

Abstract
Mixed infections with seasonal influenza A virus strains are a common occurrence and an important source of genetic diversity. Prolonged viral shedding, as observed in immunocompromised individuals, can lead to mutational accumulation over extended periods. Recently, drug resistance was reported in immunosuppressed patients infected with the 2009 pandemic influenza A (H1N1) virus within a few days after oseltamivir treatment was initiated. To better understand the evolution and emergence of drug resistance in these circumstances, we used a deep sequencing approach to survey the viral population from an immunosuppressed patient infected with H1N1/2009 influenza and treated with neuraminidase inhibitors. This patient harbored 3 genetic variants from 2 phylogenetically distinct viral clades of pandemic H1N1/2009, strongly suggestive of mixed infection. Strikingly, one of these variants also developed drug resistance de novo in response to oseltamivir treatment. Immunocompromised individuals may, therefore, constitute an important source of genetic and phenotypic diversity, both through mixed infection and de novo mutation.
Although an influenza infection is typically self-limiting and usually lasts <1 week, prolonged viral shedding is commonplace in immunosuppressed patients [1–6], such that de novo mutations can develop and accumulate over longer periods. In the case of a mixed infection, in which an individual host is infected with multiple viral variants either simultaneously or sequentially, the extended duration of influenza infection may lead to changes in the frequencies of any of the infecting variants [7] and can facilitate reassortment among them. As a consequence, immunosuppressed individuals, particularly those experiencing mixed infection, may constitute an important source of genetic and phenotypic diversity, generating variants that differ in such properties as antiviral susceptibility, virulence, and transmissibility.
In March 2009, Mexico experienced outbreaks of respiratory illness caused by a novel influenza A (H1N1) virus of swine origin [8]. Subsequently, cases of infection with this virus were found both in the United States and globally, and a pandemic was officially declared by the World Health Organization on 11 June 2009. Because the pandemic virus was demonstrated to already be resistant to the M2-ion channel blockers (amantadine and rimantadine) [9], neuraminidase inhibitors were used as first-line drugs in both prophylaxis and treatment of patients infected with H1N1/2009. Although the pandemic virus remains largely susceptible to oseltamivir and zanamivir, sporadic cases of oseltamivir resistance have been reported worldwide [10–13]. The resistance mutation in all of these cases is a histidine-to-tyrosine amino acid substitution at residue 275 of the neuraminidase (NA) protein (H274Y in N2 numbering; H275Y in N1 numbering).
Because of the rapid spread of H275Y-based oseltamivir resistance in seasonal H1N1 virus strains [14], even sporadic reports of this mutation in the pandemic H1N1/2009 virus are of significance. Oseltamivir resistance in virus strains infecting immunosuppressed patients was reported to emerge within a few days after treatment, rather than after prolonged therapy [15–17]. Clearly, this rapid selection for drug resistance is of major concern.
From the early stages of its emergence in the human population, genomic sequence data on the pandemic H1N1/2009 virus have been made rapidly available on public databases. In most cases, the sequences generated for these analyses represent consensus sequences taken from single time points during infection, thereby providing a snapshot of the dominant virus variant infecting each patient. We have previously shown that mixed infections are a common occurrence in the case of seasonal influenza virus and, thus, an important source of genetic diversity, but are not normally apparent from consensus sequencing [7]. To determine whether this is also true of the pandemic H1N1/2009 virus and what the consequences of this process might be for viral evolution, we investigated the increase of oseltamivir resistance in a longitudinal collection from an immunosuppressed patient infected with pandemic H1N1/2009 virus with use of massively parallel (deep) sequencing.
MATERIALS AND METHODS
The Study
A 15-year-old male patient received a diagnosis of T cell leukemia in December 2008 and was treated with chemotherapy. On 12 June 2009, he was hospitalized for fever (temperature, 39.7°C), chills, cough, nasal congestion, sore throat, and generalized body aches. He had no sick contacts at home, but he attended high school. His chest radiograph showed normally expanded lungs and no evidence of pneumonia, and he did not require oxygen support. A nasopharyngeal (NP) swab sample collected on 12 June tested positive for H1N1/2009 by real-time reverse-transcriptase polymerase chain reaction (RT-PCR). During 13–17 June, he received oral doses of amantadine (100 mg) and oseltamivir (75 mg) twice daily. Intravenous levofloxacin (500 mg) was also initiated and provided daily for 3 days. He became asymptomatic on 15 June and resumed his scheduled chemotherapy the following day, consisting of intrathecal methotrexate, intravenous methotrexate, leucovorin, vincristine, and a single dose of granulocyte colony-stimulating factor. He remained afebrile and was subsequently discharged on 20 June. On 23 June, the patient was hospitalized because of fever (39°C), mouth sores, decreased appetite, dehydration, and body aches. His cell counts were very low: his absolute neutrophil count was 286 cells/μL, and his absolute lymphocytic count was 923 cells/μL cefepime (2 g intravenously every 8 h) and vancomycin (1 g intravenously every 8 h) treatments were given. Another NP swab specimen collected on 25 June tested positive again for H1N1/2009 by RT-PCR. Oseltamivir treatment (75 mg twice daily) was initiated because of the progression of symptoms and a 5-day regimen was completed. The patient improved and became afebrile on day 3 of hospital admission; his absolute neutrophil count slowly improved, and results of all blood cultures performed on 23–25 June were negative. He completed 10 days of the antimicrobial regimen and was discharged on 3 July. Additional NP swab samples collected on 6 July and 5 August tested negative for H1N1/2009 by RT-PCR.
Informed Consent
The Committee on Research Involving Human Subjects at Albany Medical Center has approved the above research protocol (2636, Exemption 4 by Expedited Review under 21 CFR 56.110 and 45 CFR 46.110) on 10 September 2009. Informed consent was obtained from the patient's mother and assent from the patient through the primary provider (JCP). Approval was also obtained from the New York State Department of Health Institutional Review Board to characterize agents in specimens received from patients for diagnostic purposes (study number 02-054).
Resistance Testing
The 2 NP specimens, collected on 12 June and 25 June, were identified as positive for pandemic H1N1/2009 by real-time RT-PCR and were tested with a H275Y-mutation specific pyrosequencing assay (Centers for Disease Control and Prevention) and by partial neuraminidase (NA) gene dideoxysequencing (ie, the Sanger method). Cultured isolates of influenza virus were also grown from both specimens when inoculated onto primary Rhesus monkey kidney cells (pRhMK). Isolates were tested genotypically for neuraminidase inhibitor resistance mutations by both NA pyrosequencing and dideoxysequencing and phenotypically by the NA Star neuraminidase assay (Invitrogen). Control virus strains for the NA Star assay were kindly provided by Dr. Larisa Gubareva at the Centers for Disease Control and Prevention.
Sequencing and Assembly
Viral RNA was extracted from both specimens and amplified by multisegment RT-PCR [18]. This method enables the characterization of whole viral genomes directly from specimens rather than cultured isolates. The cDNA was randomly primed using a sequence independent single primer amplification protocol [19], amplified, and sequenced on the 454/Roche GS-FLX sequencing platform [20] using Titanium chemistry. The samples were also sequenced using the capillary (Sanger) method with the Influenza Genome Sequencing Project pipeline at the J. Craig Venter Institute in Rockville, Maryland [21]. All Influenza Genome Sequencing Project genome data are freely available on GenBank (accession numbers CY050158-CY050165 and CY050182-CY050189). The sequence reads from the GS-FLX data were sorted by bar code to separate both sequenced specimens, aligned to a reference influenza A H1N1/2009 genome, and conservatively trimmed to ensure that bar code sequence and random hexamer primer were removed.
Insertions and deletions (indels) are common in GS-FLX sequencing data and are found in proximity to homopolymer stretches. These cause misalignments and introduce errors in the reporting of single nucleotide polymorphisms. To overcome the errors from the homopolymer regions and obtain robust alignments for downstream analyses, we generated final alignments using (1) the nucmer program [22], to map each sequence read onto the consensus sequence for each segment as determined by Sanger sequencing; (2) CD-HIT-EST [23], to cluster the reads into a nonredundant set of representative reads; (3) MUSCLE [24], to align the clusters and reads; and (4) the hmmalign program from the HMMER2 suite of programs [25] (http://hmmer.wustl.edu), to build in an iterative fashion a profile hidden markov model for the final alignment of all sequence reads.
To identify the positions later used to sort the sequences into clusters for variant reconstruction, the reading frame for translation was established by including the Sanger consensus sequences in the hidden markov model alignments. For a variant nucleotide to be reported in Table 1, it had to be covered by at least 10% of the sequence reads. This conservatively high percentage ensured that variants were not determined by substitution errors; the substitution error rate on the GS-FLX platform [26] is, at best, still higher than the RNA-dependent RNA polymerase error rate associated with influenza A virus (∼.03% per nucleotide for the former vs ∼.01% for the latter). Sequence reads were sorted and clustered on the basis of the variant codons reported and assembled into variant assemblies. Reads were assembled into variants if at least 10% of the overall reads covered the position site in each grouping, with a minimum of 20× coverage at each site. Variants were reconstructed for the NA, hemagglutinin (HA), nucleoprotein (NP), and nonstructural protein 1 (NS1) genes. The other genes had insufficient sequence read coverage: the random priming methodology used did not provide homogeneous amplification across all genomic segments; thus, the redundancy of the reads was not sufficient for deep sequence analysis of certain genes.
Table 1.
Phenotypic resistance of isolates
| Residues 199, 275 | Oseltamivir IC50 (nmol/L) | Zanamivir IC50 (nmol/L) | |
| A/New York/4095/2009-RhMK | N, H | 1.1 | 1.6 |
| A/New York/4438/2009-RhMK | N, Y | 297.6 | 1.5 |
| A/California/04/2009 | D, H | 0.4 | 0.8 |
| A/Washington/29/2009 | D, Y | 52.8 | 1.1 |
NOTE. IC50, 50% inhibitory concentration; RhMK, rhesus monkey kidney cells.
Phylogenetic Analysis
Only the HA and NA variants were used in the downstream phylogenetic analyses; the NP and NS1 variants did not provide sufficient phylogenetic resolution. Sequences of the reconstructed variants for HA and NA were combined with all pandemic H1N1/2009 sequences available on GenBank sampled up to the end of July 2009, representing the period of the case in question. This resulted in data sets of 1241 sequences for HA (1698 nt) and 1137 sequences for NA (1407 nt). Phylogenetic trees for both the HA and NA were inferred using the maximum likelihood (ML) method available in the PHYML package [27] and using SPR branch-swapping. In all cases, the simple HKY85 model of nucleotide substitution was used, because the sequences in question are so similar that multiple substitutions can effectively be ignored. Support for major groupings on the phylogeny was provided by the approximate likelihood ratio test available in PHYML. As an additional check on phylogenetic accuracy, the phylogenetic analysis was repeated using the maximum likelihood method available in the GARLI program [28], again using the HKY85 substitution model. No relevant topological differences were observed. All relevant parameter values for the analyses described above are available from the authors on request.
RESULTS
Emergence of Oseltamivir Resistance
The viral samples collected on 12 June (day 1) and 25 June (day 14) were cultured in pRhMK cells. The isolates were tested for phenotypic drug susceptibility against the NA inhibitors (oseltamivir and zanamivir). Dideoxysequencing (Sanger) was performed on the primary specimens and the isolates. When the 12 June sample was tested, both the primary specimen and its corresponding isolate (A/New York/4095/2009(H1N1); abbreviated here as NY/4095) produced a wild-type (ie, oseltamivir susceptible) sequence for the NA gene at residue 275. In addition, in the neuraminidase assay the NY/4095 isolate exhibited 50% inhibitory concentration (IC50) values for both oseltamivir (1.1 nmol/L) and zanamivir (1.6 nmol/L) that indicate susceptibility to both drugs. However, when the second sample (25 June) was sequenced (A/New York/4438/2009(H1N1); abbreviated as NY/4438), 2 peaks were observed in the dideoxysequencing chromatogram at nucleotide position 823 (not shown), strongly suggesting the presence of a mixed population with a major component of wild-type virus and a minor component of H275Y mutant virus strains. Of interest, when the cultured isolate from this sample was sequenced, only the 275Y mutation was observed on the chromatogram, indicating that the oseltamivir-resistant variant had outgrown the susceptible variant. Furthermore, this isolate produced an oseltamivir IC50 value of 297.6 nmol/L in the neuraminidase assay, indicating a high level of resistance (the expected IC50 in a H275Y mutant H1N1/2009 virus is usually in the range 50–80 nmol/L). However, the IC50 value for zanamivir for this second isolate was within the normal susceptibility range (1.5 nmol/L; Table 1).
Sanger consensus sequence assemblies of both samples also revealed a unique set of residues, compared with the pandemic H1N1/2009 virus strains available in public databases: specifically, a D199N mutation in the NA, a N55D mutation in the HA, and a R626K mutation in the polymerase. Of note, the NA 199N and HA 55D mutations have not been observed in any other virus strains sampled globally, whereas the polymerase R626K mutation has, to date, only been observed in 1 other sequence (A/New York/3348/2009(H1N1); GenBank accession CY041835). NA mutation 199N was observed in both the primary specimens and the isolates. This position is of considerable interest, because it has previously been associated with the increase in oseltamivir resistance in both seasonal and H5N1 virus strains [29]. That these unique changes were seen in samples detected 14 days apart suggests that they do not represent transient deleterious mutations.
Sequence Analysis and Viral population Diversity
The results of the deep sequencing analysis are depicted in Table 2 (as nonsynonymous mutations) and highlight the proportion of sequence reads coding for certain residues for which substantial differences were observed across both samples. These deep sequencing data also reveal minor variant residues present in >10% of the sequences. To determine how these mutations are linked within each segment, we used the deep sequencing data to reconstruct portions of the variant genes. The variant codons and residues obtained from this reconstruction are shown in Table 3 and Supplemental Table 1. From this analysis, NY/4438 appears to possess 3 distinct variants. The reconstructed variants were then used in phylogenetic analyses to determine their relatedness to each other and to other pandemic H1N1/2009 virus strains.
Table 2.
List of residues from the consensus amino acid sequence (Sanger) and the deep sequencing data of the two patient specimens
| New York/4095 |
New York/4438 |
||||||||
| Ref. | Sanger | 454 | # | Sanger | 454 | # | |||
| HA | 6 | V | V | V (99%) | 68 | V | V (82%) | L (17%) | 84 |
| 55 | N | D | D (99%) | 138 | D | D (78%) | N (18%) | 142 | |
| 125 | V | V | V (99%) | 209 | V | V (79%) | M (20%) | 130 | |
| 220 | T | T | T (99%) | 533 | T | T (82%) | S (17%) | 236 | |
| NA | 106 | I | I | I (100%) | 13 | I | I (77%) | V (21%) | 130 |
| 199 | D | N | N (100%) | 24 | N | N (75%) | D (22%) | 236 | |
| 248 | D | D | D (100%) | 16 | D | D (78%) | N (21%) | 398 | |
| 275 | H | H | H (100%) | 16 | H | H (80%) | Y (19%) | 744 | |
| NP | 100 | I | I | I (98%) | 132 | I | I (71%) | V (29%) | 142 |
| NS1 | 123 | I | V | V (99%) | 555 | V | V (75%) | I (24%) | 724 |
NOTE. The residue numbering is from the first methionine of the full-length proteins. The residue in bold confers oseltamivir resistance. #, Number of sequence reads that cover that position. Ref, Dominant residue found in these positions for the global set of H1N1/2009 virus strains.
Table 3.
NA haplotype reconstruction from the sequencing data of sample A/New York/4438
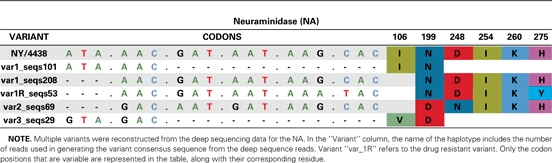 |
The sequence variants from the NY/4095 and NY/4438 specimens were compared with those pandemic H1N1/2009 virus strains circulating globally until the end of July 2009 (Figure 1, Supplementary Figure 1). The closest relatives of variant 1 were clearly the 2 Sanger consensus sequences—NY/4438 and NY/4095 (Figure 1b). At a broader phylogenetic scale, this group of virus strains clusters within the previously identified clade 7 of H1N1/2009, which dominates infections worldwide [30]. In contrast, both variants 2 and 3 are in clade 2, which was only detected during April and May 2009 [30]. Although clades 2 and 7 cocirculated in New York during the early part of the pandemic, by mid-June, when the 2 samples studied here were collected, virus strains belonging to clade 2 were no longer reported. All 3 variants are found in the later sample (NY/4438), 1 of which (variant 1) also contains a subpopulation of virus strains that possesses the H275Y NA mutation conferring oseltamivir resistance (denoted variant 1R; Figure 1b). That variants 1, 2, and 3 are so phylogenetically distinct, being more closely related to virus strains found in samples from other patients, is strongly suggestive of a diverse viral population generated by mixed infection. However, because both variants 2 and 3 are in clade 2 and differ by few mutations (Table 3), it is unclear whether they were also generated by mixed infection or represent de novo variants in this patient.
Figure 1.

A, Maximum likelihood tree of 1137 NA sequences of pandemic H1N1/2009 virus strains collected during April–July 2009 and the partially reconstructed variants in this patient. The variant 1 consensus sequences assembled from the intrahost sequences—both wild-type (WT) oseltamivir susceptible and oseltamivir resistant (H275Y; variant 1R)—are marked by red boxes, and the assemblies for variants 2 and 3 are denoted by blue boxes. B, Magnification of the section of the maximum likelihood tree containing the variant 1 assemblies. Of note, both Sanger consensus sequences (A/New York/4095/2009 and A/New York/4438/2009) cluster closely with the variant 1 assemblies. Approximate likelihood ratio test results are shown the relevant nodes, and all branch lengths are drawn to a scale of nucleotide substitutions per site.
DISCUSSION
The emergence of oseltamivir resistance in pandemic H1N1/2009 remains rare in the immunocompetent population [1, 11–13], and only ∼1% of NA sequences available in GenBank possess the H275Y mutation. In the case of immunosuppressed patients who experience prolonged viral shedding, oseltamivir treatment has apparently led to the selection of the 275Y drug resistance mutation on multiple occasions [1, 12, 16]. Here, we showed that a patient was infected with 3 variants of H1N1/2009 and that the major variant also contained a subpopulation of oseltamivir-resistant virus strains (variant 1R). This subpopulation was not strongly evident on conventional Sanger consensus sequencing, and pyrosequencing assays have been shown to be unreliable for the detection of drug-resistant mutant populations comprising <10% of the virus population [31, 32]. Therefore, we suggest that the resistance-determining H275Y mutation may be more frequent than is usually supposed but is present as a minority population in infected hosts. Indeed, in this case, H275Y was only detected by careful scrutiny of the Sanger sequencing chromatogram, confirmation by deep sequencing, and the fortuitous overgrowth in culture by the mutant.
Because the H275Y-bearing virus strains identified here were most closely related to oseltamivir-susceptible strains in the global population and because H275Y is well known to have a detrimental effect on viral fitness in the absence of drug [33], it seems likely that the H275Y mutation appeared de novo in this patient and, thus, is directly linked to the use of oseltamivir. When the primary samples were inoculated into culture, the resistant mutant outgrew the wild-type (and formerly dominant) variant. Similar observations have been made previously during Madin-Darby canine kidney cell culture [12]. Although the H275Y mutation in seasonal H1N1 influenza A spread rapidly in vivo in the absence of drug pressure, transmission of the equivalent mutant in pandemic H1N1/2009 virus strains has fortunately remained limited thus far [13].
In addition to harboring a subpopulation of oseltamivir-resistant virus strains, the patient studied here was noteworthy for the occurrence of a mixed infection: 3 variants were detected that were in 2 phylogenetically distinct clades of the pandemic H1N1/2009 virus (although it is uncertain whether variants 2 and 3 evolved de novo from one another). The occurrence of mixed infection in H1N1/2009 is clearly the precursor to intraserotype reassortment, which is commonplace in seasonal influenza A virus [34], although distinguishing bona fide reassortment from mixed infection remains an analytical challenge. In this context, it is particularly intriguing that both variants 2 and 3 belong to clade 2, which had not been previously found in samples beyond May 2009. Therefore, clade 2 either continued to circulate undetected in New York through late June or this patient was infected earlier and remained asymptomatic. Indeed, there are examples in the literature of immunosuppressed individuals with prolonged virus shedding [4, 6, 35]. In addition, it is unclear whether variants 2 and 3 were present in the initial infection along with variant 1 or occurred sequentially. Normally, individuals would be protected from a superinfection with antigenically related virus strains, as appears to be the case for pandemic H1N1/2009 virus strains [36]. However, in the case of immunosuppressed patients, it is possible that an inadequately protective immune response may be generated against the first infecting virus strain.
Finally, of note, the dominant virus variant in this patient was characterized by a number of unique mutations, including NA D199N, which has not previously been identified in pandemic H1N1/2009 influenza virus strains. Of interest, a D199E mutation in seasonal H1N1 has been associated with reduced susceptibility to oseltamivir [29], a D198G (universal numbering; equivalent to site 199) mutation in H5N1 is associated with reduced susceptibility to oseltamivir and zanamivir [37], and a D198N mutation in influenza B virus is associated with high oseltamivir resistance [5]. In light of newly characterized permissive secondary mutations that enable oseltamivir resistance in seasonal H1N1 virus strains to spread among untreated patients worldwide [36], we are currently assessing the functional significance of the D199N mutation.
Funding
This work was supported by National Institutes of Health (HHSN272200900007 to E.G., E.C.H., R.A.H., T.B.S., D.J.S., and R01 GM080533-03 to E.C.H.).
Supplementary Material
References
- 1.Centers for Disease Control and Prevention (CDC). Oseltamivir-resistant novel influenza A (H1N1) virus infection in two immunosuppressed patients - Seattle, Washington, 2009. MMWR Morb Mortal Wkly Rep. 2009;58:893–6. [PubMed] [Google Scholar]
- 2.Englund JA, Champlin RE, Wyde PR, et al. Common emergence of amantadine- rimantadine-resistant influenza A viruses in symptomatic immunocompromised adults. Clin Infect Dis. 1998;26:1418–24. doi: 10.1086/516358. [DOI] [PubMed] [Google Scholar]
- 3.Gooskens J, Jonges M, Claas EC, Meijer A, Kroes AC. Prolonged influenza virus infection during lymphocytopenia frequent detection of drug-resistant viruses. J Infect Dis. 2009;199:1435–41. doi: 10.1086/598684. [DOI] [PubMed] [Google Scholar]
- 4.Weinstock DM, Gubareva LV, Zuccotti G. Prolonged shedding of multidrug-resistant influenza A virus in an immunocompromised patient. N Engl J Med. 2003;348:867–8. doi: 10.1056/NEJM200302273480923. [DOI] [PubMed] [Google Scholar]
- 5.Ison MG, Gubareva LV, Atmar RL, Treanor J, Hayden FG. Recovery of drug-resistant influenza virus from immunocompromised patients: A case series. J Infect Dis. 2006;193:760–4. doi: 10.1086/500465. [DOI] [PubMed] [Google Scholar]
- 6.Klimov AI, Rocha E, Hayden FG, Shult PA, Roumillat LF, Cox NJ. Prolonged shedding of amantadine-resistant influenzae A viruses by immunodeficient patients: Detection by polymerase chain reaction-restriction analysis. J Infect Dis. 1995;172:1352–5. doi: 10.1093/infdis/172.5.1352. [DOI] [PubMed] [Google Scholar]
- 7.Ghedin E, Fitch A, Boyne A, et al. Mixed infection the genesis of influenza virus diversity. J Virol. 2009;83:8832–41. doi: 10.1128/JVI.00773-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraser C, Donnelly CA, Cauchemez S, et al. Pandemic potential of a strain of influenza A (H1N1): Early findings. Science. 2009;324:1557–61. doi: 10.1126/science.1176062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention (CDC). Update: Drug susceptibility of swine-origin influenza A (H1N1) viruses, April 2009. MMWR Morb Mortal Wkly Rep. 2009;58:433–5. [PubMed] [Google Scholar]
- 10.Centers for Disease Control and Prevention (CDC). Oseltamivir-resistant 2009 pandemic influenza A (H1N1) virus infection in two summer campers receiving prophylaxis–North Carolina, 2009. MMWR Morb Mortal Wkly Rep. 2009;58:969–72. [PubMed] [Google Scholar]
- 11.Baz M, Abed Y, Papenburg J, Bouhy X, Hamelin ME, Boivin G. Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. N Engl J Med. 2009;361:2296–7. doi: 10.1056/NEJMc0910060. [DOI] [PubMed] [Google Scholar]
- 12.Chen H, Cheung CL, Tai H, et al. Oseltamivir-resistant influenza A pandemic (H1N1) 2009 virus, Hong Kong, China. Emerg Infect Dis. 2009;15:1970–2. doi: 10.3201/eid1512.091057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le QM, Wertheim HF, Tran ND, van Doorn HR, Nguyen TH, Horby P. A community cluster of oseltamivir-resistant cases of 2009 H1N1 influenza. N Engl J Med. 2010;362:86–7. doi: 10.1056/NEJMc0910448. [DOI] [PubMed] [Google Scholar]
- 14.Moscona A. Oseltamivir resistance–disabling our influenza defenses. N Engl J Med. 2005;353:2633–6. doi: 10.1056/NEJMp058291. [DOI] [PubMed] [Google Scholar]
- 15.Chan PA, Connell NT, Gabonay AM, et al. Oseltamivir-resistant 2009-2010 pandemic influenza A (H1N1) in an immunocompromised patient. Clin Microbiol Infect. 2010 doi: 10.1111/j.1469-0691.2010.03212.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 16.Harvala H, Gunson R, Simmonds P, et al. The emergence of oseltamivir-resistant pandemic influenza A(H1N1) 2009 virus amongst hospitalised immunocompromised patients in Scotland, November-December, 2009. Euro Surveill. 2010;15 [PubMed] [Google Scholar]
- 17.Memoli MJ, Hrabal RJ, Hassantoufighi A, Eichelberger MC, Taubenberger JK. Rapid selection of oseltamivir- peramivir-resistant pandemic H1N1 virus during therapy in 2 immunocompromised hosts. Clin Infect Dis. 2010;50:1252–5. doi: 10.1086/651605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou B, Donnelly ME, Scholes DT, et al. Single-reaction genomic amplification accelerates sequencing vaccine production for classical and Swine origin human influenza a viruses. J Virol. 2009;83:10309–13. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Djikeng A, Halpin R, Kuzmickas R, et al. Viral genome sequencing by random priming methods. MC Genomics. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghedin E, Sengamalay NA, Shumway M, et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005;437:1162–6. doi: 10.1038/nature04239. [DOI] [PubMed] [Google Scholar]
- 22.Kurtz S, Phillippy A, Delcher AL, et al. Versatile open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Godzik A. Cd-hit: A fast program for clustering comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 24.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 26.Quinlan AR, Stewart DA, Stromberg MP, Marth GT. Pyrobayes: An improved base caller for SNP discovery in pyrosequences. Nat Methods. 2008;5:179–81. doi: 10.1038/nmeth.1172. [DOI] [PubMed] [Google Scholar]
- 27.Guindon S, Gascuel O. A simple, fast, accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 28.Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. dissertation. 2006. The University of Texas at Austin. [Google Scholar]
- 29.Deyde VM, Sheu TG, Trujillo AA, et al. Detection of molecular markers of drug resistance in the 2009 pandemic influenza A (H1N1) viruses using pyrosequencing. Antimicrob Agents Chemother. 2010;54:1102–10. doi: 10.1128/AAC.01417-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson M, Spiro D, Wentworth D, et al. The early diversification of influenza A/H1N1pdm. PLoS Curr. 2009;1:RRN1126. doi: 10.1371/currents.RRN1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deyde VM, Okomo-Adhiambo M, Sheu TG, et al. Pyrosequencing as a tool to detect molecular markers of resistance to neuraminidase inhibitors in seasonal influenza A viruses. Antiviral Res. 2009;81:16–24. doi: 10.1016/j.antiviral.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Deyde VM, Gubareva LV. Influenza genome analysis using pyrosequencing method: Current applications for a moving target. Expert Rev Mol Diagn. 2009;9:493–509. doi: 10.1586/erm.09.21. [DOI] [PubMed] [Google Scholar]
- 33.Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328:1272–5. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes EC, Ghedin E, Miller N, et al. Whole-genome analysis of human influenza A virus reveals multiple persistent lineages reassortment among recent H3N2 viruses. PLoS Biol. 2005;3:e300. doi: 10.1371/journal.pbio.0030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boivin G, Goyette N, Bernatchez H. Prolonged excretion of amantadine-resistant influenza a virus quasi species after cessation of antiviral therapy in an immunocompromised patient. Clin Infect Dis. 2002;34:E23–5. doi: 10.1086/338870. [DOI] [PubMed] [Google Scholar]
- 36.Garten RJ, Davis CT, Russell CA, et al. Antigenic genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hurt AC, Holien JK, Barr IG. In vitro generation of neuraminidase inhibitor resistance in A(H5N1) influenza viruses. Antimicrob Agents Chemother. 2009;53:4433–40. doi: 10.1128/AAC.00334-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.