

A 49-year-old woman was admitted with chronic renal failure. Prior history included poor vision, with development of photophobia at 3 years. Retinitis pigmentosa was documented, resulting in legal blindness by age 40. At the age of 20, sensorineural hearing loss was found, progressing to deafness by age 33. A diagnosis of Usher syndrome was made. At the age of 27, she developed congestive heart failure; echocardiography showed dilated cardiomyopathy. Myocardial biopsy showed interstitial fibrosis and myocellular hypertrophy (Fig.1). Mild chronic renal failure (serum creatinine 1.5 mg/dl; creatinine clearance 45) was found with negative urinalysis. At the age of 42, insulin resistance was first documented.
Fig. 1.
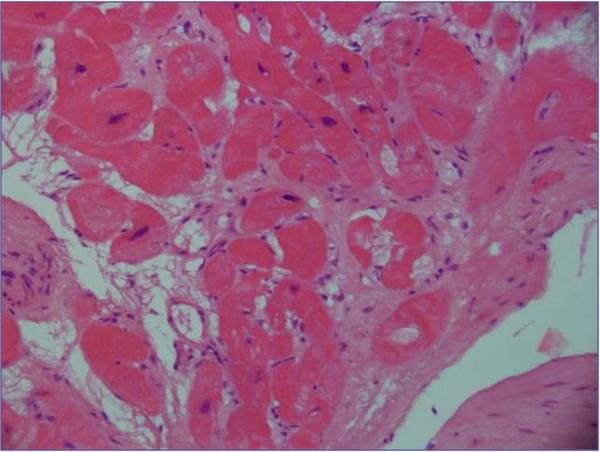
Endomyocardial biopsy showing severe interstitial fibrosis and moderate myocellular hypertrophy (Hematoxylin & Eosin; Original magnification × 20)
At admission, her weight was 63 Kg, height 145 cm, and body mass index was 30 kg/m2. Echocardiography confirmed dilated cardiomyopathy. She had normal intelligence. Serum creatinine was 1.6 mg/dl; creatinine clearance 38 ml/min; urinalysis negative; urinary osmolality 360 mosmol/kg. Ultrasonography revealed two small kidneys with a few bilateral cysts. Renal biopsy showed interstitial fibrosis, tubular atrophy and normal glomeruli (Fig. 2).
Fig. 2.
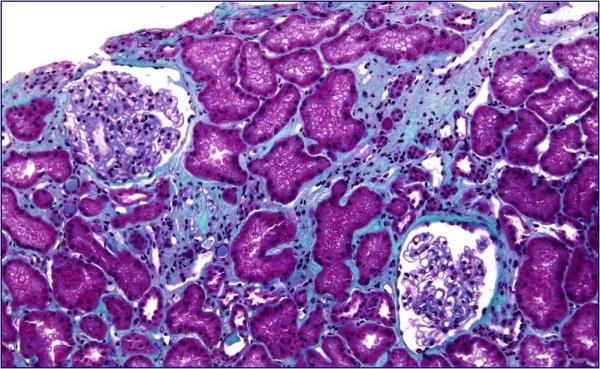
Renal biopsy showing focal areas of interstitial fibrosis and tubular atrophy (Hematoxylin & Eosin; Original magnification × 20)
Family characterization (Fig. 3) revealed unaffected parents; one phenotypically normal sister who died for breast cancer; one brother who died perinatally from congenital heart disease; one 43-year-old brother with retinitis pigmentosa, sensorineural deafness, hepatic fibrosis, and adult-onset dilated cardiomyopathy. He had chronic renal failure (serum creatinine 1.4 mg/dl; creatinine clearance 55 cc/min) with negative urinalysis. Ultrasound revealed small kidneys and hepatosplenic enlargement. Liver rigidity, measured by transient elastography (Fibroscan), was significantly increased, being indicative of hepatic fibrosis.
Fig. 3.
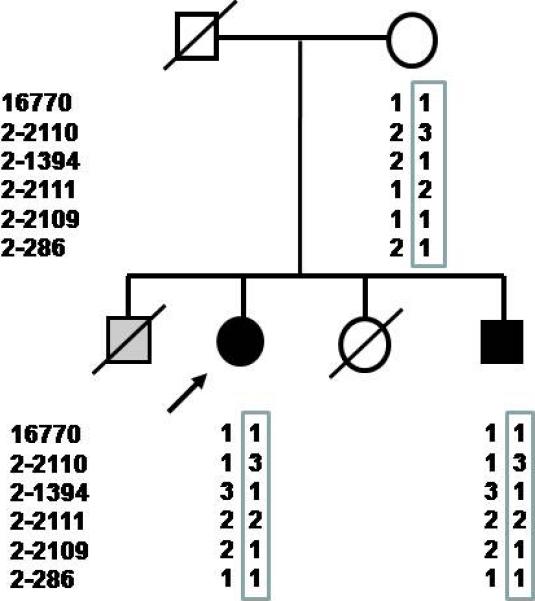
Haplotype of kindred surrounding the ALMS1 region. Arrow indicates the index case. Males and females are indicated by squares and circles, respectively; blackened circles and squares represent affected individuals; gray shading indicates a possible case of Alström syndrome with infantile congestive heart failure, but no other genetic or clinical information is available. Slash indicates deceased individuals from whom DNA was not available. Both affected individuals share a common haplotype defined by markers 16770, D2S210, D2S1394, D2S2111, D2S2109 and D2S286.
What is the likely cause of familial occurrence of retinitis pigmentosa, sensorineural deafness and nephropathy?
The Diagnosis: Alström syndrome, a rare ciliopathy with tubulo-interstitial renal involvement
The complex spectrum of phenotypes of our patients is consistent with those described for ciliopathies, genetic disorders resulting from dysfunction of cilia usually involving the kidneys (1). The combination of tubulointerstitial nephritis, retinitis pigmentosa and liver fibrosis can be observed in nephronophthisis, a ciliopathy causing the most frequent hereditary tubulointerstitial nephritis (2). However, metabolic abnormalities are not typical features of nephronophthisis, while they are part of the clinical spectrum of two pleiotropic ciliopathies, Bardet-Biedl (BBS) and Alström syndrome (AS) (1). Mutational analysis of the known variants in BBS 1-12 genes (microarray analysis; Asper Biotech, Estonia) did not reveal mutations. Sequencing analysis of all exons of the ALMS1 gene revealed in both patients a heterozygous frameshift mutation (S524fsX535) inherited from the mother. Haplotype analysis found identical haplotypes in the 2p13 ALMS1 region (Fig. 3).
AS is a rare recessive ciliopathy caused by mutations in ALMS1, a novel gene of unknown function. The major phenotypes are observed in young children; others emerge as the child develops. The diagnosis, relying primarily on clinical findings, can be confirmed by molecular testing. The diagnosis is proven when two mutations are identified. However, two disease-causing mutations are not easily identified, suggesting that some patients can harbor mutations in the promoter/intronic regions (3-5). In order to help the clinicians in making an early diagnosis, a set of age-related diagnostic criteria have been proposed (5). Our patients fulfilled the diagnostic criteria for adults AS, showing a combination of two major criteria (retinitis pigmentosa and ALMS1 mutation in one allele) and four minor criteria (sensorineural hearing loss, hepatic fibrosis, dilated cardiomyopathy, renal disease).
Renal disease develops with age in all individuals with AS. Onset occurs in early adolescence/adulthood, with polyuria and polydipsia. Renal insufficiency leading to end-stage renal disease occurs in 50% of patients. Histopathologic changes include interstitial fibrosis and tubular atrophy. Similar fibrotic infiltrations underlie many of the other clinical phenotypes, particularly cardiac, pulmonary and hepatic disease, suggesting a common pathogenic mechanism (3-5). The mechanisms of the induction of fibrosis are unknown. Further insights into the function of the ALMS1 protein will contribute to elucidate this process, increasing the knowledge of the molecular mechanisms involved in fibrogenesis.
Tubulointerstitial nephritis due to AS is a challenging diagnosis. A high index of suspicion should be maintained in patients presenting with familial recessive tubulointerstitial renal disease, retinal degeneration and sensorineural hearing loss. Presence of hepatic, cardiac involvement and metabolic abnormalities should raise the level of suspicion.
References
- 1.Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrandt F, Zhou W. Nephronophthisis-Associated Ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- 3.Marshall JD, Bronson R, Collin G, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med . 2005;165:675–83. doi: 10.1001/archinte.165.6.675. [DOI] [PubMed] [Google Scholar]
- 4.Minton JA, Owen KR, Ricketts CJ, et al. Syndromic obesity and diabetes: changes in body composition with age and mutation analysis of ALMS1 in 12 United Kingdom kindreds with Alström syndrome. J Clin Endocrinol Metab. 2006;91:3110–3116. doi: 10.1210/jc.2005-2633. [DOI] [PubMed] [Google Scholar]
- 5.Marshall JD, Beck S, Maffei P, Naggert JK. Alström Syndrome. Eur J Hum Genet. 2007;15:1193–1202. doi: 10.1038/sj.ejhg.5201933. [DOI] [PubMed] [Google Scholar]