

Abstract
There is considerable evidence that alterations in striatal medium-sized spiny neurons (MSSNs) giving rise to the direct (D1 receptor-expressing) and indirect (D2 receptor-expressing) pathways differentially contribute to the phenotype of Huntington's disease (HD). To determine how each subpopulation of MSSN is functionally affected, we examined spontaneous excitatory postsynaptic currents (sEPSCs) and dopamine (DA) modulation in two HD mouse models, the YAC128 and the BACHD (a bacterial-artificial chromosome). These mice also expressed enhanced green fluorescent protein (EGFP) under the control of the promoter for either DA D1 or D2 receptors to identify neurons. In early symptomatic YAC128 and BACHD mice, glutamate transmission was increased in both D1 and D2 MSSNs, but in different ways. D1 cells displayed increased sEPSC frequencies and decreased paired-pulse ratios (PPRs) while D2 cells displayed larger evoked glutamate currents but no change in sEPSC frequencies or PPRs. D1 receptor modulation of sEPSCs was absent in D1-YAC128 cells at the early symptomatic stage but was restored by treating the slices with tetrabenazine. In contrast, in fully symptomatic YAC128 mice, glutamate transmission was decreased specifically in D1 cells, and D1 receptor modulation was normal in D1-YAC128 cells. Behaviorally, early symptomatic mice showed increased stereotypies that were decreased by tetrabenazine treatment. Together, these studies support differential imbalances in glutamate and DA transmission in direct and indirect pathway MSSNs. Stereotypic behavior at an early stage could be explained by increased glutamate activity and DA tone in direct pathway neurons, whereas hypokinesia at later stages could result from reduced input onto these neurons.
Introduction
Huntington's disease (HD) is caused by the mutation of a gene inducing expansion of CAG triplets on the huntingtin (htt) protein (The Huntington's Disease Collaborative Research Group, 1993). It is a neurodegenerative disease characterized by a progression of motor symptoms manifesting as chorea in early stages and as akinesia in later stages. Symptoms also include depression, anxiety, irritable or aggressive behavior, and apathy (Bird et al., 1980; Bonelli and Hofmann, 2007). Tetrabenazine (TBZ), a monoamine-depleting agent is the only drug providing significant benefit in the treatment of chorea associated with HD.
The most obvious neuropathological changes are found in the striatum showing loss of medium-sized spiny neurons (MSSNs) (Vonsattel et al., 1985). MSSNs expressing enkephalin are preferentially lost in postmortem brains of fully symptomatic patients, but also in early symptomatic and presymptomatic brains whereas MSSNs expressing substance P are relatively spared (Reiner et al., 1988; Albin et al., 1992; Richfield et al., 1995). The specific vulnerability of MSSNs in HD has important implications because the two types of MSSNs project to different output structures playing a role in motor planning and execution as well as in the control of cognition and motivation (Graybiel et al., 1994). Direct pathway MSSNs express mostly dopamine (DA) D1-like receptors, substance P, and dynorphin, and project preferentially to the substantia nigra pars reticulata (SNr) and to the internal segment of the globus pallidus (GPi). Indirect pathway MSSNs preferentially express D2-like receptors and met-enkephalin, and project to the external segment of the GP, which connects to the subthalamic nucleus, which in turn projects to GPi and SNr (Gerfen, 1992; Bolam et al., 2000).
MSSNs receive two main afferent inputs: glutamate inputs from the cortex and the thalamus, and DA inputs from substantia nigra pars compacta. DA released from the nigrostriatal DA pathway modulates activity of the direct and indirect striatal output pathways, facilitating voluntary movement or inhibiting unwanted movement, respectively (Crossman, 2000). In HD, glutamate and DA transmission is altered, which is likely to induce an imbalance in activity of the direct and indirect pathways and to contribute to the motor, cognitive, and psychiatric symptoms of HD.
We previously showed that MSSNs display increased glutamate currents early in the disease and progressively lose their cortical connections as the behavioral phenotype develops in mouse models of HD (Cepeda et al., 2004; Graham et al., 2009; Joshi et al., 2009). However, the time course of specific alterations in the two subpopulations of MSSNs has never been studied. To determine how each subpopulation of MSSN is functionally affected, we examined excitatory postsynaptic currents (EPSCs) and DA modulation in two mouse models, the YAC128 and the BACHD, expressing a reporter gene under the control of the promoter for either DA D1 or D2 receptors (Gong et al., 2003). YAC128 mice were examined at an early symptomatic stage (1.5 months) and at two symptomatic stages (6 and 12 months) (Slow et al., 2003). BACHD mice were examined only at an early symptomatic stage of 2 months (Gray et al., 2008). As we observed alterations in DA modulation suggestive of an increased DA tone in HD mice, we also tested the effect of DA depletion using tetrabenazine in vitro and in vivo.
Materials and Methods
Animals.
All experimental procedures were performed in accordance with the United States Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of California, Los Angeles. Every effort was made to minimize pain and discomfort. Experiments were conducted in two mouse models of adult-onset HD crossbred to FVB/N mice expressing EGFP under the control of the D1 or D2 promoter obtained from our colony at UCLA. One mouse model carried full-length mutant human htt, including 128 CAG repeats, on a yeast-artificial chromosome (YAC128, line 53) (Slow et al., 2003). YAC128 and wild-type (WT) littermates expressing D1- or D2-EGFP were obtained from our colonies at UCLA at ages 1.5, 6, and 12 months. The other mouse model carried full-length mutant human htt, including 97 CAG repeats, on BACHD (Gray et al., 2008). BACHD and WT littermates expressing D1- or D2-EGFP were obtained from our colonies at 2 months.
Cell visualization.
EGFP-positive cells were visualized in slices using a 40× water-immersion lens. The microscope (Olympus BX50WI) was equipped with differential interference contrast optics and fluorescence. For infrared videomicroscopy, a halogen lamp and an infrared filter (790 nm, Ealing Optics) were used. For fluorescence, labeled cells were excited with ultraviolet light. The light source consisted of a mercury lamp (100 W) and filtered light (450–480 nm) was detected with a videocamera (QICAM-IR Fast 1394, QImaging) optimized to detect EGFP fluorescence and infrared light. Images were digitized and saved using the QCapture Pro software (version 5, QImaging). Once a viable MSSN was identified with infrared videomicroscopy, the filter was switched to fluorescence to determine whether it was labeled with EGFP. The digitized infrared image was superimposed over the fluorescence image, and electrophysiological recordings proceeded only if the cell identified with infrared light showed a complete overlap with EGFP fluorescence.
Experiments in slices.
Detailed procedures have been published (Cepeda et al., 1993; André et al., 2010). Mice were deeply anesthetized with halothane and killed. The brains were dissected and immediately placed in oxygenated ice-cold low-calcium artificial CSF (ACSF) containing (in mm): 130 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 5 MgCl2, 1 CaCl2, and 10 glucose. Coronal slices (350 μm) were cut and transferred to an incubating chamber containing ACSF (with 2 mm CaCl2 and 2 mm MgCl2) oxygenated with 95% O2-5% CO2 (pH 7.2–7.4, 290–310 mOsm, 25 ± 2°C). After 1 h incubation, slices were visualized and EGFP-expressing cells were selected for recordings. The patch pipette contained the following solution (in mm): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 1 MgCl2, 5 MgATP, 5 EGTA, 10 HEPES, 0.5 GTP, 10 phosphocreatine, 0.1 leupeptin (pH 7.25–7.3, osmolality, 280–290 mOsm) for voltage-clamp recordings or K-gluconate solution containing (in mm): 112.5 K-gluconate, 4 NaCl, 17.5 KCl, 0.5 CaCl2, 1 MgCl2, 5 ATP (potassium salt), 1 NaGTP, 5 EGTA, 10 HEPES (pH 7.2, 270–280 mOsm) for recordings in current-clamp mode. Access resistances were <20 MΩ. Spontaneous postsynaptic currents were recorded in standard ACSF at room temperature. Membrane current was filtered at 1 kHz and digitized at 100–200 μs using Clampex 10.2 (gap-free mode). Cells were voltage-clamped at −70 mV to determine basic membrane properties and to examine spontaneous glutamate receptor-mediated EPSCs. Bicuculline methiodide (Bic) (10 μm) was applied to block GABAA receptor-mediated currents. At that voltage, in the presence of Bic, spontaneous synaptic activity is mediated by activation of AMPA receptors as addition of CNQX completely abolishes the events (data not shown). Baseline activity was recorded for 3–5 min after 10–15 min in Bic and 5–10 min after addition of D1 or D2 receptor agonists [R-(+)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide (SKF81297), 5–10 μm or quinpirole, 5–10 μm], D2 receptor antagonist (remoxipride, 10 μm), tetrodotoxin (TTX, 1 μm) or TBZ (10 μm).
Synaptic stimulation.
To evoke synaptic currents, a monopolar stimulating electrode (glass-pipette filled with ACSF, impedance 1.5–1.8 MΩ) was placed in the corpus callosum, 200–300 μm from the recorded cell. Cell membranes were voltage-clamped at −70 mV in the presence of Bic. Test stimuli (500 ms duration) were applied every 20 s and averaged over three consecutive trials. Test stimuli were applied at increasing stimulus intensities (0.05–0.30 mA) to assess input–output functions. Subsequently, stimulation intensities were set to evoke responses at 30% of maximal amplitude. Paired-pulse stimuli were used to assess probability of release and were applied at intervals between 25 and 400 ms.
Drugs.
Stocks of Bic (20 mm, Tocris Bioscience), D1 receptor agonist SKF81297 (1 mm, Sigma), D2 receptor agonist quinpirole (10 mm, Sigma), and D2 receptor antagonist remoxipride (10 mm, Tocris Bioscience) were all dissolved in H2O. TBZ (10 mm, Tocris Bioscience) was dissolved in 100% DMSO for stock solution, and final concentration for electrophysiology was 0.1%. For in vivo injections, TBZ (1 mg/ml) was dissolved in a mixture of DMSO/NaCl (50:50).
Immunohistochemistry and EGFP counts.
D1-WT (n = 3 at 1.5 months, n = 4 at 12 months), D1-YAC128 (n = 3 at 1.5 months, n = 4 at 12 months), D2-WT (n = 5 at 1.5 months, n = 4 at 12 months), and D2-YAC128 mice (n = 8 at 1.5 months, n = 4 at 12 months) were deeply anesthetized and transcardially perfused with cold PBS followed by 4% paraformaldehyde (PFA) dissolved in PBS. Brains were then removed and postfixed for 3–5 h in 4% PFA, cryoprotected overnight in 20 and 30% sucrose in PBS and frozen. Immunofluorescence staining was performed on free-floating sections (30 μm) with the mouse monoclonal antibody against NeuN (dilution 1:2000, MAB377, Millipore Bioscience Research Reagents) overnight at 4°C in 50 mm Tris-saline, pH 7.4, 4% goat serum (Vector Laboratories). Sections were washed and incubated with a goat antibody conjugated to Cy3 (1:500; Jackson ImmunoResearch) in Tris-saline containing 4% normal goat serum and 0.05% Triton for 30 min at room temperature. After 3 washings, sections were mounted on gelatin-coated slides, air-dried, coverslipped with glycerol. Immunofluorescent sections were visualized with a confocal laser scanning microscope (LSM 510 META, Zeiss). Pictures of single confocal sections (917 × 917 pixels) were acquired at a depth of eight bits, using a water-immersion 40× lens and a pinhole set at 1 Airy unit. The region of interest was selected on the NeuN image in the dorsal striatum, pictures were then taken for each fluorophore using the same parameters. For each animal, 10 pictures (0.625 mm2 total) were taken (2–4 per section on 3 adjacent sections) in the striatum. NeuN (red)- and EGFP (green)-positive profiles were counted by two investigators blind to genotype, using Adobe Photoshop CS4 software.
Data analysis and statistics.
In the text, values are presented as means ± SEMs. Data analyses were performed with Origin (Microcal Software) and pClamp 10.2. Group means for all measures were compared using Student's t tests (for two group comparisons) and one-way (to compare the effect of age on spontaneous activity in WT cells), two-way (to compare the effect the effect of age on spontaneous activity in WT and YAC/BACHD cells), and two-way repeated-measures (RM) ANOVAs (to compare the effect of age on evoked—at different amplitudes or different intervals—in WT and YAC/BACHD cells) followed by Bonferroni t tests (multiple group comparisons) using SigmaStat software (SPSS). Differences were considered statistically significant when p < 0.05. Spontaneous synaptic currents were analyzed offline using the automatic detection protocol within the Mini Analysis Program (Justin Lee, Synaptosoft, 1999) and subsequently checked manually for accuracy. Event analyses were done blind to genotype. The threshold amplitude for the detection of an event (5 pA) was set above the root mean square background noise level (1–2 pA at V hold = −70 mV). Amplitude–frequency, cumulative amplitude, cumulative frequency, and interevent interval distributions were constructed to evaluate differences in events at each amplitude and interval.
Behavior.
Mice were housed in a room with controlled photoperiod (lights on from 6:00 A.M. to 6:00 P.M.). Behavioral studies took place between 7:00 P.M. and 10:00 P.M. in the YAC128 mice. In BACHD mice, behavior was videotaped, and stereotypic repetitive behaviors such as grooming and sniffing were examined. For that, we performed all behavior in the light, between 10:00 A.M. and 1:00 P.M. in BACHD mice. Floor sniffing was defined as olfactory investigation of the floor while the animal was moving its vibrissae. Grooming (face cleaning and fur licking) was scored in seconds. Locomotor and exploratory behaviors were examined in a novel square (12 × 12 inches) open field (Truscan 2.0, Coulbourn Instruments) for 15 min in YAC128 male mice at an early stage (1.5 months) and at two later stages (6 and 12 months), and in BACHD male mice at an early stage (2 months). Distance traveled and stereotypies (number of coordinate changes less than ±0.999 beam spaces in each dimension and back to the original point that do not exceed 2 s apart) were observed. Stereotypy measurement provides a measure of repetitive behaviors which do not contribute to location changes but cause beam breaks thus contributing to overall activity counts (Presti et al., 2004).
To determine appropriate TBZ dosage, a group of BACHD mice were injected with 2.5 or 5 mg/kg TBZ, returned to their cage and placed in the open field 90 min after injection. We decided to use 2.5 mg/kg as it did not completely block locomotion, whereas mice injected with 5 mg/kg displayed almost complete immobility for up to 2 h (see Results). To determine the effect of TBZ with time, another group of BACHD mice were habituated in the open field for 30 min, and baseline activity was recorded for 15 min. Mice (n = 3) were then injected with 2.5 mg/kg TBZ and replaced in the open field, and activity was recorded for 15 min at 5, 25, 40, and 60 min after injection. Once the dosage and timing were determined, we tested the effect of TBZ on stereotypies. Before TBZ treatment, BACHD and WT mice were tested for 15 min in the open field for 5 consecutive days to stabilize activity, and open field activities from days 3, 4 and 5 were averaged. TBZ (2.5 mg/kg) was injected subcutaneously on days 6 and 7, and mice were returned to their cage then placed in the open field 40 min after TBZ injection. Open field activities from days 6 and 7 were averaged and compared with those before TBZ injection.
Results
Glutamate synaptic transmission changes with age in WT mice
To measure glutamate synaptic transmission, we examined frequency of sEPSCs in the presence of the GABAA blocker Bic. In WT animals, both D1 and D2 cells showed significant differences with age, from 1.5 to 12 months (Fig. 1). Interestingly, glutamate transmission changed in opposite directions in the two types of MSSNs. In D1-WT cells, sEPSC frequency increased with age (one-way ANOVA F (2,69) = 7.17, p = 0.002). Frequency was the lowest at 1.5 months and significantly different from frequency at 6 (t = 2.81, p = 0.019) and 12 months (t = 3.23, p = 0.006). There was no difference in EPSC frequency between 6 and 12 months in D1-WT cells (t = 0.81, p = 1.0). In D2-WT cells, sEPSC frequency decreased with age (one-way ANOVA F (2,62) = 4.82, p = 0.011). Frequency was similar at 1.5 and 6 months (t = 0.81, p = 1.0) while it was significantly lower at 12 months compared with 1.5 (t = 2.61, p = 0.034) and 6 months (t = 2.84, p = 0.017).
Figure 1.

Frequency of sEPSCs varies differentially with age in D1 and D2 cells in WTs. A, Traces show sEPSCs in D1- and D2-WT cells at 1.5, 6, and 12 months. B, Graph shows that the mean frequency of sEPSCs increases with age in D1 while it decreases with age in D2 cells. At 1.5 months, sEPSC frequency was significantly higher in D2 cells, whereas at 12 months, it was significantly higher in D1 cells. C, Cumulative probability distributions of interevent intervals in D1-WT cells show that intervals significantly decrease at 6 and 12 months compared with 1.5 months. D, Cumulative probability distributions of interevent intervals in D2-WT cells show that intervals significantly increase at 12 months compared with 1.5 and 6 months. Numbers of cells per group in parentheses. *p < 0.05 and **p < 0.01 in this and other figures.
Glutamate transmission is increased at 1.5 months and decreased at 12 months in D1-YAC128 cells
In contrast to D1-WT cells, D1-YAC128 cells showed a decrease of frequency with age, with the highest frequency at 1.5 months (Fig. 2 A; one-way ANOVA F (2,29) = 4.61, p = 0.016). Frequency at 1.5 months was significantly higher than at 12 months in D1-YAC128 cells (t = 2.84, p = 0.022). There were also differences between D1-WT and D1-YAC128 cells with age (one-way ANOVA F (2,108) = 6.73, p = 0.002). At 1.5 months, D1-YAC128 cells displayed a higher frequency than D1-WT cells (t = 2.32, p = 0.022) while at 12 months, it was the reverse (t = 1.74, p = 0.005). Frequencies at 6 months were similar in D1-YAC and D1-WT cells (t = 0.42, p = 0.67). At 1.5 months, cumulative interevent interval histograms suggested a significantly increased probability of release onto D1-YAC128 cells, while at 12 months, cumulative interevent interval histograms suggested a decreased probability of release onto D1-YAC128 cells (Fig. 2 A). Miniature EPSCs were recorded in the presence of TTX at 1.5 months to determine whether the increased glutamatergic transmission was action potential-dependent. In the presence of TTX, mEPSC frequency was still higher in D1-YAC128 than in D1-WT cells suggesting an increase in spontaneous glutamate release onto YAC128 cells that was action potential-independent (Fig. 2 B).
Figure 2.

Biphasic alterations of excitatory synaptic transmission in D1-YAC128 cells. A, Mean sEPSC frequency and cumulative probability distributions of interevent interval histograms for sEPSCs show that D1-YAC128 cells display higher sEPSC frequencies at 1.5 months, while at 12 months D1-YAC128 cells display lower sEPSC frequencies. B, Mean mEPSC frequency (inset) and cumulative probability distribution of interevent intervals for mEPSCs at 1.5 months show that D1-YAC128 cells display a significantly higher mEPSC frequency. C, PPRs were significantly decreased in D1-YAC128 cells at 1.5 months, indicating increased glutamate release probability. In contrast, PPRs were increased in D1-YAC128 cells at 12 months, indicating decreased probability of glutamate release.
To provide further evidence that the differences in EPSC frequencies were due to altered glutamate release, we examined glutamate responses evoked by corticostriatal afferents (eEPSCs) and measured paired-pulse ratios (PPRs). In D1-WT cells, PPRs decreased from 1.5 to 12 months (two-way RM ANOVA F (1,98) = 28.7, p < 0.001, t test p < 0.01 at 25, 50, and 100 ms intervals), suggesting the initial probability of glutamate release onto D1-WT cells is increased in older mice (Fig. 2 C). At 1.5 months, PPRs were higher in D1-WT cells than in D1-YAC128 cells at 25, 50, and 100 ms intervals (two-way RM ANOVA F (1,109) = 13.91, p < 0.001, t test p < 0.05 at 25, 50, and 100 ms intervals). In contrast, at 12 months, D1-YAC128 cells displayed higher PPRs (two-way RM ANOVA F (1,114) = 12.39, p < 0.001, p < 0.05 at 25, 50, and 100 ms intervals). These findings suggest that the initial probability of glutamate release onto D1-YAC128 cells is higher than on D1-WT cells at 1.5 months, which could explain the higher frequencies of EPSCs in D1-YAC128 cells at that age. In contrast, at 12 months, probability of release onto D1-YAC128 cells is lower, correlating with a lower frequency of sEPSCs.
Glutamate release is not significantly altered in D2-YAC128 cells
Although mean sEPSC frequency tended to be lower with age in D2-YAC128 cells, the difference failed to reach significance (Fig. 3 A; one-way ANOVA F (2,39) = 2.52 p = 0.094). However, the t test between 1.5 and 12 months showed a decrease of sEPSC frequency in D2-YAC128 cells with age (t 25 = 2.68 p = 0.013). There was no significant genotype difference between D2-WT and D2-YAC128 cells with age (two-way ANOVA F (2,98) = 0.29, p = 0.74). However, the cumulative interevent interval distribution exhibited a significant rightward shift. To investigate further if the rightward shift could indicate a decrease in glutamate release, we measured mEPSCs and PPRs (Fig. 3 A). In the presence of TTX, mEPSC frequency was similar in D2-WT and D2-YAC128 cells at 1.5 months (Fig. 3 B). There were no differences in PPRs between D2-WT and D2-YAC128 cells at 1.5 (two-way RM ANOVA F (1,109) = 0.06 p = 0.79) or at 12 months (two-way RM ANOVA F (1,99) = 0.28 p = 0.6) (Fig. 3 C) suggesting glutamate release onto cells of the indirect pathway is not altered in YAC128.
Figure 3.

Excitatory synaptic transmission in D2-YAC128 cells. A, Mean sEPSC frequency was not different in D2-YAC128 and D2-WT cells at any age. Cumulative probability distributions of interevent intervals only showed slight differences at 12 months when D2-YAC128 cells displayed significantly fewer sEPSCs, at the longer intervals only. B, Mean mEPSC frequency (inset) and cumulative probability distribution of interevent intervals for mEPSCs were not different at 1.5 months for D2-YAC128 and D2-WT cells. C, PPRs were not different in D2-YAC128 cells compared with D2-WT cells at each age tested.
Evoked glutamate currents are increased in D2-YAC128 cells at 1.5 months and decreased in D1-YAC128 cells at 12 months
In WT cells, eEPSCs also differed with age. In D1-WT cells, eEPSCs were larger at 12 months than at 1.5 months (two-way RM ANOVA F (1,139) = 5.092 p = 0.033) (Fig. 4 A,B). In D2-WT cells, eEPSC peak amplitude did not change with age (two-way RM ANOVA F (1,129) = 0.418 p = 0.524). In addition, eEPSCs were larger in D1-WT cells than in D2-WT cells, both at 1.5 and 12 months (two-way RM ANOVA F (3,269) = 5.084, p = 0.004). D1-YAC128 cells displayed similar currents as D1-WT cells at 1.5 months (Fig. 4 C; two-way RM ANOVA F (1,139) = 0.381, p = 0.542). In contrast, D2-YAC128 cells had larger eEPSCs compared with D2-WT cells at that age (two-way RM ANOVA F (1,75) = 9.326 p = 0.005) (Fig. 4 D). At 12 months, D1-YAC128 cells displayed smaller currents than D1-WT while D2-YAC128 and D2-WT cells displayed similar currents (Fig. 4 C,D).
Figure 4.

Mean amplitude of evoked EPSCs is increased in D2-YAC128 cells at 1.5 months and decreased at 12 months in D1-YAC128 cells. A, Traces show evoked EPSCs in WT (black) and YAC128 (red) at 1.5 and 12 months. Numbers above traces are stimulus intensities. B, Input–output functions show evoked EPSCs at the two ages in WT cells. Currents increase significantly with age in D1 cells but show no change in D2 cells. C, At 1.5 months, evoked EPSC amplitudes are similar in D1-YAC128 compared with D1-WT cells, whereas at 12 months, evoked EPSC amplitudes are decreased in D1-YAC128 cells compared with D1-WT cells. D, At 1.5 months, evoked EPSC amplitudes are increased in D2-YAC128 compared with D2-WT cells while at 12 months, they are similar.
Glutamate transmission in BACHD mice
At 2 months, similar to D1-YAC128 cells at 1.5 months, D1-BACHD cells displayed a significant decrease in PPR (two-way RM ANOVA F (1,89) = 15.6, p = 0.001) at 25 (t = 3.8, p < 0.001) and 50 (t = 3.57, p < 0.001) ms (Fig. 5 B) suggesting an increased probability of glutamate release onto cells of the direct pathway in BACHD mice at an early stage. D2-BACHD cells did not show any difference in PPR compared with their WT littermates (two-way RM ANOVA F (1,83) = 0.19, p = 0.662), again a result similar to findings in D2-YAC128 cells at the presymptomatic stage (Fig. 5). Alterations in eEPSC amplitudes were also similar in BACHD and YAC128 D1 and D2 cells: eEPSC peak amplitudes were similar in D1-BACHD and D1-WT cells (F (1,89) = 1.18, p = 0.293) while D2 cells from BACHD mice showed larger currents than in WT mice (F (1,69) = 7.75, p = 0.034) (Fig. 5 C).
Figure 5.

Evoked EPSCs and PPRs in BACHD mice at 2 months. A, PPRs are significantly decreased in D1-BACHD compared with D1-WT cells at 2 months. In contrast, PPRs are similar in D2-BACHD and D2-WT cells. B, Evoked EPSC amplitudes are significantly increased in D2-BACHD compared with D2-WT cells, whereas they are similar in D1-BACHD and D1-WT cells.
D1 and D2 modulation of glutamate synaptic activity and cell firing in YAC128
The D1 receptor agonist SKF81297 (5 and 10 μm) increased sEPSC frequency (paired t test, t 20 = 5.86, p < 0.001) in D1-WT cells as published previously (André et al., 2010). This effect was similar at 1.5, 6, and 12 months. In D1-YAC128 cells, the D1 receptor agonist did not change the frequency of sEPSCs (t 9 = 0.96, p = 0.361) at 1.5 months (significantly different from D1-WT cells, t 20 = 3.23, p = 0.004). However, SKF81297 increased sEPSC frequency in D1-YAC128 cells at 6 and 12 months, and this effect was similar to that observed in D1-WT cells (Fig. 6 A). The D2 receptor agonist quinpirole (10 μm) decreased sEPSC frequency at 1.5 (t 12 = 5.34, p < 0.001) and 6 (t 11 = 3.24, p = 0.008) months in D2-WT cells (Fig. 6 B,C). At 1.5 (t 11 = 0.30, p = 0.765) and 6 (t 12 = 0.72, p = 0.480) months, quinpirole had no significant effect on sEPSC frequency in D2-YAC128 cells. At 12 months, the D2 receptor agonist had no effect on sEPSC frequency in D2-WT cells (data not shown), suggesting a floor effect because of the low sEPSC frequencies. Therefore, we tested the D2 antagonist remoxipride (10 μm) at 12 months and found that it increased sEPSC frequency in D2-WT cells (t 10 = 2.68, p = 0.023) while it had no effect in D2-YAC128 cells (t 8 = 0.69, p = 0.509) (Fig. 6 B,C).
Figure 6.

DA modulation of sEPSCs in D1 and D2 cells is altered in YAC128 mice. A, Traces show that at 1.5 months, the D1 receptor agonist SKF81297 increased sEPSC frequency in D1-WT cells, whereas it had no effect in D1-YAC128 cells. In contrast, at 12 months, SKF81297 increased the frequency of sEPSCs. B, Traces show that at 1.5 months, the D2 receptor agonist quinpirole decreases sEPSCs in D2-WT cells but not in D2-YAC128 cells. At 12 months, the D2 receptor antagonist remoxipride increases sEPSC frequency in D2-WT cells but not in D2-YAC128 cells. C, Bar graphs show the percentage changes in sEPSC frequency in D1 and D2 cells in WT and YAC128 mice at three ages.
We also examined the effects of DA receptor agonists on cell excitability in current clamp in YAC128 mice at 1.5 months (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In D1 and D2 cells for WTs or YAC128 mice, there were no differences in resting membrane potential, current–voltage relationship or rheobase. In contrast, the rheobase was significantly higher in D1-WT compared with D2-WT cells (data not shown) as previously published (Cepeda et al., 2008; Gertler et al., 2008). In D1-WT cells, SKF81297 (5 μm) increased excitability by increasing the spike frequency (p = 0.03) while it had no significant effect in D1-YAC128 cells (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). In D2-WT cells, quinpirole (10 μm) decreased excitability by decreasing spike frequency (p < 0.01) while it had no significant effect in D2-YAC128 cells (supplemental Fig. 1G,H, available at www.jneurosci.org as supplemental material).
Our data suggest complex biphasic anomalies in DA neurotransmission, with loss of D1 and D2 receptor function in early symptomatic mice. D1 receptor function in fully symptomatic mice appears similar to that of WTs. This early alteration in DA neurotransmission might be induced by abnormal DA concentrations as elevated striatal DA concentrations decrease DA receptor function (Giros et al., 1996; Dumartin et al., 2000; Wu et al., 2007). In addition, in HD patients, TBZ is the only drug that alleviates chorea. TBZ ultimately depletes DA stores, suggesting that decreasing DA might be beneficial for hyperkinetic symptoms. Most studies report a decrease in DA release in symptomatic HD (Hickey et al., 2002; Johnson et al., 2007), but it remains unknown whether striatal DA tone is increased in early HD and contributes to some of the hyperkinetic symptoms. To test whether DA function is rescued in early HD, we examined the effect of TBZ in early HD in vitro and in vivo.
TBZ restores the effect of exogenously applied D1 receptor agonist and PPRs
In YAC128 slices from 1.5 month mice, we bath-applied TBZ (10 μm) after establishing baseline glutamate sEPSC frequency for 15 min in D1-WT and D1-YAC128 cells (sEPSC frequency was quantified for 3 min before TBZ application). In D1-WT cells, TBZ had biphasic effects, increasing sEPSC frequency at 5 min (+25.3 ± 6.0%, paired t test, t 8 = 5.58 p < 0.01, 8 cells) (Fig. 7 A). At 10 min, the increase (+14.2 ± 6.9%) was no longer significant. sEPSC frequency returned to baseline values by 20 min, then gradually decreased. The decrease was significant after 40 min (−13.3 ± 3.8%, t 8 = 2.68 p = 0.028) and 65 min (−18.6 ± 4.1%, t 8 = 2.50 p = 0.037). In D1-YAC128 cells, TBZ did not induce an increase in sEPSC frequency. The effect of TBZ at 5 min was significantly different between D1-WT and D1-YAC128 cells (t 15 = 3.65 p = 0.002). In D1-YAC128 cells, TBZ induced a decrease that was significant by 20 min (−16.8 ± 5.5, p = 0.016, 7 cells). The decrease in TBZ was significantly higher in D1-YAC128 than in D1-WT cells at 25, 40, 50, and 65 min (F (1,6302) = 36.63, p < 0.001). To test whether the decrease in TBZ could be nonspecific and due to a run-down of the cell health during these long recordings, we assessed the effect of DMSO on sEPSC frequency. In D1-WT cells, DMSO did not change sEPSC frequency even after 60 min indicating that changes in sEPSC frequency are specific to TBZ.
Figure 7.

A, Effect of TBZ on sEPSC frequency in D1-WT and D1-YAC128 cells at 1.5 months. In D1-WT cells, TBZ induced a transient but significant increase in sEPSC frequency at 5 min while at 40, 50, and 65 min, TBZ induced a significant decrease in sEPSC frequency. In D1-YAC128 cells, TBZ did not increase sEPSC frequency but induced a larger decrease than in D1-WT cells at 25, 40, 50, and 65 min. B, In YAC128 slices (1.5 months) incubated for 2–4 h in TBZ, the D1 receptor agonist SKF81297 (5 μm) induced a significant increase in frequency of sEPSCs in D1 cells while in YAC128 incubated in DMSO, SKF81297 had no effect. The effect of SKF81297 was significantly different between DMSO and TBZ treatment. C, In 2-month-old BACHD mice, in TBZ-incubated slices, D1 cells showed PPRs higher than those in DMSO-incubated slices, similar to PPRs in D1-WT cells. D, Traces and graphs show that sEPSC frequency is higher in D1-BACHD cells (2 months) than in D1-WT cells. In addition, the D1 agonist SKF81297 increased sEPSC frequency in D1-WT cells but not in D1-BACHD cells. E, Amplitude–frequency histogram shows that in D1-WT cells, the D1 antagonist SCH23390 (20 μm) had no effect on sEPSC frequency. F, In contrast, in D1-BACHD cells, the D1 antagonist decreased sEPSC frequency. Inset shows that the percentage change induced by SCH23390 is significantly different in D1-WT and D1-BACHD cells. *p < 0.05, **p < 0.01, ***p < 0.001, effect of TBZ compared with control (0 min in TBZ). # p < 0.05 and ### p < 0.001 in D1-YAC128 (or D1-BACHD) compared with D1-WT cells. *p < 0.05 in TBZ-treated vs DMSO-treated slices.
We then tested the effect of the D1 receptor agonist in a different group of cells in slices from YAC128 at 1.5 months incubated in TBZ (10 μm) or in DMSO (0.1%) for 2–4 h (Fig. 7 B). In YAC128 slices incubated in TBZ, SKF81297 (5 μm) significantly increased sEPSC frequency (t 8 = 2.67, p = 0.028) while in YAC128 slices incubated in DMSO, the D1 receptor agonist had no significant effect (t 9 = 1.04, p = 0.326). The difference between the two treatments was statistically significant (t 17 = 2.65, p = 0.017).
As in D1-YAC128, sEPSC frequency was significantly higher in D1-BACHD cells at 2 months (Fig. 7 D; t 18 = 2.781, p = 0.012), and the D1 agonist SKF81297 had no effect in D1-BACHD cells. To test whether the increased PPRs could be a result of increased presynaptic glutamate release probability due to elevated DA tone, we examined PPRs in D1 cells in BACHD slices that were incubated in TBZ (10 μm) or in DMSO (0.1%) for 2–4 h (Fig. 7 C), and we tested the effect of a D1 antagonist on sEPSCs. In BACHD slices incubated in TBZ, PPRs were significantly higher than in BACHD slices incubated in DMSO (t 23 = 2.883, p = 0.008). The D1 antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH23390) (20 μm) did not have any effect in D1-WT cells (Fig. 7 E), suggesting DA tone is low in WT animals. However, in D1-BACHD cells, SCH23390 had a significant effect and decreased sEPSC frequency by −15.9 ± 6.5% (Fig. 7 F). Effect of SCH23390 was significantly larger in D1-BACHD compared with D1-WT cells (t 18 = 2.858, p = 0.01).
Locomotor and repetitive behaviors in HD mice
We measured locomotor activity in the dark during the dark phase for 15 min in a novel open-field test in WT and YAC128 mice at 1.5, 6, and 12 months. In WT mice, locomotor activity slightly decreased with age, this was reflected by decreased distance traveled at 12 months compared with 1.5 and 6 months (F (2,55) = 7.38, p = 0.001) (Fig. 8 A). In YAC128 mice, distance traveled also decreased with age, and this was significant at 12 compared with 6 months (F (2,55) = 7.38, p = 0.001). YAC128 mice at 1.5 months did not show any differences in locomotion compared with WTs while symptomatic 6- and 12-month-old YAC128 mice displayed decreased distance traveled (Fig. 8 A). The number of stereotypic movements measured by the open field test decreased with age in YAC128 mice (t = 3.9, p < 0.001 at 6 vs 1.5 months) while it did not change with age in WT mice. YAC128 mice at 1.5 (t = 4.5, p < 0.001) and 6 (t = 2.8, p = 0.006) months showed more stereotypies than WT mice while at 12 (t = 1.7, p = 0.1) months, stereotypies were similar in WT and YAC128 mice (Fig. 8 B).
Figure 8.

Locomotor activity in YAC128 and BACHD mice. A, YAC128 mice at 6 and 12 months displayed decreased distance traveled during 15 min in the open-field apparatus compared with WT mice. In WT and YAC128 mice, the distance traveled slightly decreased with age. In WT mice at 12 months, distance traveled was lower than that at 1.5 and 6 months. In YAC128 mice, distance was significantly decreased at 12 compared with 6 months. B, YAC128 mice at 1.5 and 6 months old showed more stereotypies than WT mice, whereas at 12 months, stereotypies were similar in WT and YAC128 mice. The number of stereotypic movements decreased with age in YAC128 mice. C, In 2-month-old BACHD mice, distance traveled was significantly lower than in the WT mice. D–F, BACHD mice display more stereotypies (D), more grooming (E), and more sniffing (F) than WT mice. G, TBZ (2.5 mg/kg) decreased distance traveled during 15 min in the open-field apparatus in 2-month-old WT and BACHD mice, and the percentage decrease was similar in WT and BACHD mice. H, TBZ did not have any effect on stereotypies in WT mice, while it decreased them in BACHD mice. *p < 0.05 and **p < 0.01 in YAC128 (or BACHD) compared with WT mice at each age. # p < 0.05 in WT mice at different ages. § p < 0.05 in YAC128 mice at different ages. ⋀ p < 0.05 before and after TBZ treatment.
In BACHD compared with WT mice, the distance traveled was significantly lower at 2 months (t 35 = 2.1, p = 0.042; Fig. 8 C). This decrease in locomotor activity was due to the occurrence of increased stereotypies (t 33 = 2.0, p = 0.48) detected by the open field test, and visually, BACHD mice spent significantly more time grooming (t 23 = 2.3, p = 0.027) and floor sniffing (t 23 = 2.1, p = 0.043) (Fig. 8 D–F). Approximately 40% of the BACHD mice (5/13) spent >20% of the time grooming and when those mice were excluded, the distance traveled was no longer different in the BACHD (4116 ± 144 cm) compared with the WT group (4324 ± 233 cm, p > 0.05). The intensive grooming was not present in WT mice (n = 12).
We chose to use 2.5 mg/kg TBZ for in vivo studies because it decreased distance traveled by ∼50% after 90 min while mice injected with 5 mg/kg displayed almost total immobility (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). At this concentration, the maximal effect for TBZ was obtained 40 min postinjection (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material). At 40 min, TBZ decreased distance traveled similarly (t 9 = 0.12, p = 0.90) in WT and BACHD mice (Fig. 8 G). However, TBZ's effect on stereotypies was significantly different in WT and BACHD mice (Fig. 8 H). TBZ did not have any significant effect on stereotypies in WT mice while it significantly decreased stereotypies in BACHD mice to WT levels. The percentage change induced by TBZ was significantly different between WT and BACHD mice (t 8 = 3.41 p = 0.009).
Expression of EGFP
Five to eight hundred neurons were counted for each animal. Neuron densities (NeuN-positive cells/mm2) were similar between WT and YAC128 mice at 1.5 and 12 months (Fig. 9). This does not exclude there is a loss of cells as reported by others (Slow et al., 2003; Van Raamsdonk et al., 2005b; Tang et al., 2007). We did not perform stereology as our focus was to determine the ratio of EGFP-expressing neurons. We found there were no differences in EGFP expression between WT and YAC128 mice at any age (two-way ANOVA F (3,33) = 0.122, p = 0.947). The only difference was in the ratio of neurons expressing D1- or D2-EGFP as reported before (Bertran-Gonzalez et al., 2008). At 1.5 months, there were more neurons expressing D1-EGFP (D1-WT: 53.8 ± 0.7 vs D2-WT: 48.2 ± 1.4%, t 5 = 3.68, p = 0.014; D1-YAC128: 54.4 ± 1.0 vs D2-YAC128: 47.7 ± 0.5%, t 8 = 7.46 p < 0.001). At 1 year, the difference was not significant but was in the same direction (D1-WT: 51.5 ± 1.8 vs D2-WT: 46.5 ± 2.1%, t 6 = 2.14, p = 0.075; D1-YAC128: 50.6 ± 2.4 vs D2-YAC128: 45.2 ± 2.0%, t 7 = 1.923 p = 0.096).
Figure 9.
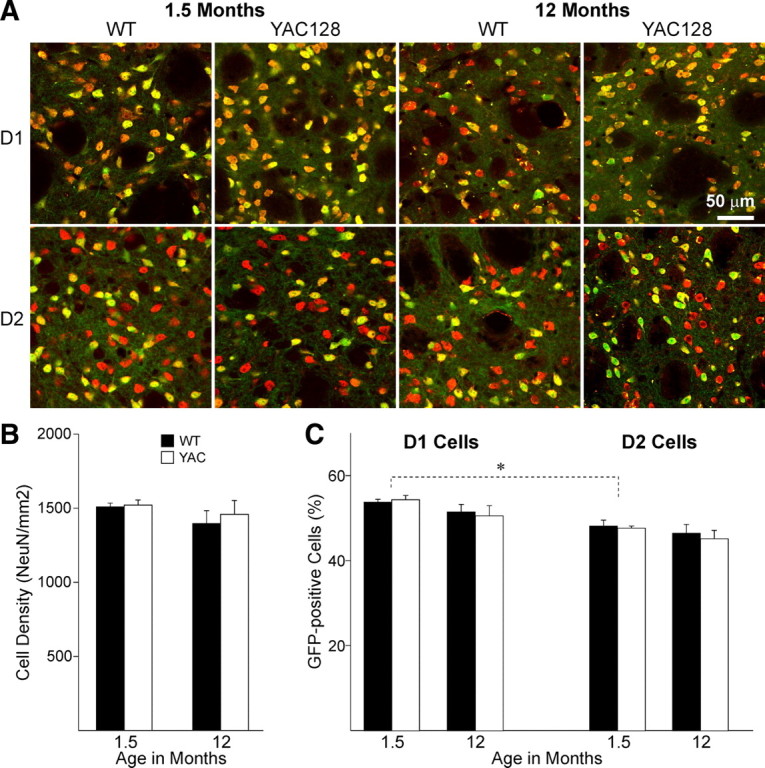
A, Photomicrographs of immunofluorescent staining for NeuN (red) combined with EGFP in D1 (top row) or D2 cells (bottom row), at 1.5 (2 left columns) and 12 (2 right columns) months in the dorsal striatum of WT and YAC128 mice. Pictures show overlay of EGFP and NeuN, in which cells expressing both appear yellow. B, Bar graph of neuronal (NeuN) densities at both ages do not show any differences between WT and YAC128 mice. C, Bar graph shows that the ratio of EGFP-positive neurons does not change between WT and YAC128 mice at any age. There are more D1-EGFP-positive than D2-EGFP-positive neurons at 1.5 months. *p < 0.05 between D1- and D2-EGFP neurons.
Discussion
We show differences in glutamate transmission to direct and indirect pathways that could explain behavioral changes in two models of HD. At the early stage, glutamate release was increased onto D1 cells while it was unaltered onto D2 cells in HD mice. At the late stage, glutamate transmission was decreased onto D1 cells only. We also observed biphasic alterations in D1 receptor agonist function: modulation induced by activation of D1 receptors was absent at the early stage while it was present at later stages. Our data suggest that some alterations at the early stage are mediated via elevated DA neurotransmission as depleting DA restored the D1 receptor agonist effect and decreased stereotypies.
Glutamate transmission changes with age in WT mice
In young mice, sEPSC frequency was higher in D2-WT than in D1-WT cells, as reported previously (Kreitzer and Malenka, 2007; Cepeda et al., 2008), suggesting that excitatory synapses onto D2 MSSNs exhibit higher release probability than those onto D1 MSSNs or that D2 cells receive more excitatory inputs. Conversely, glutamate transmission was higher in D1 than in D2 MSSNs in older mice, indicating age-dependent changes in striatal glutamate inputs. sEPSC frequency increased with age in D1-WT cells while it decreased in D2-WT cells. In addition, PPRs decreased and amplitude of evoked glutamate currents increased with age in D1-WT cells. These results show that glutamate transmission is higher onto direct than on indirect pathway MSSNs in older mice, which could have important implications on output structure activity controlling motor function. In HD, symptoms change with age, and our data in WT mice show it is essential to determine changes in glutamate transmission in older mice to understand the progression of neuropathophysiological changes in HD.
Glutamate transmission is increased at an early stage in HD
In early symptomatic YAC128 and BACHD mice, sEPSCs and mEPSCs were increased while PPRs were decreased in D1 cells. Because the PPR is independent of the number of release sites (Zucker, 1989; Cummings et al., 2009), the changes are likely attributable to altered probability of release. Low PPRs at short intervals indicate depletion of neurotransmitter from the presynaptic terminal and an increased probability of release of an immediately releasable store (Zucker, 1989; Choi and Lovinger, 1997). The lack of changes in current amplitudes also suggest that postsynaptic currents are unaltered and that differences in frequencies and in PPRs occur presynaptically. Differences in PPRs contrast with another study reporting increased facilitation in 26-d-old YAC128, in which the authors suggest that reduced glutamate release occurs in indirect pathway MSSNs (Milnerwood and Raymond, 2007). Discrepancies in findings might be explained by the fact that D1 and D2 MSSNs were not separated and that the study was performed an at earlier stage while ours is performed later, when these alterations no longer occur.
We hypothesize that tonic activation of D1 receptors underlies the increase in glutamate release in HD. We showed that one of the mechanisms by which activation of postsynaptic D1 receptors increases presynaptic glutamate release occurs through blockade of endocannabinoid release by MSSNs, thereby releasing the negative control endocannabinoids normally exert onto neighboring terminals (André et al., 2010). Increased glutamate release onto D1 cells could shift the balance of direct and indirect pathway activity, disinhibiting output collicular and thalamic structures and increasing movement (Chevalier and Deniau, 1990). Increased glutamate transmission onto direct pathway neurons is also consistent with morphological descriptions of MSSNs presenting proliferative changes such as increased number of dendrites and increased size of dendritic spines in early grade HD (Graveland et al., 1985; Ferrante et al., 1991).
D2 cells also displayed early increased glutamate transmission as shown by larger evoked currents, which could also increase activity in the indirect pathway. Activation of the indirect pathway has been shown to decrease motor activity, which we did not observe in the present study. Larger evoked currents in D2 cells are unlikely to be induced by increased glutamate release, as there were no differences in EPSC frequency, interevent intervals or PPRs. Instead, glutamate receptor composition or postsynaptic localization might be altered. For example, extrasynaptic NMDA receptor activation by glutamate spillover is higher in YAC128, inducing larger eEPSCs and excitotoxicity (Milnerwood et al., 2010). Although in our recording conditions, glutamate responses are induced mostly by activation of AMPA receptors, it is possible that extrasynaptic AMPA receptors are also altered in D2 cells, contributing to the larger eEPSCs. Consistent with this idea, AMPA currents were also larger in YAC128 mice at an early stage in dissociated MSSNs, a preparation that preserves only postsynaptic receptors (Joshi et al., 2009). Increased glutamate currents could explain the increased vulnerability of MSSNs to glutamate observed during the early stage of the disease in this model (Graham et al., 2009).
Glutamate transmission is decreased at a late stage
The greatest changes in glutamate transmission at 12 months were observed in D1-YAC128 cells, with lower sEPSC frequency, reduced evoked glutamate currents, and higher PPRs than D1-WT cells. In D2 cells, only a slight decrease in interevent intervals was observed while there was no change in evoked glutamate currents or in PPRs. These results suggest that the late stage of the disease is characterized by a loss of presynaptic and postsynaptic glutamate function, presumably through a loss of glutamate terminals and receptors. Degenerative changes have been reported in HD both in the cortex and the striatum and include loss of cortical and striatal neurons and loss of spines (Klapstein et al., 2001; Cepeda et al., 2007). Our group also reported smaller evoked NMDA and AMPA currents in symptomatic YAC128 (Graham et al., 2009; Joshi et al., 2009) and R6/2 mice (Cepeda et al., 2003). The present study demonstrates that the loss of glutamate inputs starts in direct pathway neurons.
D1 cells are more affected than D2 cells
Our results show more changes occur in D1 cells than in D2 cells, at both presymptomatic and symptomatic ages. This was surprising as we were expecting D2 cells to be more affected as they are presumed to be more vulnerable in HD (Reiner et al., 1988; Albin et al., 1992). It is important to note that we select MSSNs expressing EGFP to differentiate D1 and D2 receptor-expressing cells. However, our cell counts show that the percentage of D2-EGFP-positive neurons does not change with the phenotype or with age, indicating that cells expressing D1 and D2 receptors are not decreased in YAC128 at 12 months, as has been reported previously (Benn et al., 2007). In the present study, neuronal densities were not changed in 12 months old YAC128 mice but, as we did not count the total number of cells, it is possible that our animals displayed cell loss due to striatal volume loss. Another caveat is that the most affected indirect pathway MSSNs might not survive in our preparation, leaving only the least affected and healthiest neurons for recording.
DA function
An intriguing observation is the biphasic alterations in D1 receptor function. D1 receptor modulation was absent at the early stage of the disease, but similar to that of WTs at later stages. In 1.5 months D1-YAC128 cells, D1 receptor modulation was restored by TBZ which depletes DA stores, suggesting increased DA tone at that age. The lack of effect of the D1 agonist might be caused by decreased expression of D1 receptors at the plasma membrane as reported in hyperdopaminergia conditions (Dumartin et al., 2000). In D1-WT cells, TBZ induced a biphasic effect, first increasing sEPSC frequency at 5 min, then decreasing it at 30 min. Although TBZ blocks the vesicular monoamine transporter-2 and inhibits monoamine release resulting in monoamine depletion, the initial effect of TBZ is to induce DA accumulation in the cytoplasm by preventing cytoplasmic DA transport into vesicles (Rudnick and Clark, 1993), which increases the release of DA by reverse transport (Sulzer et al., 1993). The early increase in sEPSCs is likely to be induced by the initial release of DA while the subsequent DA depletion causes the decrease in sEPSCs. Other findings also suggest that extracellular DA concentration might be higher at an early stage as antidopaminergic treatments induced a larger effect in D1-YAC128 and D1-BACHD than in D1-WT cells.
Finally, behavioral alterations at early stages (increased stereotypies) could be indicative of elevated striatal DA concentrations as TBZ decreased them in BACHD mice but not in WT mice. Abnormal grooming has also been reported in R6/2 mice (Carter et al., 1999). l-DOPA paradoxically decreased abnormal grooming, and this effect was attributed to a defect in DA signaling (Hickey et al., 2002) as hyperdopaminergia usually induces increased grooming (Antoniou et al., 1998; Kita et al., 2000; Spielewoy, 2000). Our results are in line with observations that DA receptor antagonists and agents that decrease DA reduce chorea and motor symptoms in patients and in animal models, while dopaminergic stimulation exacerbates some (Huntington Study Group, 2006; Tang et al., 2007; Mestre et al., 2009). Most studies report decreases or no changes in DA release and in striatal DA concentrations in fully symptomatic mice (Hickey et al., 2002; Vetter et al., 2003; Ortiz et al., 2010). However there is no information on DA levels in presymptomatic mice. It is therefore possible that DA neurotransmission is increased in early HD.
In contrast to D1 receptor function that was similar to that of WTs in symptomatic animals, we found that the D2 receptor function was altered at all stages. The fact that the D2 receptor antagonist had no effect on sEPSC frequency suggests that presynaptic D2 receptors might be lost in symptomatic YAC128, consistent with the loss of corticostriatal inputs in late stages.
Correlation between behavior and glutamate transmission in the direct pathway
Stereotypic behavior was increased in YAC128 and BACHD at early stages and correlated with an increase of glutamate transmission onto direct pathway neurons. Increased grooming and sniffing could be the result of DA D1 or D2 receptor activation or through elevated DA transmission (Usiello et al., 2000; Horvitz et al., 2001). Specifically, overactivation of the direct pathway could explain increased motor stereotypies observed here.
Although hypoactivity in symptomatic animals has been reported, there is evidence that hyperactivity precedes hypoactivity in genetic HD (Carter et al., 1999; Bolivar et al., 2004; Van Raamsdonk et al., 2005a) and in lesion models (Sanberg et al., 1989; Brouillet et al., 1999). Abnormal involuntary movements, dyskinesia and chorea are hallmarks of the human pathology at early stages. As the disease progresses the initial hyperkinetic syndrome is progressively replaced by a more hypokinetic syndrome (Thompson et al., 1988) that could correspond to spine or neuron loss in late stages of the disease (Ferrante et al., 1991; Klapstein et al., 2001).
Conclusion
The present findings suggest anomalies in glutamate transmission in HD might be induced by abnormal DA transmission. Early in the progression of the disease, increased glutamate release onto D1 cells and increased stereotypies are consistent with increased DA tone. In symptomatic animals, decreased glutamate release onto D1 cells and hypoactive behavior are consistent with decreased DA tone and/or loss of corticostriatal terminals. The fact that the greatest alterations in glutamate neurotransmission were observed in D1 cells indicates that modulation of direct pathway neurons (D1 antagonists in early HD and agonists in more advanced stages) might prove useful as a strategy to treat HD motor symptoms.
Footnotes
This work was supported by National Institutes of Health Grant NS41574 and by the CHDI Foundation. X.W.Y. is supported by National Institutes of Health Grant NS049501.
References
- Albin RL, Reiner A, Anderson KD, Dure LS, 4th, Handelin B, Balfour R, Whetsell WO, Jr, Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31:425–430. doi: 10.1002/ana.410310412. [DOI] [PubMed] [Google Scholar]
- André VM, Cepeda C, Cummings DM, Jocoy EL, Fisher YE, William Yang X, Levine MS. Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur J Neurosci. 2010;31:14–28. doi: 10.1111/j.1460-9568.2009.07047.x. [DOI] [PubMed] [Google Scholar]
- Antoniou K, Kafetzopoulos E, Papadopoulou-Daifoti Z, Hyphantis T, Marselos M. D-amphetamine, cocaine and caffeine: a comparative study of acute effects on locomotor activity and behavioural patterns in rats. Neurosci Biobehav Rev. 1998;23:189–196. doi: 10.1016/s0149-7634(98)00020-7. [DOI] [PubMed] [Google Scholar]
- Benn CL, Slow EJ, Farrell LA, Graham R, Deng Y, Hayden MR, Cha JH. Glutamate receptor abnormalities in the YAC128 transgenic mouse model of Huntington's disease. Neuroscience. 2007;147:354–372. doi: 10.1016/j.neuroscience.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird ED, Spokes EG, Iversen LL. Dopamine and noradrenaline in post-mortem brain in Huntington's disease and schizophrenic illness. Acta Psychiatr Scand Suppl. 1980;280:63–73. [PubMed] [Google Scholar]
- Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. J Anat. 2000;196:527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolivar VJ, Manley K, Messer A. Early exploratory behavior abnormalities in R6/1 Huntington's disease transgenic mice. Brain Res. 2004;1005:29–35. doi: 10.1016/j.brainres.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Bonelli RM, Hofmann P. A systematic review of the treatment studies in Huntington's disease since 1990. Expert Opin Pharmacother. 2007;8:141–153. doi: 10.1517/14656566.8.2.141. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Condé F, Beal MF, Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernández-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Levine MS. Dopamine and glutamate in Huntington's disease. In: Schmidt WJ, Reith MEA, editors. Dopamine and glutamate in psychiatric disorders. Totowa, NJ: Humana; 2004. pp. 539–565. [Google Scholar]
- Cepeda C, Wu N, André VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington's disease. Prog Neurobiol. 2007;81:253–271. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, André VM, Yamazaki I, Wu N, Kleiman-Weiner M, Levine MS. Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur J Neurosci. 2008;27:671–682. doi: 10.1111/j.1460-9568.2008.06038.x. [DOI] [PubMed] [Google Scholar]
- Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci. 1990;13:277–280. doi: 10.1016/0166-2236(90)90109-n. [DOI] [PubMed] [Google Scholar]
- Choi S, Lovinger DM. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci U S A. 1997;94:2665–2670. doi: 10.1073/pnas.94.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossman AR. Functional anatomy of movement disorders. J Anat. 2000;196:519–525. doi: 10.1046/j.1469-7580.2000.19640519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DM, André VM, Uzgil BO, Gee SM, Fisher YE, Cepeda C, Levine MS. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci. 2009;29:10371–10386. doi: 10.1523/JNEUROSCI.1592-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumartin B, Jaber M, Gonon F, Caron MG, Giros B, Bloch B. Dopamine tone regulates D1 receptor trafficking and delivery in striatal neurons in dopamine transporter-deficient mice. Proc Natl Acad Sci U S A. 2000;97:1879–1884. doi: 10.1073/pnas.97.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ. Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Graham RK, Pouladi MA, Joshi P, Lu G, Deng Y, Wu NP, Figueroa BE, Metzler M, André VM, Slow EJ, Raymond L, Friedlander R, Levine MS, Leavitt BR, Hayden MR. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J Neurosci. 2009;29:2193–2204. doi: 10.1523/JNEUROSCI.5473-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Reynolds GP, Morton AJ. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington's disease. J Neurochem. 2002;81:46–59. doi: 10.1046/j.1471-4159.2002.00804.x. [DOI] [PubMed] [Google Scholar]
- Horvitz JC, Williams G, Joy R. Time-dependent actions of D2 family agonist quinpirole on spontaneous behavior in the rat: dissociation between sniffing and locomotion. Psychopharmacology (Berl) 2001;154:350–355. doi: 10.1007/s002130000677. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366–372. doi: 10.1212/01.wnl.0000198586.85250.13. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Villanueva M, Haynes CL, Seipel AT, Buhler LA, Wightman RM. Catecholamine exocytosis is diminished in R6/2 Huntington's disease model mice. J Neurochem. 2007;103:2102–2110. doi: 10.1111/j.1471-4159.2007.04908.x. [DOI] [PubMed] [Google Scholar]
- Joshi PR, Wu NP, André VM, Cummings DM, Cepeda C, Joyce JA, Carroll JB, Leavitt BR, Hayden MR, Levine MS, Bamford NS. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci. 2009;29:2414–2427. doi: 10.1523/JNEUROSCI.5687-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T, Matsunari Y, Saraya T, Shimada K, O'Hara K, Kubo K, Wagner GC, Nakashima T. Methamphetamine-induced striatal dopamine release, behavior changes and neurotoxicity in BALB/c mice. Int J Dev Neurosci. 2000;18:521–530. doi: 10.1016/s0736-5748(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for symptomatic treatment in Huntington's disease. Cochrane Database Syst Rev. 2009:CD006456. doi: 10.1002/14651858.CD006456.pub2. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load. J Physiol. 2007;585:817–831. doi: 10.1113/jphysiol.2007.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Ortiz AN, Kurth BJ, Osterhaus GL, Johnson MA. Dysregulation of intracellular dopamine stores revealed in the R6/2 mouse striatum. J Neurochem. 2010;112:755–761. doi: 10.1111/j.1471-4159.2009.06501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presti MF, Gibney BC, Lewis MH. Effects of intrastriatal administration of selective dopaminergic ligands on spontaneous stereotypy in mice. Physiol Behav. 2004;80:433–439. doi: 10.1016/j.physbeh.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Maguire-Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington's disease patients. Ann Neurol. 1995;38:852–861. doi: 10.1002/ana.410380605. [DOI] [PubMed] [Google Scholar]
- Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta. 1993;1144:249–263. doi: 10.1016/0005-2728(93)90109-s. [DOI] [PubMed] [Google Scholar]
- Sanberg PR, Calderon SF, Giordano M, Tew JM, Norman AB. The quinolinic acid model of Huntington's disease: locomotor abnormalities. Exp Neurol. 1989;105:45–53. doi: 10.1016/0014-4886(89)90170-2. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Spielewoy C, Roubert C, Hamon M, Nosten-Bertrand M, Betancur C, Giros B. Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice. Behav Pharmacol. 2000;11:279–290. doi: 10.1097/00008877-200006000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Maidment NT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J Neurosci. 2007;27:7899–7910. doi: 10.1523/JNEUROSCI.1396-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Berardelli A, Rothwell JC, Day BL, Dick JP, Benecke R, Marsden CD. The coexistence of bradykinesia and chorea in Huntington's disease and its implications for theories of basal ganglia control of movement. Brain. 1988;111:223–244. doi: 10.1093/brain/111.2.223. [DOI] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rougé-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Rogers DA, Bissada N, Vogl AW, Hayden MR, Leavitt BR. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005a;14:1379–1392. doi: 10.1093/hmg/ddi147. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005b;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- Vetter JM, Jehle T, Heinemeyer J, Franz P, Behrens PF, Jackisch R, Landwehrmeyer GB, Feuerstein TJ. Mice transgenic for exon 1 of Huntington's disease: properties of cholinergic and dopaminergic pre-synaptic function in the striatum. J Neurochem. 2003;85:1054–1063. doi: 10.1046/j.1471-4159.2003.01704.x. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wu N, Cepeda C, Zhuang X, Levine MS. Altered corticostriatal neurotransmission and modulation in dopamine transporter knock-down mice. J Neurophysiol. 2007;98:423–432. doi: 10.1152/jn.00971.2006. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]