

Abstract
Hepatocyte injury is ubiquitous in clinical practice, and the mode of cell death associated with this injury is often apoptosis, especially by death receptors. Information from experimental systems demonstrates that hepatocyte apoptosis is sufficient to cause liver hepatic fibrogenesis. The mechanisms linking hepatocyte apoptosis to hepatic fibrosis remain incompletely understood, but likely relate to engulfment of apoptotic bodies by professional phagocytic cells and stellate cells, and release of mediators by cells undergoing apoptosis. Inhibition of apoptosis with caspase inhibitors has demonstrated beneficial effects in murine models of hepatic fibrosis. Recent studies implicating Toll-like receptor 9 (TLR9) in liver injury and fibrosis are also of particular interest. Engulfment of apoptotic bodies is one mechanism by which the TLR9 ligand (CpG DNA motifs) could be delivered to this intracellular receptor. These concepts suggest therapy focused on interrupting the cellular mechanisms linking apoptosis to fibrosis would be useful in human liver diseases.
Keywords: Bcl-2 proteins, caspase inhibitors, death receptors, stellate cells, Toll-like receptor 9
Liver injury is quite common in human disease. Indeed, biomarkers of hepatocyte injury such as alanine transaminase (ALT) values are universally present in human sera. What constitutes the normal range for the serum ALT, and therefore, a ‘healthy liver’ is an unresolved topic 1. However, the mere presence of circulating ALT in man implies low-levels of hepatocyte injury. It is now widely appreciated that liver injury is accompanied by liver cell death, often by apoptosis. Indeed, hepatocyte apoptosis is ubiquitous in human liver diseases (Table 1)2. Given the remarkable regenerative capacity of the liver, loosing a few liver cells would appear to be inconsequential to health of the organ and organism. Unfortunately, prolonged hepatocellular injury results in an exuberant wound healing response, causing hepatic fibrosis, and, in its extreme form, liver cirrhosis. It is hepatic cirrhosis with its sequela of portal hypertension, end-stage liver disease and hepatocellular carcinoma that eventually compromises human life in most chronic liver diseases. One of the pressing unmet needs in clinical medicine is how to prevent, retard, or even reverse hepatic fibrosis. Therefore, important biomedical questions emanating from these observations are how does liver injury promote hepatic fibrosis, and is cell death by apoptosis a pro-fibrogenic process. This contribution to Seminars in Liver Diseases will address this issue. We will review current information linking apoptosis to fibrosis and highlight evolving mechanisms which merit further detailed study. This review will be an update since our past review on this subject 3. Current information continues to imply a direct link between hepatocyte apoptosis and liver fibrosis, thereby suggesting that anti-apoptotic therapies should also be anti-fibrogenic.
Table I. Liver Cell Apoptosis in Human Liver Diseases.
| Liver Disease | References |
|---|---|
| Chronic Viral Hepatitis | 77-82 |
| Acute Viral Hepatitis | 83 |
| Acute/Fulminant Hepatic Failure | 84, 85 |
| Alcohol-related Liver Disease | 25,86,87 |
| Non-alcoholic Steatohepatitis | 27,87 |
| Autoimmune Hepatitis | 88 |
| Primary Biliary Cirrhosis | 89 |
| Drug-induced Liver Injury | |
| HCV-related Fibrosis | 81 |
| Wilson's Disease | 90 |
| Acute Allograft Rejection | 91 |
| Hepatocarcinoma | 92, 93-97 |
Overview of Apoptosis
Apoptosis is a form of cell death characterized by membrane blebbing, shrinkage of the cell, chromatin condensation, and nuclear fragmentation 4. Fragmentation of the cell into membrane defined bodies termed apoptotic bodies is a hallmark of apoptosis; in the liver, these apoptotic bodies were termed councilman bodies for decades until their true pathogenesis could be easily verified 5. Apoptosis is mediated biochemically by activation of intracellular zymogens termed caspases, cysteine-dependent aspartate specific protease 6. These zymogens are themselves activated by proteolytic cleavage at aspartate moieties, either within special protein complexes (initiator caspases) or by other active caspases (down stream or effector caspases). The initiator caspase 8 is activated by induced proximity and dimerization of the proform within a death receptor complex 7, whereas procaspase 9 is activated by recruitment to and assembly within a large protein complex termed the apoptosome 6. The activating cleavage of effector caspases 3 and 7 by initiator caspases (i.e., caspase 8 or 9) generates a neoepitope which can be identified by immunohistochemical techniques in liver tissue specimens 8. Likewise caspase cleavage of cytokeratin 18 also generates a neoepitope which is released into the serum and can be measured by enzyme-linked immunosorbent assay (ELISA, M30 assay) as an index of hepatocyte apoptosis 9. Effector caspases are responsible for activating CAD (caspase-activated DNase) by cleaving ICAD (inhibitor of caspase-activated DNase)10. CAD activation results in DNA cleavage at internucleosomal linker regions causing the classic ladder pattern of DNA fragments (multiples of the 180 base pair nucleosomal regions). DNA cleavage during apoptosis is the basis for the TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay, a method for detecting DNA fragmentation by labeling the terminal end of nucleic acids. Thus, apoptosis can be readily identified by its biochemical biomarkers, and this information can be used to explore the relationship between hepatocyte apoptosis and liver fibrosis.
Mechanisms of Hepatocyte Apoptosis
Apoptosis is a process required to maintain tissue homeostasis and health by counterbalancing cell proliferation and eliminating damaged and/or aged cells. This is particularly crucial in an organ like the liver which is naturally exposed to toxins and viruses 11. Any alteration in the balance between proliferation and cell death, by either excessive or insufficient apoptosis, invariably leads to a pathologic condition. In the liver, massive hepatocyte apoptosis results in acute liver failure, whereas persistent hepatocyte apoptosis is often associated with fibrogenesis, chronic liver dysfunction, and even cancer development 12.
Apoptosis can be triggered by a variety of intra- and extra-cellular stimuli. The intracellular stimuli, such as DNA damage, generally result in mitochondrial outer membrane permeabilization (MOMP) and release of pro-apoptotic factors, including cytochrome c, second mitochondria-derived activator of caspases (SMAC)/direct IAP-binding protein with low PI (DIABLO), and apoptosis-inducing factor (AIF), from the intermembrane space into the cytosol. This signaling cascade, known as the mitochondrial (or intrinsic) pathway of apoptosis, is initiated by the activation of pro-apoptotic BH3-only (i.e., Bid, Bim, Bad, PUMA, Noxa) and multi-domain (Bax and Bak) members of the Bcl-2 family, which are responsible for the induction of MOMP 13, and antagonized by the anti-apoptotic members of the same family (Bcl-2, Bcl-xL, Mcl-1) 14. Released cytochrome c associates with apoptotic protease activating factor 1 (Apaf-1) to form the apoptosome, a large multimeric complex which recruits procaspase 9 and facilitates its autoactivation 6. Caspase 9 then cleaves and activates caspase 3 and 7, which, in turn, proceed to degrade several cellular substrates, resulting in the morphological changes associated to apoptosis. At the same time, endogenous cellular inhibitors of apoptosis proteins (IAPs), normally inhibiting accidentally activated caspases, are neutralized by SMAC/DIABLO, which is released from the mitochondria together with cytochrome c 15.
The extracellular stimuli signal through the engagement of death receptors on the plasma membrane by their cognate ligands, and the formation of a large death-inducing signaling complex (DISC)16. This pathway is referred to as the extrinsic pathway of apoptosis. Four of these death receptors, Fas, tumor necrosis factor receptor 1 (TNF-R1) and death receptor 4 and 5 (DR4 and DR5, also know as TNF-related apoptosis-inducing ligand receptor 1 and 2, TRAIL-R1 and TRAIL-R2), as well as their ligands, Fas ligand (FasL), TNF-α and TRAIL, are abundantly expressed in the liver 17, and their signaling cascades have been extensively studied over the years. Despite some differences in adaptors and other proteins recruited to their respective DISC, one common event occurring after the stimulation of all death receptors is the recruitment of the adaptor Fas-associated protein with death domain (FADD) and procaspase 8, which results in its autoactivation 7. Subsequently, caspase 8 can either directly cleave and activate caspase 3 and 7 (type I cells, such as lymphocytes) similarly to caspase 9, or can engage the mitochondrial pathway by cleaving the BH3-only protein Bid (type II cells, such as hepatocytes), whose truncated fragment translocates to the mitochondrial outer membrane causing MOMP 18,19. Therefore, the intrinsic and extrinsic pathways are not mutually exclusive, with Bid mediating the crosstalk between the two pathways in type II cells.
Because of the ubiquitous expression of death receptors and ligands in liver cells, apoptosis in the liver is generally mediated by the extrinsic pathway. In particular, activation of Fas and TNF-R1 is associated with hepatocyte apoptosis in a wide variety of liver diseases, including viral hepatitis, fulminant hepatic failure, cholestatic liver disease, alcoholic hepatitis, non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH), Wilsons' disease and ischemia-reperfusion injury 20. For example, during viral infection, the liver damage is only marginally caused by a cytopathic effect of the virus itself, but rather due to the infiltrating FasL-expressing cytotoxic T lymphocytes (CTL) which eliminate the infected hepatocytes by engaging Fas on the hepatocyte surface. CTL also induce hepatocyte apoptosis via the TNF-TNF-R1 system, and secretion of the cytotoxins perforin and granzyme 21. In cholestasis, elevated intracellular concentrations of toxic bile salts result in increased Fas density on the plasma membrane and ligand-independent activation of the receptor 22,23, as demonstrated by the absence of liver injury in Fas-deficient mice, but not FasL-deficient mice, after bile duct ligation (a model of extrahepatic cholestasis). Toxic bile salts are also known to up-regulate DR5 expression, therefore increasing sensitivity to TRAIL-mediated apoptosis 24. Elevated Fas and FasL are also features of alcoholic liver injury 25. Moreover, alcohol promotes Kuppfer cells activation and TNF-α production, and increases the sensitivity of hepatocytes to TNF-α-mediated apoptosis 26. Fas-mediated hepatocyte apoptosis is also increased in patients with NASH, and correlates with the progression of the disease from simple steatosis to steatohepatitis 27. However, liver injury during NASH is not only due to activated death receptors. Free fatty acids (FFAs) accumulating in the liver as a consequence of insulin resistance also induce cell death, a process referred to as lipoapoptosis. In particular, excessive FFAs accumulate in the endoplasmic reticulum (ER), resulting in ER stress. Saturated FFAs induce Bim expression and phosphorylation 28, JNK-mediated PUMA expression and Bax activation 29,30, resulting in MOMP as well as lysosomal permeabilization 31. Moreover, concentrations of FFAs too low to induce lipoapoptosis have been shown to up-regulate DR5 expression and increase sensitivity of hepatocytes to TRAIL-mediated apoptosis 32.
Apoptosis and Liver Fibrosis
Several human studies have linked apoptosis to fibrosis. For example, the magnitude of apoptosis correlates with stage of fibrotic disease in non-alcoholic steatohepatitis (NASH)27,33, and in fibrosis progression in recurrent hepatitis C (HCV) following liver transplantation33. Although these human studies are correlative, experimental models more mechanistically link hepatocyte apoptosis to fibrosis. Mice deficient in Fas display reduced hepatic fibrosis following bile duct ligation 34. Reduction of hepatocyte apoptosis with a caspase inhibitor also repressed hepatic fibrosis in this rodent model of cholestatic liver injury 8. Although these observations could be explained by the potential pro-inflammatory effects of Fas-mediated liver injury, which require caspases 35,36, other data suggest hepatocyte apoptosis alone is sufficient to elicit a pro-fibrogenic response in the liver. The anti-apoptotic members of the Bcl-2 family of proteins are the guardians of the mitochondrial pathway of cell death 37. The hepatocyte depends upon two such proteins for survival, Bcl-xL and Mcl-1 38. Hepatocyte-specific deletion of either protein results in hepatocyte apoptosis, elevation of the serum ALT values and hepatic fibrosis 39,40. These data are quite remarkable because they appear to be pure models of hepatocyte apoptosis yet they recapitulate many facets of human liver disease. Moreover, these findings also suggest the hepatocyte is constitutively undergoing pro-apoptotic stress, even in the absence of disease, an observation consistent with the presence of ALT in the serum of even healthy human subjects. Thus, the question is not if apoptosis contribute to progressive liver injury, but how? There are two broad mechanisms by which apoptotic hepatocytes beget hepatocyte fibrosis: 1) engulfment of apoptotic bodies is pro-fibrogenic; 2) apoptotic cells release pro-fibrogenic mediators. These concepts are not mutually exclusive and likely both are operational in human liver disease.
Apoptotic Bodies and Their Engulfment
Apoptotic bodies are eliminated from the body by cellular engulfment 41. If not eliminated, the cellular membrane defining the apoptotic body becomes permeant, releasing cellular constituents into the extracellular space eliciting an inflammatory response, a phenomenon termed secondary necrosis 41. In massive liver injury, the ability of phagocytic cells to identify and clean up all the apoptotic bodies is likely overwhelmed, and this process could explain liver failure and inflammation despite an initial apoptotic stimulus, such as liver injury by Fas stimulation 42. In order to undergo engulfment, the apoptotic cell must generate two cells signals, a “find me” signal and an “eat me” signal. One recognized “eat me” signal is the translocation of phosphatidylserine from the inner (cytoplasmic) leaflet of the plasma membrane to the outer (cell surface) leaflet, which results in engulfment of the apoptotic bodies by phagocytic cells 10. In the liver, both hepatic stellate cells (HSC), the pericytes of the sinusoids, and Kupffer cells, the resident macrophages of the liver, can phagocytose apoptotic bodies (Fig. 1) 43-45.
Figure 1. Cellular mechanisms linking hepatocyte apoptosis to liver fibrogenesis.
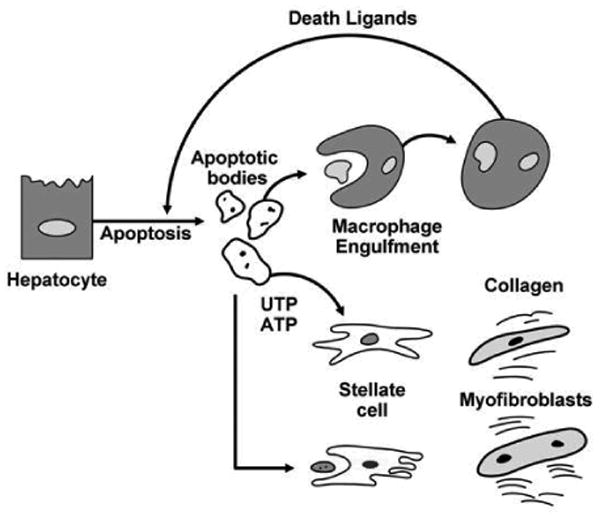
Engulfment of apoptotic bodies by Kupffer cells enhances their expression of death ligands which in a feed-forward loop further promote hepatocyte apoptosis and generation of apoptotic bodies. Stellate cell or myofibroblast engulfment of apoptotic bodies enhances their expression of collagen type 1 and TGFβ1, promoting development of fibrosis and cirrhosis. Apoptotic cells also release the nucleotides UTP and ATP, which bind P2Y2 purinergic receptors on myofibroblasts further promoting collagen generation.
Apoptotic Bodies and The “Find Me” Signal
Recent information suggests that apoptotic cells release the nucleotides ATP and UTP into the extracellular space 46. These chemical mediators have been termed the scent of death 47. ATP and UTP bind to purinergic receptors on macrophages and HSC, especially the P2Y2 receptor 46. The binding to macrophages serve as a chemoattractant recruiting these professional phagocytic cells to the site of the dying cell. As P2Y2 receptors are also present on HSC 48, release of nucleotides by apoptotic hepatocytes is likely an additional mechanism by which apoptosis promotes hepatic fibrosis. Indeed, ATP and UTP released from apoptotic cells may function to recruit HSC to sites of liver injury and promote their activation to myofibroblasts (Fig. 1). Other chemoattractants released from apoptotic cells include fractalkine, adenosine and the lipid lysophosphatidylcholine41,49. Both compounds also serve as ligands for G-protein coupled receptors and also likely contribute to HSC activation in liver injury. Although a role for lysophosphatidyl choline has not been explored in liver fibrosis, fractalkine has been linked to hepatic inflammation and fibrosis and merits further attention as a chemokine promoting liver fibrosis by apoptotic cells 50,51.
Apoptotic Bodies and Liver Indigestion
Apoptosis in development is a non-inflammatory process responsible for removing excess cells in a spatial-temporal sequence. In contrast, apoptosis in pathologic conditions is not controlled and can be deleterious to the organ and the entire organism 52. Apoptotic bodies and their engulfment are a major source of this hepatic “indigestion”. For example, hepatocyte inflammation is a major driving force for hepatic fibrogenesis 53. Apoptosis contributes to inflammation by promoting Kupffer cell activation. Following engulfment of apoptotic bodies, Kupffer cells express the death ligands TNF-α, TRAIL and FasL 43,54 (Fig. 1) which may exert a pro-inflammatory effect in this context 55. All these death ligands have cognate receptors on hepatocytes and may induce death receptor-mediated apoptosis further aggravating liver injury. Thus, engulfment of apoptotic bodies by Kupffer cells is likely an important pro-fibrogenic mechanism in liver disease.
In the liver, as in other organs, activated myofibroblasts generate collagen and are responsible for organ scarring. Myofibroblasts are derived from portal fibroblasts and hepatic stellate cells 56,57. Whatever their lineage, activated myofibroblasts are phagocytic and capable of engulfing apoptotic bodies 44,45,58,59. When activated myofibroblast cell lines engulf apoptotic bodies derived from hepatocytes, they produce profibrogenic cytokines (such as TGF-β) and type I collagen 44. The engulfment of apoptotic bodies also results in up-regulation of NADPH oxidase, an enzyme which generates oxygen free radicals 59. These data help link apoptosis to oxidative stress, which is frequently implicated as a mechanism contributing to hepatic injury.
If engulfment of apoptotic bodies is pro-fibrogenic, what in the apoptotic bodies is responsible for this phenomenon? Apoptotic bodies contain micronuclei, a source of DNA. Unmethylated cytosine-phosphate guanosine (CpG)-DNA motifs are recognized by Toll-like receptor 9 (TLR 9) (Fig. 2). Indeed, denatured DNA from apoptotic cells is a ligand for TLR 9 60,61 and TLR 9-deficient mice display reduced hepatic injury and fibrosis following liver injury by carbon tetrachloride, bile duct ligation, acetaminophen, and a model of fatty liver disease 60-62. The engulfment of the apoptotic DNA would permit access of the CpG-DNA ligand to the intracellular TLR9 where it would, in turn, activate a variety of signaling cascades resulting in myofibroblast generation of collagen 1 and the pro-fibrogenic cytokine TGF-β 63 (Fig. 2). Although TLR9 is thought to be restricted to dendritic cells in humans, Watanabe et al. demonstrated that, in human stellate cells, CpG oligonucleotides induced up-regulation of TGF-β and collagen 1 mRNA, and that these effects were blocked by TLR9 antagonists 60. These very important data link apoptosis to activation of the innate immune response within myofibroblasts.
Figure 2. Activation of TLR9 by apoptotic body engulfment.
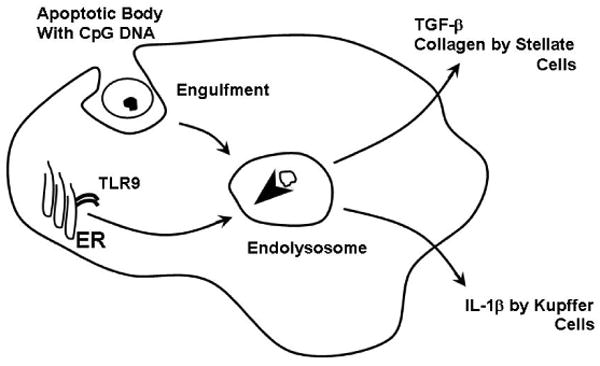
The apoptotic bodies contain micronuclei with CpG DNA motifs. Following engulfment, the apoptotic bodies are translocated into an endolysosome. This process triggers TLR9 trafficking from the endoplasmic reticulum (ER) to the endolysosome. The lower pH of the endolysosome promotes TLR9 activation. The active receptor complex in stellate cells results in transforming growth factor-beta (TGF-β) and collagen I production,60 whereas in Kupffer cells it promotes interleukin-1β, a proinflammatory cytokine, generation 98
Apoptosis and Anti-Fibrogenic Strategies for Human Liver Disease
The most direct therapeutic strategy for repressing liver fibrosis is to eliminate the cause for the liver injury. This has proven clinically possible for viral hepatitis where direct and indirect anti-viral therapies are quite effective. However, effective treatments do not exist for many liver diseases such as primary sclerosing cholangitis, NASH, alcohol-mediated hepatitis, and patients with HCV or HBV unresponsive to antiviral therapies. For such patients, anti-apoptotic and anti-fibrogenic strategies have a hepatoprotective role. Two therapeutic strategies emanating from this review include caspase inhibitors and TLR9 receptor antagonists (Fig. 3).
Figure 3. Therapeutic strategies to minimize liver disease progression.
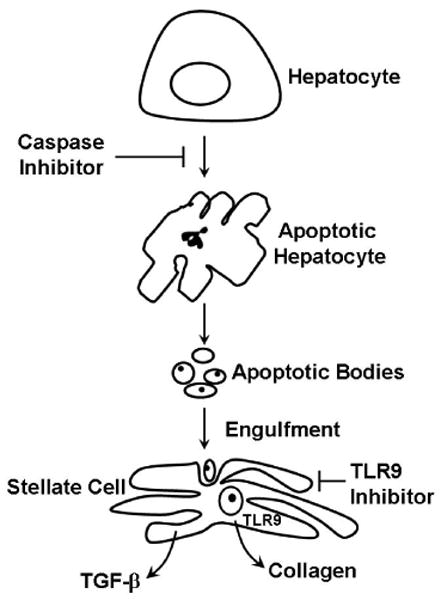
Apoptosis can be linked to hepatic fibrosis through engulfment of apoptotic bodies with subsequent activation of TLR9 in stellate cells. Based on these concepts, inhibition of death receptor-mediated apoptosis by caspase inhibitors should attenuate hepatic fibrosis. Likewise, TLR9 inhibitors should prevent stellate cell activation despite engulfment of apoptotic bodies thereby also reducing hepatic fibrogenesis.
Small molecule caspase inhibitors are attractive for the treatment of human liver diseases. Indeed, caspase inhibitors attenuate hepatic injury and fibrosis in pre-clinical models of cholestasis and non-alcoholic liver disease 8,64. More importantly, in a human trial a pan-caspase inhibitor reduced serum ALT values in patients with HCV without affecting viral replication 65,66. This agent also attenuates liver ischemia/reperfusion injury in rodents and man during organ transplantation 67,68. Although there is a concern that apoptosis inhibition by caspase inhibitors would promote carcinogenesis, we view this as unlikely. There are two major apoptotic pathways, an extrinsic or death receptor-mediated pathway and an intrinsic or mitochondrial pathway, as described above. Although the mitochondrial pathway is characterized by caspase 9 activation, caspase 9 deletion does not prevent cell death 69. Indeed, once a sufficient number of mitochondria have displayed MOMP, cell death is caspase-independent 70. Since most oncogenic processes elicit a DNA damage response which engages the mitochondrial pathway of cell death 71, cell death and elimination of the potentially oncogenic cell should ensue whether or not caspases are inhibited. In contrast, the extrinsic pathway is truly dependent on caspase 8 and, perhaps, caspase 10 72. For example, the genetic absence of caspase 8 (mice do not express caspase 10) attenuates liver injury in experimental murine models mediated by death ligands 72,73. Thus, caspase inhibition should only reliably inhibit death receptor-mediated apoptosis, a common mechanism of cell death in human liver diseases 17,74. As knockout murine models of various death receptors do not develop spontaneous cancers, caspase inhibitors should prove to be safe in man. However, more animal data in this regard would be reassuring.
There is considerable interest in developing short oligonucleotides rich in guanine residues as inhibitors of TLR9 antagonists as immunomodulatory agents in immune-mediated diseases such as systemic lupus erythematosis 75,76. Many of these compounds are salutary in murine models of this disease. They have yet to be applied broadly to animal models of fibrosis, and we await these studies with anticipation.
Acknowledgments
The secretarial help of Erin Nystuen-Bungum is gratefully acknowledged. We are indebted to prior post-doctoral fellows who helped pioneer the relationship between apoptosis and fibrosis in the liver, especially Dr. Ali Canbay who now has an independent laboratory program at University of Duisburg-Essen, Essen, Germany and Dr. Natalie Torok, who also has an independent laboratory program at University of California Davis Medical Center, Sacramento, CA. I also am appreciative of the expert aid of Steve Bronk in providing the graphic figures.
This work was supported by RO1 grants from the NIH, DK41876 and DK 63947, and the Mayo Foundation.
Abbreviations Used
- AIF
Apoptosis-inducing factor
- ALT
Alanine transaminase
- ATP
Adenine triphosphate
- Bcl-2
B-cell lymphoma-2
- Bcl-xL
B-cell lymphoma-2 like protein
- CAD
Caspase-activated DNase
- Caspase
Cysteine-dependent aspartate specific protease
- CpG
Cytosine-phosphate guanosine
- DNA
Deoxyribonucleic acid
- ELISA
Enzyme-linked immunosorbent assay
- HBV
Hepatitis B
- HCV
Hepatitis C
- ICAD
Inhibitor of caspase-activated DNase
- Mcl-1
Myeloid cell leukemia-1
- NASH
Non-alcoholic steatohepatitis
- P2Y2
Purinergic 2 Y2
- SMAC/DIABLO
Second mitochondria-derived activator of caspases/Direct IAP-binding protein with low PI (DIABLO)
- TLR9
Toll-like receptor 9
- TNF
Tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- UTP
Uridine triphosphate
References
- 1.Kim WR, Flamm SL, Di Bisceglie AM, Bodenheimer HC. Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology. 2008;47(4):1363–1370. doi: 10.1002/hep.22109. [DOI] [PubMed] [Google Scholar]
- 2.Rust C, Gores GJ. Apoptosis and liver disease. Am J Med. 2000;108(7):567–574. doi: 10.1016/s0002-9343(00)00370-3. [DOI] [PubMed] [Google Scholar]
- 3.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39(2):273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 4.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grassi A, Susca M, Ferri S, et al. Detection of the M30 neoepitope as a new tool to quantify liver apoptosis: timing and patterns of positivity on frozen and paraffin-embedded sections. Am J Clin Pathol. 2004;121(2):211–219. doi: 10.1309/UK62-1LFJ-4FX0-7KDE. [DOI] [PubMed] [Google Scholar]
- 6.Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284(33):21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oberst A, Pop C, Tremblay AG, et al. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010;285(22):16632–16642. doi: 10.1074/jbc.M109.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther. 2004;308(3):1191–1196. doi: 10.1124/jpet.103.060129. [DOI] [PubMed] [Google Scholar]
- 9.Wieckowska A, Zein NN, Yerian LM, et al. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44(1):27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 10.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 11.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90(3):1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134(6):1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 15.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 16.Guicciardi ME, Gores GJ. Life and death by death receptors. Faseb J. 2009;23(6):1625–1637. doi: 10.1096/fj.08-111005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faubion WA, Gores GJ. Death receptors in liver biology and pathobiology. Hepatology. 1999;29(1):1–4. doi: 10.1002/hep.510290101. [DOI] [PubMed] [Google Scholar]
- 18.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94(4):481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 20.Akazawa Y, Gores GJ. Death receptor-mediated liver injury. Seminars in liver disease. 2007;27(4):327–338. doi: 10.1055/s-2007-991510. [DOI] [PubMed] [Google Scholar]
- 21.Chisari FV. Cytotoxic T cells and viral hepatitis. J Clin Invest. 1997;99(7):1472–1477. doi: 10.1172/JCI119308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sodeman T, Bronk SF, Roberts PJ, Miyoshi H, Gores GJ. Bile salts mediate hepatocyte apoptosis by increasing cell surface trafficking of Fas. Am J Physiol Gastrointest Liver Physiol. 2000;278(6):G992–999. doi: 10.1152/ajpgi.2000.278.6.G992. [DOI] [PubMed] [Google Scholar]
- 23.Faubion WA, Guicciardi ME, Miyoshi H, et al. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest. 1999;103(1):137–145. doi: 10.1172/JCI4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higuchi H, Bronk SF, Taniai M, Canbay A, Gores GJ. Cholestasis increases tumor necrosis factor-related apoptotis-inducing ligand (TRAIL)-R2/DR5 expression and sensitizes the liver to TRAIL-mediated cytotoxicity. J Pharmacol Exp Ther. 2002;303(2):461–467. doi: 10.1124/jpet.102.040030. [DOI] [PubMed] [Google Scholar]
- 25.Natori S, Rust C, Stadheim LM, et al. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol. 2001;34(2):248–253. doi: 10.1016/s0168-8278(00)00089-1. [DOI] [PubMed] [Google Scholar]
- 26.Nanji AA, Jokelainen K, Fotouhinia M, et al. Increased severity of alcoholic liver injury in female rats: role of oxidative stress, endotoxin, and chemokines. Am J Physiol Gastrointest Liver Physiol. 2001;281(6):G1348–1356. doi: 10.1152/ajpgi.2001.281.6.G1348. [DOI] [PubMed] [Google Scholar]
- 27.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125(2):437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 28.Barreyro FJ, Kobayashi S, Bronk SF, et al. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem. 2007;282(37):27141–27154. doi: 10.1074/jbc.M704391200. [DOI] [PubMed] [Google Scholar]
- 29.Cazanave SC, Mott JL, Elmi NA, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284(39):26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281(17):12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 31.Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290(6):G1339–1346. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56(8):1124–1131. doi: 10.1136/gut.2006.118059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meriden Z, Forde KA, Pasha TL, et al. Histologic predictors of fibrosis progression in liver allografts in patients with hepatitis C virus infection. Clin Gastroenterol Hepatol. 8(3):289–296. 296, e281–288. doi: 10.1016/j.cgh.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canbay A, Higuchi H, Bronk SF, et al. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123(4):1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 35.Jaeschke H. Inflammation in response to hepatocellular apoptosis. Hepatology. 2002;35(4):964–966. doi: 10.1053/jhep.2002.0350964. [DOI] [PubMed] [Google Scholar]
- 36.Faouzi S, Burckhardt BE, Hanson JC, et al. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J Biol Chem. 2001;276(52):49077–49082. doi: 10.1074/jbc.M109791200. [DOI] [PubMed] [Google Scholar]
- 37.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 37(3):299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cazanave SC, Gores GJ. The liver's dance with death: two Bcl-2 guardian proteins from the abyss. Hepatology. 2009;50(4):1009–1013. doi: 10.1002/hep.23188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takehara T, Tatsumi T, Suzuki T, et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127(4):1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 40.Hikita H, Takehara T, Shimizu S, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology. 2009;50(4):1217–1226. doi: 10.1002/hep.23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140(5):619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 42.Nagata S. Apoptosis by death factor. Cell. 1997;88(3):355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 43.Canbay A, Feldstein AE, Higuchi H, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38(5):1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 44.Canbay A, Taimr P, Torok N, et al. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83(5):655–663. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 45.Jiang JX, Mikami K, Venugopal S, Li Y, Torok NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51(1):139–148. doi: 10.1016/j.jhep.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gregory C. Cell biology: Sent by the scent of death. Nature. 2009;461(7261):181–182. doi: 10.1038/461181a. [DOI] [PubMed] [Google Scholar]
- 48.Dranoff JA, Ogawa M, Kruglov EA, et al. Expression of P2Y nucleotide receptors and ectonucleotidases in quiescent and activated rat hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2004;287(2):G417–424. doi: 10.1152/ajpgi.00294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mehal W, Imaeda A. Cell death and fibrogenesis. Seminars in liver disease. 30(3):226–231. doi: 10.1055/s-0030-1255352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bourd-Boittin K, Basset L, Bonnier D, et al. CX3CL1/fractalkine shedding by human hepatic stellate cells: contribution to chronic inflammation in the liver. J Cell Mol Med. 2009;13(8A):1526–1535. doi: 10.1111/j.1582-4934.2009.00787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wasmuth HE, Zaldivar MM, Berres ML, et al. The fractalkine receptor CX3CR1 is involved in liver fibrosis due to chronic hepatitis C infection. J Hepatol. 2008;48(2):208–215. doi: 10.1016/j.jhep.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 52.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiener PA, Davis PM, Starling GC, et al. Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J Exp Med. 1997;185(8):1511–1516. doi: 10.1084/jem.185.8.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L) Science. 1998;282(5394):1714–1717. doi: 10.1126/science.282.5394.1714. [DOI] [PubMed] [Google Scholar]
- 56.Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology. 51(4):1438–1444. doi: 10.1002/hep.23405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang JX, Mikami K, Shah VH, Torok NJ. Leptin induces phagocytosis of apoptotic bodies by hepatic stellate cells via a Rho guanosine triphosphatase-dependent mechanism. Hepatology. 2008;48(5):1497–1505. doi: 10.1002/hep.22515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhan SS, Jiang JX, Wu J, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43(3):435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 60.Watanabe A, Hashmi A, Gomes DA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46(5):1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 61.Imaeda AB, Watanabe A, Sohail MA, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119(2):305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gabele E, Muhlbauer M, Dorn C, et al. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem Biophys Res Commun. 2008;376(2):271–276. doi: 10.1016/j.bbrc.2008.08.096. [DOI] [PubMed] [Google Scholar]
- 63.Torok NJ. Apoptotic cell death takes its toll. Hepatology. 2007;46(5):1323–1325. doi: 10.1002/hep.21968. [DOI] [PubMed] [Google Scholar]
- 64.Witek RP, Stone WC, Karaca FG, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50(5):1421–1430. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 65.Masuoka HC, Guicciardi ME, Gores GJ. Caspase inhibitors for the treatment of hepatitis C. Clin Liver Dis. 2009;13(3):467–475. doi: 10.1016/j.cld.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46(2):324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 67.Hoglen NC, Anselmo DM, Katori M, et al. A caspase inhibitor, IDN-6556, ameliorates early hepatic injury in an ex vivo rat model of warm and cold ischemia. Liver Transpl. 2007;13(3):361–366. doi: 10.1002/lt.21016. [DOI] [PubMed] [Google Scholar]
- 68.Baskin-Bey ES, Washburn K, Feng S, et al. Clinical Trial of the Pan-Caspase Inhibitor, IDN-6556, in Human Liver Preservation Injury. Am J Transplant. 2007;7(1):218–225. doi: 10.1111/j.1600-6143.2006.01595.x. [DOI] [PubMed] [Google Scholar]
- 69.Ekert PG, Read SH, Silke J, et al. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J Cell Biol. 2004;165(6):835–842. doi: 10.1083/jcb.200312031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tait SW, Parsons MJ, Llambi F, et al. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 18(5):802–813. doi: 10.1016/j.devcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1(1):19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 72.Kaufmann T, Jost PJ, Pellegrini M, et al. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity. 2009;30(1):56–66. doi: 10.1016/j.immuni.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zender L, Hutker S, Liedtke C, et al. Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc Natl Acad Sci U S A. 2003;100(13):7797–7802. doi: 10.1073/pnas.1330920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoon JH, Gores GJ. Death receptor-mediated apoptosis and the liver. J Hepatol. 2002;37(3):400–410. doi: 10.1016/s0168-8278(02)00209-x. [DOI] [PubMed] [Google Scholar]
- 75.Lenert PS. Classification, mechanisms of action, and therapeutic applications of inhibitory oligonucleotides for Toll-like receptors (TLR) 7 and 9. Mediators Inflamm. 2010:986596. doi: 10.1155/2010/986596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Santiago-Raber ML, Baudino L, Izui S. Emerging roles of TLR7 and TLR9 in murine SLE. J Autoimmun. 2009;33(3-4):231–238. doi: 10.1016/j.jaut.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 77.Fiore G, Piazzolla G, Galetta V, et al. Liver tissue expression of CD80 and CD95 antigens in chronic hepatitis C: relationship with biological and histological disease activities. Microbios. 1999;97(386):29–38. [PubMed] [Google Scholar]
- 78.Tagashira M, Yamamoto K, Fujio K, et al. Expression of perforin and Fas ligand mRNA in the liver of viral hepatitis. J Clin Immunol. 2000;20(5):347–353. doi: 10.1023/a:1006668013276. [DOI] [PubMed] [Google Scholar]
- 79.Ibuki N, Yamamoto K, Yabushita K, et al. In situ expression of Granzyme B and Fas-ligand in the liver of viral hepatitis. Liver. 2002;22(3):198–204. doi: 10.1046/j.0106-9543.2002.00tes.x. [DOI] [PubMed] [Google Scholar]
- 80.Ehrmann J, Jr, Galuszkova D, Ehrmann J, et al. Apoptosis-related proteins, BCL-2, BAX, FAS, FAS-L and PCNA in liver biopsies of patients with chronic hepatitis B virus infection. Pathol Oncol Res. 2000;6(2):130–135. doi: 10.1007/BF03032363. [DOI] [PubMed] [Google Scholar]
- 81.Pianko S, Patella S, Ostapowicz G, Desmond P, Sievert W. Fas-mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption, and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J Viral Hepat. 2001;8(6):406–413. doi: 10.1046/j.1365-2893.2001.00316.x. [DOI] [PubMed] [Google Scholar]
- 82.Tang TJ, Kwekkeboom J, Laman JD, et al. The role of intrahepatic immune effector cells in inflammatory liver injury and viral control during chronic hepatitis B infection. J Viral Hepat. 2003;10(3):159–167. doi: 10.1046/j.1365-2893.2003.00412.x. [DOI] [PubMed] [Google Scholar]
- 83.Rivero M, Crespo J, Fabrega E, et al. Apoptosis mediated by the Fas system in the fulminant hepatitis by hepatitis B virus. J Viral Hepat. 2002;9(2):107–113. doi: 10.1046/j.1365-2893.2002.00338.x. [DOI] [PubMed] [Google Scholar]
- 84.Tagami A, Ohnishi H, Hughes RD. Increased serum soluble Fas in patients with acute liver failure due to paracetamol overdose. Hepatogastroenterology. 2003;50(51):742–745. [PubMed] [Google Scholar]
- 85.Ryo K, Kamogawa Y, Ikeda I, et al. Significance of Fas antigen-mediated apoptosis in human fulminant hepatic failure. Am J Gastroenterol. 2000;95(8):2047–2055. doi: 10.1111/j.1572-0241.2000.02268.x. [DOI] [PubMed] [Google Scholar]
- 86.Tagami A, Ohnishi H, Moriwaki H, Phillips M, Hughes RD. Fas-mediated apoptosis in acute alcoholic hepatitis. Hepatogastroenterology. 2003;50(50):443–448. [PubMed] [Google Scholar]
- 87.Ribeiro PS, Cortez-Pinto H, Sola S, et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99(9):1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 88.Fox CK, Furtwaengler A, Nepomuceno RR, Martinez OM, Krams SM. Apoptotic pathways in primary biliary cirrhosis and autoimmune hepatitis. Liver. 2001;21(4):272–279. doi: 10.1034/j.1600-0676.2001.021004272.x. [DOI] [PubMed] [Google Scholar]
- 89.Tinmouth J, Lee M, Wanless IR, et al. Apoptosis of biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. Liver. 2002;22(3):228–234. doi: 10.1046/j.0106-9543.2002.01595.x. [DOI] [PubMed] [Google Scholar]
- 90.Strand S, Hofmann WJ, Grambihler A, et al. Hepatic failure and liver cell damage in acute Wilson's disease involve CD95 (APO-1/Fas) mediated apoptosis. Nat Med. 1998;4(5):588–593. doi: 10.1038/nm0598-588. [DOI] [PubMed] [Google Scholar]
- 91.Tannapfel A, Kohlhaw K, Ebelt J, et al. Apoptosis and the expression of Fas and Fas ligand (FasL) antigen in rejection and reinfection in liver allograft specimens. Transplantation. 1999;67(7):1079–1083. doi: 10.1097/00007890-199904150-00027. [DOI] [PubMed] [Google Scholar]
- 92.Strand S, Hofmann WJ, Hug H, et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells--a mechanism of immune evasion? Nat Med. 1996;2(12):1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]
- 93.Nagao M, Nakajima Y, Hisanaga M, et al. The alteration of Fas receptor and ligand system in hepatocellular carcinomas: how do hepatoma cells escape from the host immune surveillance in vivo? Hepatology. 1999;30(2):413–421. doi: 10.1002/hep.510300237. [DOI] [PubMed] [Google Scholar]
- 94.Ito Y, Monden M, Takeda T, et al. The status of Fas and Fas ligand expression can predict recurrence of hepatocellular carcinoma. Br J Cancer. 2000;82(6):1211–1217. doi: 10.1054/bjoc.1999.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ito Y, Takeda T, Umeshita K, et al. Fas antigen expression in hepatocellular carcinoma tissues. Oncol Rep. 1998;5(1):41–44. [PubMed] [Google Scholar]
- 96.Lee SH, Shin MS, Lee HS, et al. Expression of Fas and Fas-related molecules in human hepatocellular carcinoma. Hum Pathol. 2001;32(3):250–256. doi: 10.1053/hupa.2001.22769. [DOI] [PubMed] [Google Scholar]
- 97.Okano H, Shiraki K, Inoue H, et al. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest. 2003;83(7):1033–1043. doi: 10.1097/01.lab.0000079328.76631.28. [DOI] [PubMed] [Google Scholar]
- 98.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 139(1):323–334. e327. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]