alexa-594 (Invitrogen). Data were acquired with a Multiclamp 700B and PCLAMP 10 software (Molecular Devices) at 10 kHz. Current traces were low-pass filtered at 1 kHz and events having amplitude of >2× rms noise were detected using MiniAnalysis (Synaptosoft). Kinetic measurements were performed on scaled, mean EPSC traces using a monoexponential decay function.
Results
Neuronal activity regulates PICK1 protein level in cultured neurons
It is well established that chronic changes in synaptic activity result in global remodeling and reorganization of postsynaptic proteins (Ehlers, 2003). However, the effect of chronic changes in neuronal activity on PICK1 expression has not been examined. Bidirectional homeostatic scaling can be modeled in primary cultured neurons wherein the synaptic strength is scaled up by prolonged suppression of neuronal activity, or scaled down by elevated neuronal activity (O'Brien et al., 1998; Turrigiano et al., 1998). We found that 48 h of TTX treatment, which blocks all evoked neuronal activity, significantly reduced PICK1 expression (73.1 ± 6.8% of control) (Fig. 1A,B). In contrast, chronic bicuculline treatment, which elevates neuronal firing, had no effect on PICK1 expression (93.2 ± 5.4% of control) (Fig. 1A,B). The reduction of PICK1 protein level was apparent 24 h after TTX incubation and persisted for as long as 72 h. Interestingly, short-term treatment with TTX (up to 6 h) did not have any significant effect on PICK1 protein level (Fig. 1C,D). The time scale by which PICK1 protein level is downregulated by chronic TTX treatment coincides with the accumulation of surface AMPAR in response to decreased synaptic activity (O'Brien et al., 1998; Turrigiano et al., 1998; Sutton et al., 2006), suggesting a potential role for PICK1 in regulating the trafficking of AMPARs during synaptic scaling.
Figure 1.

Synaptic inactivity decreases PICK1 protein level. A, Western blot analysis of PICK1 from total protein extracts of mouse cortical neurons treated for 48 h with control solution (Ctrl), TTX (2 μm), or bicuculline (Bic, 40 μm). B, Quantification of PICK1 protein levels after normalizing to α-tubulin. Data represent mean ± SEM of band intensities normalized to control values of untreated neurons (ANOVA, *p < 0.05; **p < 0.01; n = 15). C, D, Western blot analysis (C) and quantification (D) of TTX-dependent decrease of PICK1 protein over time. Data represent mean ± SEM of band intensities normalized to control values of neurons at 0 h time point (Mann–Whitney test, **p < 0.01; n = 6). E, Western blot analysis of PICK1 from lysates of cortical neurons treated for 48 h with TTX in the presence of either the vehicle control DMSO (0.1%), the proteosome inhibitor lactacystin (Lac; 0.5 μm) or the lysosomal inhibitor leupeptin (Leu; 50 μg/ml). F, Quantification of PICK1 protein levels after normalizing to β-actin. Data represent mean ± SEM of band intensities normalized to control values of untreated neurons (ANOVA, **p < 0.01; ***p < 0.001; n = 4).
To gain insight into the mechanism behind reduced PICK1 level following chronic synaptic inactivity, cultured cortical neurons were incubated with TTX for 48 h in the presence or absence of pharmacological agents that block the proteasomal and the lysosomal degradation pathways. As expected, cortical neurons treated with TTX in the presence of 0.1% DMSO (vehicle) led to a significant decrease in PICK1 protein level (53.9 ± 5.9% of control) (Fig. 1E,F). This effect was largely prevented by coincubation with the lysosomal inhibitor leupeptin (50 μg/ml, 85.6 ± 2.9% of control) (Fig. 1E,F), but not by the proteosamal inhibitor lactacystin (0.5 μm, 59.4 ± 7.0% of control) (Fig. 1E,F). These data suggest that the degradation of PICK1 during activity deprivation is mediated by the lysosomal degradation pathway.
TTX-induced synaptic scaling is occluded in PICK1 knock-out neurons
To examine the role of PICK1 in synaptic scaling, we treated cultured cortical neurons derived from PICK1 knock-out and WT littermates with TTX or bicuculline (Bic) for 48 h before mEPSC recording. WT neurons exhibited normal synaptic scaling up and scaling down of mEPSC amplitudes upon TTX and bicuculline treatment, respectively (control, 14.45 ± 0.82 pA; TTX, 23.96 ± 2.57 pA; Bic, 9.89 ± 0.69 pA) (Fig. 2A–C). Strikingly, PICK1 knock-out neurons showed significantly higher basal mEPSC amplitudes and thus, TTX-induced synaptic scaling appeared to be occluded (control, 20.13 ± 1.43 pA; TTX, 21.39 ± 2.25 pA) (Fig. 2A–C). In contrast, PICK1 knock-out neurons showed a robust synaptic scaling down of mEPSC amplitudes following chronic bicuculline treatment (control, 20.13 ± 1.43 pA; Bic, 10.84 ± 1.12 pA) (Fig. 2A–C). As expected, neither TTX nor bicuculline treatment changed mEPSC frequency in PICK1 knock-out (KO) or WT neurons (WT: control, 2.20 ± 0.42 Hz; TTX, 3.00 ± 0.88 Hz; Bic, 2.29 ± 0.50 Hz; KO: control, 2.66 ± 0.75 Hz; TTX, 4.22 ± 0.88 Hz; Bic, 3.48 ± 1.18 Hz) (Fig. 2A,D). In addition, we did not observe any change in mEPSC decay kinetics in any condition (WT: control, 5.18 ± 0.35 ms; TTX, 4.78 ± 0.62 ms; Bic, 6.38 ± 0.73 ms; KO: control, 5.32 ± 0.60 ms; TTX, 4.74 ± 0.49 ms; Bic, 5.73 ± 0.45 ms), indicating that bidirectional synaptic scaling involves the accumulation and removal of GluA2-containing AMPARs on synapses. Together, these data suggest that PICK is involved in the unidirectional scaling up of AMPAR-mediated mEPSC upon chronic synaptic inactivity.
Figure 2.
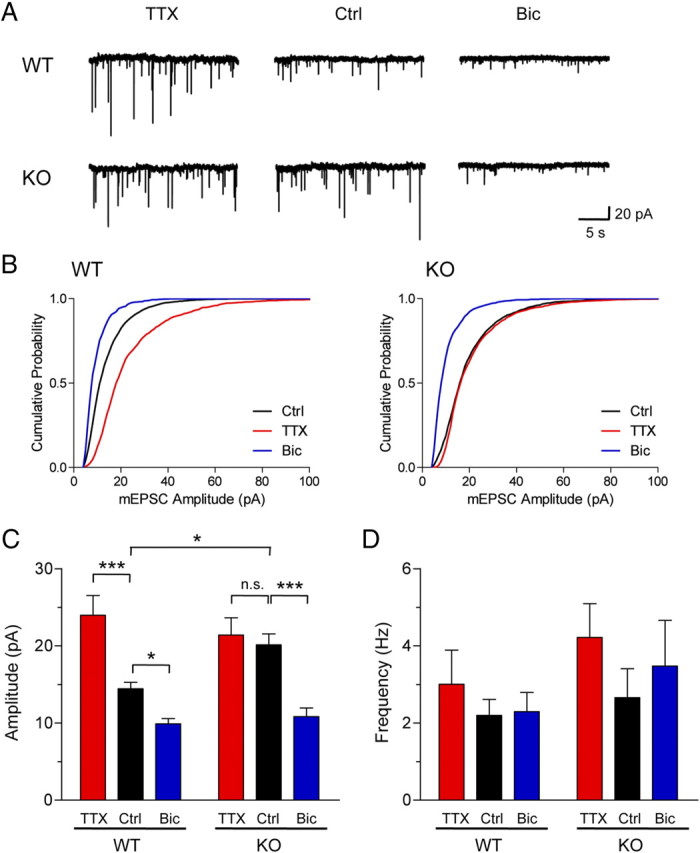
Synaptic scaling during decreased network activity is occluded in PICK1 knock-out neurons. A, B, Representative whole-cell recording sample traces of mEPSC events (A) and cumulative distribution plots of mEPSC amplitude (B) from cultured cortical neurons derived from PICK1 knock-out and WT littermates after treatment with control solution (Ctrl), TTX (2 μm), or bicuculline (Bic, 40 μm) for 48 h. C, D, Quantification of mean mEPSC amplitude (C) and frequency (D) for each population. Data represent mean ± SEM (ANOVA, *p < 0.05; ***p < 0.001; n = 8 neurons per group).
PICK1 knockdown occludes TTX-induced synaptic scaling
To ascertain that the effects we observed in PICK1 knock-out neurons were not due to the loss of PICK1 function during neuronal development or the disruption of PICK1 presynaptic function, which may lead to abnormal network activity, we transiently transfected PICK1 shRNA construct to acutely knockdown PICK1 expression in a sparse population of cortical neurons. The PICK1 shRNA#1 efficiently reduced myc-PICK1 expression in heterologous cells and endogenous PICK1 expression in neurons after 3 d of expression (7 d in the case of lentiviral-mediated knockdown) (Fig. 3A–C). We further characterized PICK1 shRNA#1 using two independent assays. Knockdown of PICK1 in cultured hippocampal neurons accelerated GluA2 recycling following NMDA receptor activation, resembling the phenotype seen in PICK1 knock-out neurons (Lin and Huganir, 2007) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In addition, PICK1 knockdown in young cortical neurons resulted in a significantly reduced number of distal dendritic processes, consistent with results from a previous study (Rocca et al., 2008) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 3.
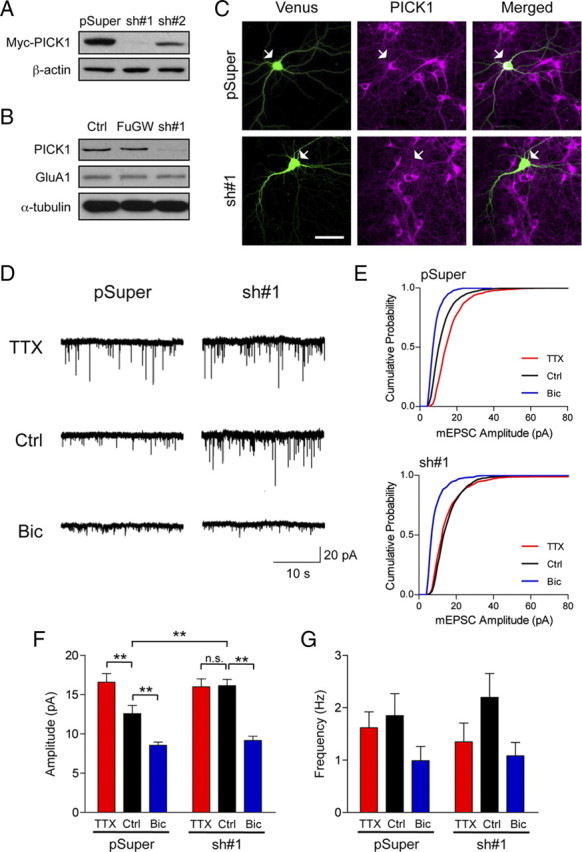
PICK1 knockdown occludes synaptic scaling during decreased network activity. A, PICK1 shRNAs decrease expression of Myc-PICK1 in HEK293 cells. B, Lysates from cultured cortical neurons either uninfected or infected with GFP (FuGW) or PICK1-shRNA#1::GFP (sh#1) lentivirus were subjected to immunoblot analysis with anti-PICK1, anti-GluA1, and anti-α-tubulin antibodies. C, Cortical neurons (DIV 8) expressing Venus or a bicistronic Venus::PICK1-shRNA#1 vector for 3 d were fixed and stained with anti-PICK1 antibody. The neuron expressing PICK1-shRNA showed a significant decrease in endogenous PICK1 staining (bottom panel) compared with the vector control-transfected neuron (top panel). Scale bar, 50 μm. D, E, Representative whole-cell recording sample traces of mEPSC events (D) and cumulative distribution plots of mEPSC amplitude (E) from cultured cortical neurons transfected with either pSuper-Venus (pSuper) or pSuper-Venus-PICK1 shRNA (sh#1) after treatment with control solution (Ctrl), TTX (2 μm), or bicuculline (Bic, 40 μm) for 48 h. F, G, Quantification of mean mEPSC amplitude (F) and frequency (G) for each population. Data represent mean ± SEM (ANOVA, **p < 0.01; n = 7–10 neurons per group). n.s., Not significant.
To directly investigate the cell autonomous effect of PICK1 knockdown in synaptic scaling, we treated cultured cortical neurons that had been transfected with either pSuper-Venus or pSuper-Venus-PICK1 shRNA#1 for 24 h with TTX or bicuculline for 48 h before mEPSC recording. Neurons transfected with pSuper-Venus exhibited normal synaptic scaling up and scaling down of mEPSC amplitudes upon TTX and bicuculline treatment, respectively (control, 12.57 ± 1.05 pA; TTX, 16.58 ± 1.10 pA; Bic, 8.54 ± 0.41 pA) (Fig. 3D–F). Consistent with our findings in PICK1 knock-out neurons, PICK1 knockdown also resulted in significantly higher basal mEPSC amplitudes and occluded TTX-induced synaptic scaling (control, 16.13 ± 0.80 pA; TTX, 15.99 ± 1.00 pA) (Fig. 3D–F). In addition, PICK1 knockdown neurons showed a robust synaptic scaling down of mEPSC amplitudes following chronic bicuculline treatment (control, 16.13 ± 0.80 pA; Bic, 9.17 ± 0.52 pA) (Fig. 3D–F). Again, we did not observe any significant changes in mEPSC frequency following TTX and bicuculline treatment in pSuper or PICK1 sh#1 transfected neurons (pSuper: control, 1.85 ± 0.42 Hz; TTX, 1.62 ± 0.30 Hz; Bic, 0.99 ± 0.27 Hz; sh#1: control, 2.19 ± 0.46 Hz; TTX, 1.35 ± 0.36 Hz; Bic, 1.08 ± 0.25 Hz) (Fig. 3D,G). Moreover, we did not observe any change in mEPSC decay kinetics in any condition (pSuper: control, 4.51 ± 0.33 ms; TTX, 4.46 ± 0.23 ms; Bic, 5.52 ± 0.52 ms; sh#1: control, 4.21 ± 0.23 ms; TTX, 4.36 ± 0.22 ms; Bic = 4.67 ± 0.33 ms), again indicating that bidirectional synaptic scaling involves the accumulation and removal of GluA2-containing AMPARs at synapses. Together, the lack of TTX-induced synaptic scaling in both the PICK1 knockdown and PICK1 knock-out neurons provides important complementary results establishing the involvement of PICK1 cell autonomously in postsynaptic neurons in unidirectional scaling up of AMPAR-mediated mEPSC.
Altered surface AMPAR subunit abundance in PICK1 knock-out neurons
To assess the contribution of PICK1 to the trafficking of AMPARs during synaptic scaling, we compared the surface expression of GluA1, GluA2, and GluA3 in PICK1 knock-out neurons using a biotinylation assay. Under basal conditions, we observed a change in surface AMPAR levels in PICK1 knock-out neurons. The levels of surface GluA2 and surface GluA3 in PICK1 knock-out neurons were significantly higher than in neurons derived from WT littermates (surface/total ratio: GluA2, 203 ± 31% of WT; GluA3, 171 ± 24% of WT) (Fig. 4A,B). In addition, we observed a slight but significant decrease in the level of surface GluA1 expression (surface/total ratio: 74 ± 11% of WT) (Fig. 4A,B). The decrease in surface GluA1 may be due to a compensatory mechanism, since a reciprocal increase in surface GluA1 expression has also been observed in neurons overexpressing GluA2 siRNA (Gainey et al., 2009). These altered surface AMPAR levels are most likely due to impaired receptor trafficking events rather than protein synthesis or degradation defects, as there was no significant difference in the total expression levels of GluA1, GluA2, and GluA3 subunits in PICK1 knock-out neurons (GluA1, 94 ± 7% of WT; GluA2, 109 ± 14% of WT; GluA3, 85 ± 7% of WT) (Fig. 4A,C). These data indicate that the increase in surface GluA2 and GluA3 levels may account for the increase in basal synaptic transmission observed in PICK1 knock-out neurons (Fig. 2C).
Figure 4.

Surface AMPAR protein levels are altered in PICK1 knock-out neurons. A, Cultured cortical neurons from PICK1 knock-out or WT littermates were subjected to surface biotinylation assay. The relative amount of surface and total AMPARs was assessed by Western blot using specific antibodies against GluA1, GluA2, and GluA3. PICK1 blot confirmed the genotype of the cultured neurons. B, C, Quantification of the surface:total ratio (B) and total (C) GluA1, GluA2, and GluA3 after normalizing against α-tubulin. Data represent mean ± SEM of band intensities normalized to control values of WT neurons (Mann–Whitney test, *p < 0.05; n = 12).
PICK1 knock-out neurons lack an increase in surface GluA2 subunit following TTX treatment
Next, we examined the surface expression of GluA1, GluA2, and GluA3 subunits following TTX and bicuculline treatment. Consistent with electrophysiological recordings, WT neurons exhibited robust increase and decrease in both surface GluA1 and GluA2 expression after TTX and bicuculline treatment, respectively (TTX: GluA1, 156 ± 13% of control; GluA2, 133 ± 9% of control; Bic: GluA1, 64 ± 5% of control; GluA2, 65 ± 8% of control) (Fig. 5A,B). Strikingly, adaptation of surface GluA2 expression, but not GluA1, was impaired in PICK1 knock-out neurons in response to TTX treatment (GluA1, 146 ± 10% of control; GluA2, 96 ± 8% of control) (Fig. 5A,B). The inability of GluA2 subunit to scale up is likely due to an occlusion effect since basal surface GluA2 expression was already elevated in PICK1 knock-out neurons (Fig. 4A,B). The normal increase in surface GluA1 expression upon TTX treatment indicates a crucial role of PICK1 in selectively controlling GluA2 trafficking. This is consistent with a recent finding that demonstrates a requirement of AMPAR GluA2 subunit for TTX-induced synaptic scaling (Gainey et al., 2009). In agreement with our electrophysiological recording, no defect of AMPAR trafficking was observed in PICK1 knock-out neurons in response to bicuculline treatment (GluA1, 57 ± 6% of control; GluA2, 67 ± 6% of control) (Fig. 5A,B).
Figure 5.

The increase in surface GluA2 AMPAR subunit following chronic synaptic inactivity is impaired in PICK1 knock-out neurons. A, Cultured cortical neurons from PICK1 knock-out or WT littermates were treated with control solution (Ctrl), TTX (2 μm), or bicuculline (Bic, 40 μm) and were subjected to surface biotinylation assay 48 h later. The relative amount of surface and total AMPARs was assessed by Western blot analyses using specific antibodies against GluA1 and GluA2 subunits after normalizing against α-tubulin. B–D, Quantification of the surface (B), total (C), and surface:total ratio (D) of GluA1 and GluR2 AMPAR subunits in PICK1 WT and knock-out neurons. Data represent mean ± SEM of band intensities normalized to control values of untreated neurons (ANOVA, *p < 0.05; **p < 0.01; ***p < 0.001; n = 18–24).
To determine whether the lack of an increase in GluA2 surface expression was due to changes in total protein levels, we next examined the total expression of AMPAR subunits following TTX and bicuculline treatment. In WT neurons, chronic TTX treatment selectively increased the total GluA1 level, but not the total expression of GluA2 subunit (GluA1, 139 ± 13% of control; GluA2, 91 ± 8% of control) (Fig. 5A,C). Conversely, bicuculline treatment led to significant reduction in total AMPAR expression (GluA1, 67 ± 6% of control; GluA2, 70 ± 8% of control) (Fig. 5A,C). We found no significant difference between PICK1 knock-out and WT neurons in changes in the total expression of AMPAR subunits upon TTX and bicuculline treatment (PICK1 KO, TTX: GluA1, 118 ± 5% of control; GluA2, 91 ± 6% of control; Bic: GluA1, 63 ± 6% of control; GluA2, 78 ± 5% of control) (Fig. 5A,C). Moreover, surface-to-total-ratio analyses revealed a distinct mechanism of regulation for GluA1 and GluA2 during TTX-induced synaptic scaling. In both PICK1 knock-out and WT neurons, GluA1 total and surface levels change at a similar rate in response to TTX treatment (WT, 121 ± 9% of control; KO, 126 ± 10% of control) (Fig. 5D). In contrast, the surface-to-total-GluA2 ratio becomes significantly higher upon TTX treatment in WT but not in PICK1 KO neurons (WT, 156 ± 9% of control; KO, 115 ± 13% of control) (Fig. 5D). Altogether, these data suggest that loss of PICK1 function occludes TTX-induced synaptic scaling by specifically impairing the trafficking of GluA2-containing AMPARs.
Chronic activity blockade causes aberrant trafficking of GluA2-containing AMPARs in PICK1 knock-out neurons
Unlike surface GluA2, the increase in surface GluA1 induced by TTX treatment is not impaired in PICK1 knock-out neurons. Surprisingly, however, mEPSC amplitudes failed to scale up in PICK1 knock-out neurons. This result, along with a lack of change in mEPSC decay kinetics combined with the lack of surface GluA2 increases, suggests that these newly inserted GluA1-containing receptors are most likely extrasynaptic Ca2+-permeable GluA1 homomers. To test this hypothesis, we combined surface biotinylation and GluA2/3 immunodepletion assays from neuronal lysates and examined the fraction of total and surface homomeric GluA1 AMPARs remaining. Two-rounds of immunoprecipitation effectively pulled down >98% of GluA2 and GluA3, leaving ∼15% of total GluA1 in the unbound fraction in all conditions (Fig. 6A,B). As expected, we did not observe any significant changes in the amount of surface GluA1 homomers following TTX treatment in WT neurons (control, 3.9 ± 0.4%; TTX, 3.4 ± 0.6%) (Fig. 6A,C), suggesting that synaptic inactivity drives incorporation of GluA1/2 heteromers to the plasma membrane. Under basal condition, the levels of surface GluA1 homomers were significantly lower in the PICK1 knock-out neurons (WT, 3.9 ± 0.4%; KO, 2.7 ± 0.3%) (Fig. 6A,C), consistent with our previous study (Clem et al., 2010). Surprisingly, the level of surface GluA1 homomers was not altered following activity deprivation in PICK1 knock-out neurons (control, 2.7 ± 0.3%; TTX, 2.0 ± 0.4%) (Fig. 6A,C). This suggests that the TTX-induced increase in surface GluA1 in PICK1 knock-out neurons represents an increase in surface GluA1/2 heteromers.
Figure 6.

Synaptic inactivity does not induce accumulation of homomeric GluA1 AMPARs on to plasma membrane. A, Cultured cortical neurons from PICK1 knock-out or WT littermates were treated with control solution (Ctrl) or TTX (2 μm) and were subjected to surface biotinylation assay 48 h later. Neuronal lysates were then subjected to two-rounds of GluA2/3 immunodepletion before Neutravidin chromatography steps. I, Input; T, total unbound fraction; S, surface unbound fraction following GluA2/3 immunodepletion. Surface fraction was loaded at 17× input concentration. B, C, Quantification of the surface (B) and total (C) GluA1, GluA2, and GluA3 AMPAR subunits in PICK1 WT and knock-out neurons. Data represent mean ± SEM of band intensities normalized to control values of untreated neurons (Student's t test, *p < 0.05; n = 6).
How can we account for the lack of change in surface GluA2 subunit in the PICK1 knock-out neurons following chronic activity deprivation? Since PICK1 knock-out neurons have increased levels of surface GluA2/3 AMPAR complexes, we hypothesize that during chronic activity deprivation, the increase in surface GluA1/2 heteromers is counterbalanced by the removal of heteromeric GluA2/3 receptors from the plasma membrane. Since nearly all of GluA2 subunits form heteromeric complexes with either GluA1 and GluA3 subunits (Wenthold et al., 1996; Lu et al., 2009), we performed GluA1 immunodepletion assays from neuronal lysates and examined the fraction of total and surface heteromeric GluA2/3 AMPARs remaining following chronic activity blockade in both PICK1 WT and knock-out neurons. Two-rounds of immunoprecipitation effectively depleted >98% of total GluA1 from neuronal lysates in all conditions (Fig. 7A,B). Interestingly, under basal condition total GluA2 and GluA3 in the unbound fraction following GluA1 immunodepletion were significantly higher in the PICK1 knock-out neurons compared with the neurons prepared from the WT littermates (WT: GluA2, 47.4 ± 3.0%; GluA3, 56.3 ± 3.0%; KO: GluA2, 59.6 ± 1.4%; GluA3, 70.8 ± 2.1%) (Fig. 7A,B), suggesting a possible role of PICK1 in AMPAR assembly or stability. Consistent with our previous finding, levels of surface GluA2 and GluA3 subunits were also significantly higher in the PICK1 knock-out neurons (WT: GluA2 = 3.9 ± 0.5%, GluA3 = 16.1 ± 2.2%; KO: GluA2 = 6.4 ± 0.4%, GluA3 = 34.9 ± 5.0%) (Fig. 7A,C). We found no significant differences in surface GluA2 or GluA3 levels following activity deprivation in WT neurons, indicating the absence of heteromeric GluA2/3 AMPARs insertion to the plasma membrane (control: GluA2, 3.9 ± 0.5%; GluA3, 16.1 ± 2.2%; TTX: GluA2, 4.9 ± 0.6%; GluA3, 21.6 ± 1.9%) (Fig. 7A,C). In PICK1 knock-out neurons, however, the level of surface GluA2/3 heteromers decreased significantly, suggesting internalization of these receptors following synaptic inactivity (control: GluA2, 6.4 ± 0.4%; GluA3, 34.9 ± 5.0%; TTX: GluA2, 3.9 ± 1.0%; GluA3, 19.6 ± 4.7%) (Fig. 7A,C). Altogether, our data suggest that loss of PICK1 function leads to a complex disruption of proper trafficking of heteromeric GluA2/3 AMPARs, which underlies the occlusion of TTX-induced synaptic scaling in cultured neurons.
Figure 7.

Synaptic inactivity causes aberrant trafficking of GluA2-containing AMPARs in PICK1 knock-out neurons. A, Cultured cortical neurons from PICK1 knock-out or WT littermates were treated with control solution (Ctrl) or TTX (2 μm) and were subjected to surface biotinylation assay 48 h later. Neuronal lysates were then subjected to two-rounds of GluA1 immunodepletion before Neutravidin chromatography steps. I, Input; T, total unbound fraction; S, surface unbound fraction following GluA1 immunodepletion. Surface fraction was loaded at 13× input concentration. B, C, Quantification of the surface (B) and total (C) GluA1, GluA2 and GluA3 AMPAR subunits in PICK1 WT and knock-out neurons. Data represent mean ± SEM of band intensities normalized to control values of untreated neurons (Student's t test, *p < 0.05; **p < 0.01; n = 7).
Discussion
Synaptic scaling is a form of homeostatic plasticity by which a neuron adjusts it synaptic strength to maintain stable neuronal output during changes in network activity (Turrigiano, 2008). This mechanism can be achieved through a cell-wide adjustment of AMPARs at excitatory synapses (O'Brien et al., 1998; Turrigiano et al., 1998; Wierenga et al., 2005; Shepherd et al., 2006). However, the subunit composition of newly inserted or removed receptors during synaptic scaling is complex and currently controversial. Some studies have reported that both GluA1 and GluA2 subunits contribute to synaptic scaling (O'Brien et al., 1998; Wierenga et al., 2005; Cingolani et al., 2008; Gainey et al., 2009; Sun and Wolf, 2009), but others have reported that synaptic scaling operates mainly on the GluA1 subunit (Thiagarajan et al., 2005; Sutton et al., 2006; Aoto et al., 2008). This discrepancy depends on factors such as basal activity or developmental stage of particular types of neuronal culture, choices of pharmacological treatment, as well as durations of the treatment. In this study, we chose to induce bidirectional synaptic scaling in young cultured cortical neurons with prolonged TTX or bicuculline treatment, as this system has been shown to target GluA1 and GluA2 heteromeric receptors (Wierenga et al., 2005; Wierenga et al., 2006; Ibata et al., 2008; Gainey et al., 2009). Consistent with previous studies, we observed bidirectional changes in both surface GluA1 and GluA2 levels that led to synaptic scaling of AMPAR-mediated mEPSC amplitudes, but not mEPSC frequency, suggesting an exclusive postsynaptic locus of expression. In addition, we did not observe any change in mEPSC decay kinetics, nor did we see any increase in surface homomeric GluA1 receptors following prolonged activity blockade, further confirming the involvement of GluA2-containing AMPARs during homeostatic synaptic scaling in our system.
Several studies have shown that PICK1 regulates the surface expression of GluA2-containing AMPARs either by facilitating their activity-dependent removal from the plasma membrane or by stabilizing them in intracellular pools, both of which result in a net decrease in surface GluA2 level (Kim et al., 2001; Perez et al., 2001; Terashima et al., 2004; Lin and Huganir, 2007; Rocca et al., 2008). Consistent with the proposed role of PICK1, we saw an increase in basal surface GluA2 and GluA3 accompanied by a slight compensatory decrease of surface GluA1 in PICK1 knock-out neurons. Interestingly, we also saw an increase in total heteromeric GluA2/3 receptors in PICK1 knock-out neurons, indicating a possible role for PICK1 in regulating the assembly of AMPAR in the biosynthesis pathway (Greger et al., 2002). Alternatively, PICK1 may also be involved in determining the stability of GluA2-containing AMPARs. In addition, we observed elevated basal synaptic transmission, suggesting that the increase in GluA2-containing AMPARs, most likely in the form of GluA2/3 heteromers, occurred at synapses. Indeed, previous studies have reported synaptic potentiation of basal transmission in hippocampal slices when GluA2-EVKI peptides, which disrupt PICK1-GluA2 interaction, are included in the patch pipette (Kim et al., 2001; Yao et al., 2008). Conversely, overexpression of PICK1 leads to a decrease in surface GluA2 level but not surface GluA1 in hippocampal neurons (Terashima et al., 2004).
In the present study, we found compelling evidence that PICK1 plays a cell autonomous role in TTX-induced synaptic scaling of postsynaptic neurons. Chronic synaptic inactivity reduces PICK1 protein levels in a time course that coincides with the accumulation of surface AMPARs. Consistent with the role for PICK1 in stabilizing intracellular pools of GluA2-containing AMPARs, we propose a model whereby the reduction in PICK1 level during prolonged synaptic inactivity relieves the intracellular retention, thus promoting the forward trafficking and accumulation of GluA2-containing receptors to the plasma membrane (Fig. 8A). Based on our model, one would expect that loss of PICK1 function would mimic the TTX-induced scaling of surface AMPARs and mEPSC amplitudes (Fig. 8B). Strikingly, TTX-induced accumulation of surface GluA1 subunit was normal, whereas the adaptation of GluA2 subunits was impaired in the PICK1 knock-out neurons. Our GluA2/3 immunodepletion experiments suggest that these newly inserted GluA1-containing receptors are not Ca2+-permeable GluA1 homomers; instead they accumulate on the extrasynaptic plasma membrane as GluA1/2 heteromers. Moreover, GluA1 immunodepletion experiments in PICK1 KO neurons revealed an abnormal removal of surface GluA2/3 heteromers upon TTX treatment. These two pieces of evidence may account for the lack of a net increase in surface GluA2 levels upon TTX treatment. Thus, alteration in AMPAR subunit composition and aberrant trafficking GluA2-containing AMPARs underlie the occlusion of AMPAR-mediated mEPSC increase following activity deprivation in PICK1 KO neurons.
Figure 8.
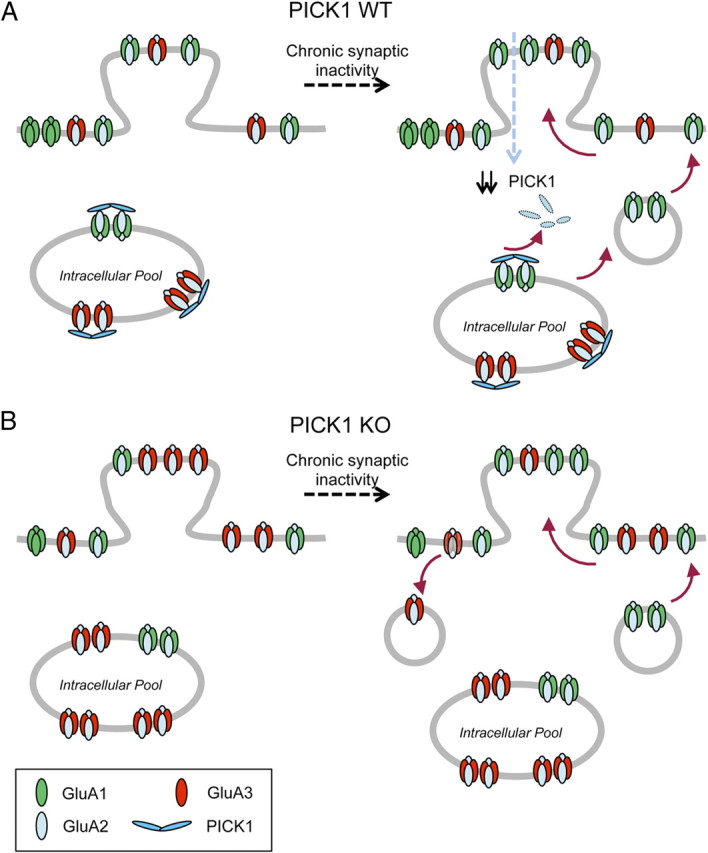
Model of PICK1 regulation of GluA2-containing AMPARs during synaptic scaling. A, Under basal conditions, PICK1 maintains pools of GluA2-containing AMPARs intracellularly. Chronic synaptic inactivity results in graded reduction in total PICK1 protein level, relieving the intracellular retention of GluA2-containing AMPARs in the form of GluA1/2 heteromers, which can then be incorporated into the plasma membrane. By lateral diffusion, these receptors are then inserted into and accumulate at the synaptic plasma membrane, presumably through the interaction between GluA2 and NSF. B, In the PICK1 knock-out neurons, there is a change in AMPAR subunit composition resulting in increased levels of total and surface GluA2/3 heteromers and elevated basal AMPAR-mediated mEPSC amplitudes. In the absence of PICK1, activity deprivation induces aberrant removal of GluA2/3 heteromers in exchange for the newly inserted GluA1/2 heteromers on the plasma membrane.
A prominent current theory of homeostatic plasticity holds that a global negative feedback mechanism compensates for long-term perturbations of network activity. But how might these seemingly opposing types of plasticity coexist in a neuron? A recent study demonstrated a differential requirement for GluA1 and GluA2 subunits in LTP and synaptic scaling, respectively, implying that these two types of plasticity may involve two molecularly distinct pathways for AMPAR trafficking (Gainey et al., 2009). Here, we present yet another piece of evidence to support this notion. Previous studies have demonstrated the crucial role of PICK1 in hippocampal and cerebellar LTD (Steinberg et al., 2006; Terashima et al., 2008; Volk et al., 2010). By analogy, both LTD and bicuculline-induced synaptic scaling require the removal of AMPARs from synapses. Surprisingly, we found that the bicuculline-induced synaptic scaling was intact in PICK1 knock-out neurons, as shown by a robust reduction in surface AMPARs and mEPSC amplitudes. Our data argue against the role of PICK1 in AMPAR internalization from the plasma membrane, a common feature for both types of plasticity. Instead, we favor a role for PICK1 in stabilizing internalized GluA2-containing AMPARs in intracellular pools, which may explain the LTD deficit in PICK1 knock-out mouse (Steinberg et al., 2006; Terashima et al., 2008; Volk et al., 2010). In the case of bicuculline-induced synaptic scaling, the internalized receptors are destined for degradation by either the proteasomal or lysosomal pathways, as shown by the decrease in total GluA1 and GluA2 protein levels (Fig. 5A,C). Together, our data strongly support the idea of a differential requirement of two molecularly distinct AMPAR trafficking pathways in Hebbian and homeostatic plasticity. These mechanisms may ensure that the two types of plasticity may coexist in neurons without interference.
Footnotes
This work was supported by grants from the National Institutes of Health and the Howard Hughes Medical Institute (to R.L.H.). V.A. is supported by fellowships from the International Human Frontier Science Program (LT00399/2008-L) and the Australian National Health and Medical Research Council (ID. 477108). R.L.C. is supported by a National Research Service Award fellowship from the National Institutes of Health (F32 MH087037–01). We thank Dr. Akira Sawa for providing pSuper-Venus construct. We are grateful to Drs. Gareth Thomas and Lenora Volk for critical reading of the manuscript. We thank Min Dai, Yilin Yu, Monica Coulter, and Richard Johnson for technical support.
Under a licensing agreement between Millipore Corporation and The Johns Hopkins University, R.L.H. is entitled to a share of royalties received by the University on sales of products described in this article. R.L.H. is a paid consultant to Millipore Corporation. The terms of this arrangement are being managed by The Johns Hopkins University in accordance with its conflict-of-interest policies.
References
- Aoto J, Nam CI, Poon MM, Ting P, Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60:308–320. doi: 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, Goda Y. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58:749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Anggono V, Huganir RL. PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J Neurosci. 2010;30:6360–6366. doi: 10.1523/JNEUROSCI.6276-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Dev KK, Nishimune A, Henley JM, Nakanishi S. The protein kinase C alpha binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology. 1999;38:635–644. doi: 10.1016/s0028-3908(98)00230-5. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG. Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci. 2009;29:6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir RL. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron. 2005;45:903–915. doi: 10.1016/j.neuron.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Goel A, Lee HK. Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J Neurosci. 2007;27:6692–6700. doi: 10.1523/JNEUROSCI.5038-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34:759–772. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Han EB, Stevens CF. Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc Natl Acad Sci U S A. 2009;106:10817–10822. doi: 10.1073/pnas.0903603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118:152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci U S A. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson SB, Turrigiano GG. Potentiation of cortical inhibition by visual deprivation. Nature. 2006;443:81–84. doi: 10.1038/nature05079. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008;10:259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DT, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Sun X, Wolf ME. Nucleus accumbens neurons exhibit synaptic scaling that is occluded by repeated dopamine pre-exposure. Eur J Neurosci. 2009;30:539–550. doi: 10.1111/j.1460-9568.2009.06852.x. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Takamiya K, Mao L, Huganir RL, Linden DJ. The glutamate receptor-interacting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J Neurosci. 2008;28:5752–5755. doi: 10.1523/JNEUROSCI.0654-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, Henley JM, Collingridge GL, Isaac JT. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J Neurosci. 2004;24:5381–5390. doi: 10.1523/JNEUROSCI.4378-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Volk L, Kim CH, Takamiya K, Yu Y, Huganir RL. Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc Natl Acad Sci U S A. 2010;107:21784–21789. doi: 10.1073/pnas.1016103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25:2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Walsh MF, Turrigiano GG. Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol. 2006;96:2127–2133. doi: 10.1152/jn.00107.2006. [DOI] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Xu J, Xia J. Structure and function of PICK1. Neurosignals. 2006;15:190–201. doi: 10.1159/000098482. [DOI] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]