

Abstract
Cyclin-dependent kinase 5 (CDK5), a neuronal kinase that functions in migration, has been found to be activated in some human cancers where it has been implicated in promoting metastasis. In this study, we investigated the role of CDK5 in pancreatic cancers where metastatic disease is most common at diagnosis. CDK5 was widely active in pancreatic cancer cells. Functional ablation significantly inhibited invasion, migration and anchorage-independent growth in vitro, and orthotopic tumor formation and systemic metastases in vivo. CDK5 blockade resulted in profound inhibition of Ras signaling through its critical effectors RalA and RalB. Conversely, restoring Ral function rescued the effects of CDK5 inhibition in pancreatic cancer cells. Our findings identify CDK5 as a pharmacologically tractable target to degrade Ras signaling in pancreatic cancer.
Keywords: Pancreatic cancer, cyclin-dependent kinase 5 (CDK5), p35, RalA, RalB, metastasis
Introduction
Pancreatic cancer is one of the most lethal human malignancies, with an estimated 34,290 deaths in the United States in 2008 (1). Despite extensive efforts aimed at developing improved therapeutic strategies, the dismal five-year overall survival rate of ∼4% has not significantly improved over the last five decades. Therefore, the quest for potent targeted therapeutic approaches for pancreatic cancer remains an imperative clinical issue.
Cyclin-dependent kinase 5 (CDK5) has ∼60% structural identity with cyclin-dependent kinases. Nevertheless, its moniker as a “cyclin-dependent kinase” is somewhat misleading; CDK5 has no known cell cycle or mitotic function, and it is not activated by cyclins (2). Instead, CDK5 is activated by association with either of two obligate CDK5-specific activator proteins, p35 (CDK5R1) or p39 (CDK5R2) (3, 4). Thus, while CDK5 is ubiquitously expressed, the restricted expression pattern of the obligate activator proteins confines physiologic CDK5 function to a limited repertoire of tissues, most prominently in cells of neuronal origin; expression of either p35 or p39 is synonymous with CDK5 kinase activity. Homozygous deletion of the Cdk5 gene in mice has shed light on the critical functions of CDK5 in brain development (5). Specifically, Cdk5-/- mice die perinatally, with a remarkable inversion of the six layers of the cerebral cortex, suggesting a global abnormality of neuronal migration. Misexpression of the CDK5 activator p35 (and more specifically, its proteolytic C-terminal fragment p25) leads to dysregulation of CDK5 function, and promotes neurofibrillary tangles and plaque formation, both hallmarks of Alzheimer disease (6, 7).
Given the numerous parallels between neuronal migration during embryogenesis and the migration of cancer cells from their primary site during metastasis (8, 9), a requirement for CDK5 in the latter phenomenon might be envisioned. Over the past decade, the compendium of extra-neuronal functions of CDK5 has expanded (10). We previously identified an association between CDK5 and human cancer, with the demonstration that inhibition of CDK5 activity inhibits prostate cancer metastases in experimental models (11). These observations were predominantly empirical, and the intracellular mechanisms linking CDK5 activity to cancer progression remained essentially unknown. Here we demonstrate that CDK5 blockade not only inhibited pancreatic cancer migration in vitro and metastases in vivo, but also downregulated anchorage independent growth and primary tumor engraftment in xenograft models, suggesting a more pervasive requirement for sustained CDK5 signaling on pancreatic tumorigenesis. We identify a novel Ras-CDK5-Ral signaling axis in pancreatic cancer that provides a unique therapeutic opportunity to target mutant Ras in cancer cells through inhibition of a key downstream effector pathway. Given the central role of Ral GTPases in both tumor initiation and tumor progression in Ras-driven neoplasia (12, 13), this study establishes CDK5 as a bona fide therapeutic target in pancreatic cancer, a malignancy with greater than 90% frequency of KRAS2 gene mutations (14-16).
Materials and Methods
Cell lines and constructs
Pancreatic cancer cell lines (PK-9, SW1990, Su86.86, BxPC-3, MIAPaCa-2, PANC-1, AsPc-1, CFPAC-1) were obtained from the American Type Culture Collection, while the low-passage cell lines (Pa16C, Pa03C, Pa20C, Pa18C, and Pa04C) were generated at our institution, as recently described (14). All cancer cell lines were cultured as previously described (17). The generation and culture of hTERT-immortalized HPNE cells has previously been described (18).
RNA extraction, reverse transcription and quantitative real-time PCR
RNA extraction from tissue samples or cultured cells was done as described previously (19).
Migration and wound healing assays
In vitro wound healing and migration assays were performed as previously described (11, 19).
Soft agar assays
Soft agar assays were performed as described previously (19).
Western blotting
Western blot analyses were performed as described previously (11).
Kinase assays
CDK5 in vitro kinase activity was determined as previously described (11).
Immunofluorescence
Immunofluorescence analysis was performed as previously described (20) with minor modifications.
Ral, Rho and Rac activation assays
RalA-GTP and RalB-GTP levels were determined using the RalA or RalB activation assay kit (both Upstate, Temecula, CA, USA) following the standard procedure recommended by the manufacturer.
Subcutaneous and orthotopic xenografts
Generation of subcutaneous and orthotopic xenografts of pancreatic cancer has been described previously (19, 21-24).
Statistical analysis
Kruskal-Wallace analysis and chi-square test were performed using SPSS version 15.0.1.1 (SPSS Inc., Chicago, IL, USA) for Microsoft Windows, two-tailed t-test and Mann-Whitney-U-test were performed using Prism (GraphPad Software Inc., San Diego, CA, USA) version 5.01. P<0.05 was regarded to be statistically significant. Bar diagrams were generated using Prism version 5.01 and show means and standard errors if not otherwise indicated.
Results
The CDK5 obligate activator p35 is commonly expressed in human pancreatic cancer cells
Western blot analysis showed p35 protein expression in all thirteen tested pancreatic cancer cell lines as well as in the non-neoplastic human pancreatic hTERT-HPNE cells. Higher expression of p35 was observed in 9/13 pancreatic cancer cell lines, while the remaining four cell lines showed similar basal expression as compared to hTERT-HPNE (Figure 1A). Expression of p35 transcripts was confirmed at the mRNA level in 8/8 pancreatic cell lines tested (Figure 1B).
Figure 1. Expression of CDK5 and the CDK5 activator protein, p35, in pancreatic cancer cells.

(A) Expression of both CDK5 and its activator protein p35 was observed in all 13 cancer cell lines examined, although in three of them (SW1990, Su86.86, BxPC-3) the expression levels of p35 were miminal, comparable to the immortalized non-malignant human pancreatic cell line hTERT-HPNE. (B) RT-PCR analysis revealed expression of p35 mRNA transcripts in 8/8 tested pancreatic cancer cell lines. No-RT preparations served as negative controls.
Blockade of endogenous CDK5 function inhibits pancreatic cancer cell migration and anchorage independent growth
To determine the phenotypic impact of CDK5 blockade in pancreatic cancer, we generated MIAPaCa-2 cells with enforced expression of a dominant-negative CDK5 (dnCDK5) protein. The dnCDK5 construct is a D144N kinase-dead mutant that is expressed in a bi-directional vector, pBI-EGFP, which also expresses enhanced green fluorescent protein (EGFP) (11). The pBI-EGFP dnCDK5 and pBI-EGFP empty vectors were stably selected in MIAPaCa-2 cells and two independent clones (‘CDK5-dn1’ and ‘CDK5-dn2’) with high level of expression were picked (Figure 2A). Blockade of endogenous CDK5 had little to no effect on cell number over 96 hours, as assessed by the MTT assay. In contrast, there was a profound inhibition of cell migration at 48 and 72 hours, as assessed by an in vitro modified Boyden chamber assay, in the two dnCDK5 clones (Figure 2B). Wound healing assays performed in vector versus dnCDK5-expressing MIAPaCa-2 cells confirmed the inhibitory effects of CDK5 blockade on cell motility (Supplementary Figure 1A and B). Of note, siRNA-mediated CDK5-knockdown also significantly reduced migration of BxPC-3 cells (not shown). These results were similar to those observed in prostate cancer cells with knockdown of endogenous CDK5 function (11). Remarkably, we additionally observed a dramatic inhibition of anchorage independent growth in the MIAPaCa-CDK5dn clones compared to the vector-only clones (Figure 2C). This inhibition of anchorage independent growth suggested that CDK5 might play a role in pancreatic tumorigenesis beyond what has been reported in the context of prostate cancer, where no significant effects on clonogenicity or tumor formation were observed. Abrogation of CDK5 function by expression of the dominant negative CDK5 allele caused significant changes in cell morphology as demonstrated by immunofluorescence labeling of the cell skeleton (Figure 2D). While vector transfected cells mostly showed an almost spindle-like shape with a clearly defined leading edge and microtubule-organizing centers (MTOC) between the leading edge and nucleus, MIAPaCa-2 cells stably expressing the CDK5-dn allele were more round in shape and mostly lacked a clearly defined leading edge and MTOC.
Figure 2. Dominant negative form of CDK5 inhibits migration and anchorage independent growth of pancreatic cancer cells.
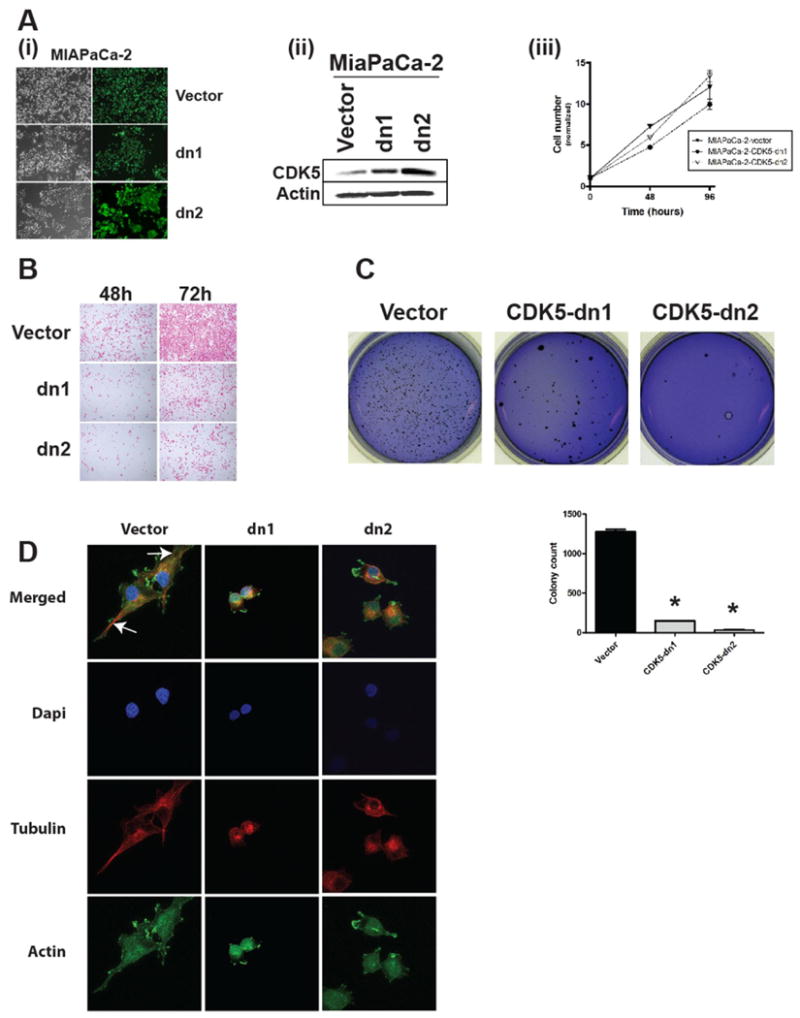
(A, B) Cells expressing the pBI-EGFP vector (either empty or expressing D144N dnCDK5) were confirmed by (A) EGFP expression and (B) Western blot analysis for CDK5 levels in MIAPaCa-2 cells. (C) Inhibition of CDK5 activity had no effect on MIAPaCa-2 cell growth on plastic, as assessed by MTS assay in empty versus the two dnCDK5-expressing clones (CDK5-dn1 and CDK5-dn2). (D) In contrast, modified Boyden chamber assays showed marked reduction of in vitro migration in both dnCDK5 clones compared to vector-transfected cells at 48 and 72 hours, respectively. (E) Colony formation in soft agar was also significantly reduced in both dnCDK5 clones compared to vector-transfected cells. Bar diagrams show means and standard errors of the respective colony counts; * indicates P<0.05 as compared to vector-transfected controls. (F) Immunofluorescence staining demonstrated striking differences in cell morphology in the dnCDK5 versus vector-transfected MIAPaCa-2 cells. While vector-transfected cells exhibit clear polarity with microtubule-organizing centers (MTOC) (arrows) between the nucleus and leading edge of the cell, dnCDK5-expressing MIAPaCa-2 cells lack polarity and exhibit a rounded cell shape.
To corroborate these observations in additional pancreatic cancer models, we used five pancreatic cancer cell lines with activating KRAS2 mutations (CFPAC1, PANC-1, Pa16C, Pa03C and Pa20C), besides MIAPaCa-2 cells. Further, we utilized a complementary genetic strategy for antagonizing CDK5 function, RNA interference (RNAi) by enforced expression of a lentiviral short hairpin RNA (shRNA) directed against CDK5. Stable CDK5 RNAi was validated by reduction in endogenous protein level compared to scrambled shRNA expressing cells (Figure 3A). In vitro modified Boyden chamber assays confirmed that, analogous to the dnCDK5-expressing MIAPaCa-2 cells, CDK5 RNAi decreases the migratory abilities of pancreatic cancer cells to a variable extent in five of six of tested samples (Figure 3B and Supplementary Figure 2). Cell viability (MTS) assays confirmed the absence of any significant reduction in net cell growth upon CDK5 knockdown (Supplementary Figure 3). We then assessed the consequence of CDK5 RNAi on anchorage independent growth. Similar to the effects seen in CDK5-dn MIAPaCa-2 cells, we observed significant inhibition of anchorage independent growth upon RNAi-mediated CDK5 knockdown in four of six cell lines (Figure 3C and Supplementary Figure 2B). Thus, in the majority of KRAS2-mutant pancreatic cancer cell lines, loss of CDK5 function was associated with no significant effects on cell viability, while inhibiting cell migration as well as anchorage independent growth.
Figure 3. Knockdown of CDK5 expression by RNA-mediated interference in KRAS2 mutant pancreatic cancer cells blocks migration and inhibits anchorage independent growth.
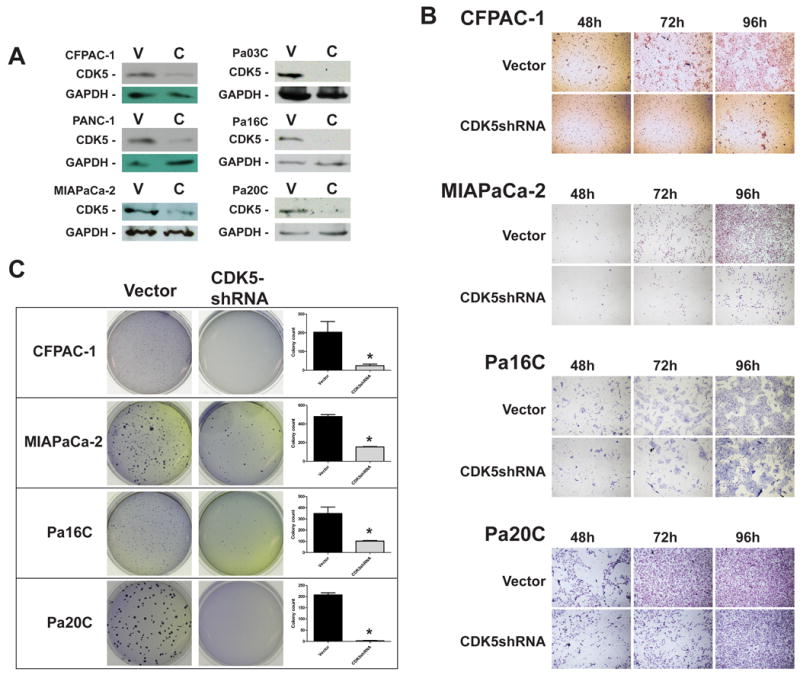
(A) Western blot analysis confirmed successful knockdown of CDK5 expression in CFPAC-1, PANC-1, MIAPaCa-2, Pa03C, Pa16C and Pa20C cells transfected with lentiviral CDK5-shRNA (designated ‘C’) as compared to lentiviral vector transfected cells (‘V’). (B) Modified Boyden chamber assays confirmed variable decrease of in vitro migration upon CDK5 downregulation, with the most pronounced effects observed in CFPAC-1 and MIAPaCa-2 cells. (C) CDK5 RNAi also significantly inhibited colony formation in soft agar in all four KRAS2 mutant cell lines. Bar diagrams show means and standard errors of the respective colony counts; * indicates P<0.05 as compared to empty vector-transfected controls.
In parental MIAPaCa-2 cells, immunoprecipitation-kinase assays confirmed significant CDK5 enzymatic activity in pancreatic cancer cells, and this activity could be blocked by exposure to the CDK inhibitor roscovitine (Supplementary Figure 4A) or by expression of a CDK5dn allele (Supplementary Figure 4B). Consistent with the effects observed with genetic inhibition of CDK5 function, roscovitine treatment decreased the number of migrating cells in an in vitro Boyden chamber assay (Supplementary Figure 5).
Inhibition of CDK5 function inhibits tumor growth and metastasis in an orthotopic xenograft model of human pancreatic cancer
Having established a role for sustained CDK5 activity on cell motility and anchorage independent growth of pancreatic cancer cells, we next examined effects of CDK5 inhibition in an in vivo orthotopic xenograft model. Two independent MIAPaCa-2 clones expressing a dominant negative allele of CDK5 (‘dn1’ and ‘dn2’) were used for these studies, and four mice were injected per group. At six weeks post-injection, the mice were euthanized and a comprehensive necropsy was performed for assessment of primary and metastatic tumor load. The orthotopic primary xenografts established from both independent dnCDK5 clones were significantly smaller compared to the vector -transfected MIAPaCa-2 xenografts (Figures 4A and B). Other than a reduction in overall viable cell density, there were no histological differences in the tumor microarchitecture between xenografts from vector- versus CDK5-dn transfected MIAPaCa-2 cells (Figure 4C). Further, necropsy confirmed a significant reduction in frequency of metastases to regional lymph nodes, spleen, liver, kidneys, lungs, or intestine in both sets of mice harboring dnCDK5 clones, compared to vector-transfected MIAPaCa-2 xenografts (Figure 4D; Supplementary Figure 5D) (8/48 (17%) versus 11/24 (46%) affected organ sites; P=0.008). Ascites was observed in 4/4 (100%) of mice in the control group, but only in 2/8 (25%) of mice carrying dnCDK5 xenografts (NS; P=0.061). Similar results were obtained, when shRNA-mediated knockdown of CDK5 in Pa20C cells was used as a complementary orthotopic xenograft model system (Supplementary Table 1 and Supplementary Figure 6). Thus, consistent with the phenotypes observed in vitro, loss of CDK5 function in pancreatic cancer cells is associated with significantly decreased tumorigenicity and systemic metastases in vivo.
Figure 4. CDK5 inhibition inhibits primary tumor growth and systemic metastases in an orthotopic xenograft model of pancreatic cancer.
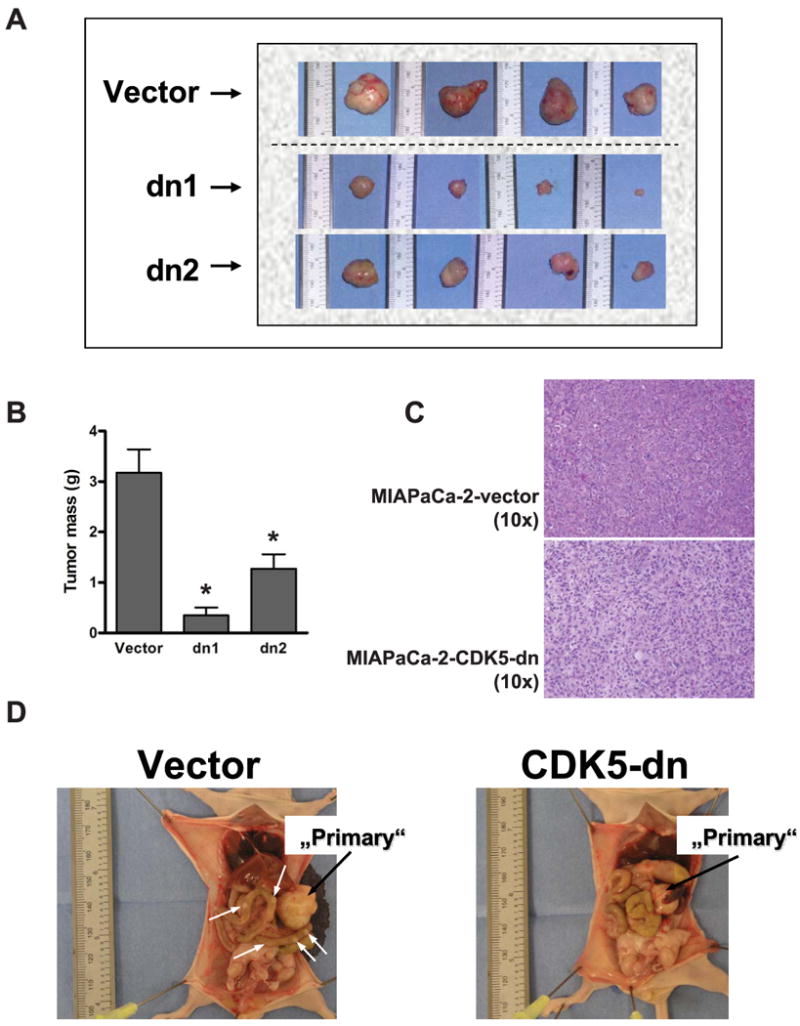
(A) Orthotopic xenografts were generated in athymic mice by intra-pancreatic injection of MIAPaCa-2 cells expressing empty pBI-EGFP vector, or dnCDK5 (clones dn1 and dn2). The resulting primary tumors were universally smaller in both dnCDK5 clones compared to vector-transfected cells. (B) The average primary tumor mass (grams) was significantly lower in the two cohorts with loss of CDK5 function compared to vector-transfected cells. (C) Other than a decrease in viable cell density, the reduction in primary tumor mass was not accompanied by any obvious changes in the tumor architecture or extent of differentiation, as assessed by H&E staining on light microscopy. (D) Reduction in primary tumor mass in the dnCDK5 clones was accompanied by inhibition of systemic metastases compared to vector-transfected cells. (White arrows show metastatic foci; * indicates p<0.05 as compared to vector transfected controls.)
CDK5 blockade inhibits the Ral effector pathway central to oncogenic Ras signaling
These data showed that in pancreatic cancer, loss of CDK5 function interferes with the malignant phenotype, affecting both tumor formation (anchorage independent growth in vitro and orthotopic engraftment in vivo) and tumor progression (cell migration in vitro and systemic metastases in vivo). It was important to begin to identify the signal transduction pathways underlying this reduced tumorigenicity. Previously, Counter and colleagues have demonstrated the critical and divergent roles of the Ral effector pathway in oncogenic Ras-induced neoplasia, using pancreatic cancer as a prototype (12, 13). Specifically, these investigators showed that loss of RalA function can inhibit tumorigenicity across a spectrum of KRAS2-mutant pancreatic cancer cell lines, while sustained RalB activity appeared to be a critical determinant of tumor progression and metastasis. Those studies demonstrated that the Ral pathway is a central effector of Ras-mediated malignant properties in pancreatic cancer. Therefore, we explored whether CDK5 function might be important for activity of the Ras-Ral effector pathway axis in pancreatic cancer, as a basis for the observed phenotypes in our model system. Indeed, we found that the activated forms of both Ral proteins, RalA-GTP and RalB-GTP, were profoundly reduced in the dnCDK5-expressing MIAPaCa-2 clones, across multiple independent experiments (Figure 5A). Rho-GTP and Rac-GTP levels were also found to be decreased in the CDK5-dn clones. Similarly, striking downregulation of the activated Ral-GTP moieties was seen upon RNAi knockdown of CDK5 in five of the six tested KRAS2-mutant pancreatic cancer cell lines (Figure 5B). Of note, Pa03C, which demonstrated neither loss of anchorage independent growth nor decrease in migration, retained activation of both Ral proteins, underscoring the importance of this effector pathway in the observed phenotype(s). We also examined the effects of roscovitine on Ral-GTP levels, and marked reduction of activated RalA-GTP was observed in all three examined KRAS2-mutant lines (MIAPaCa-2, CFPAC-1, and Panc-1), while RalB-GTP was downregulated in all but MIAPaCa-2 cells (Figure 5C). Thus, these experiments confirmed that in the majority of pancreatic cancer cells with constitutively activated Ras signaling, CDK5 inhibition can block the activation of a critical effector pathway implicated in tumor initiation and progression.
Figure 5. Genetic or pharmacological blockade of CDK5 function is associated with Ral pathway inhibition in multiple KRAS2 mutant pancreatic cancer cells.
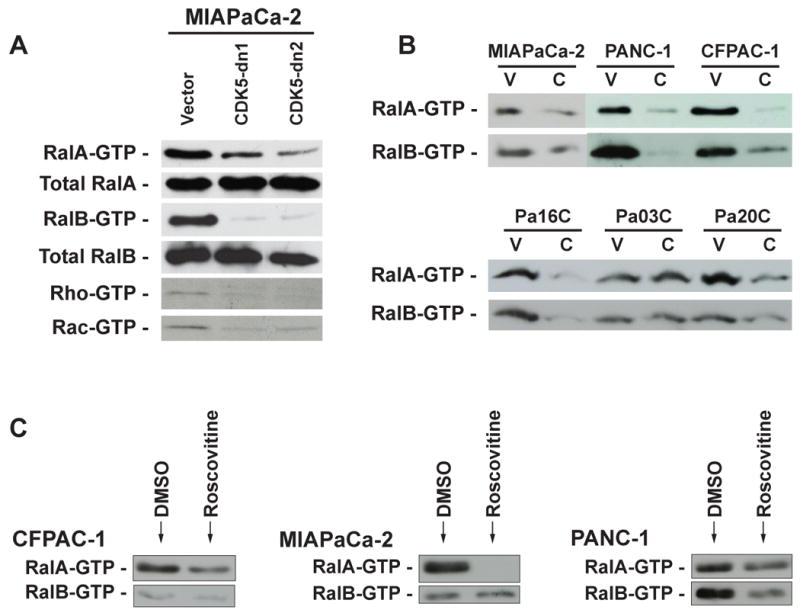
(A) Reduction in the endogenous level of activated RalA-GTP, RalB-GTP, Rho-GTP and Rac-GTP was observed in MIAPaCa-2 cells expressing dnCDK5 as compared to vector transfected cells. (B) RalA and RalB activation assays of six KRAS2 mutant pancreatic cancer cell lines transfected with CDK5shRNA (‘C’) compared to vector-only transfected controls (‘V’) confirms downregulation of the active GTP-bound protein in five of six cell lines, with Pa03C being the exception. (C) RalA and RalB activation assays in a subset of three pancreatic cancer cell lines treated with roscovitine in vitro for 30 minutes as compared to DMSO-treated cells demonstrates reduction of GTP-bound RalA in all three cell lines, and reduction of GTP-bound RalB in two of three cell lines.
In order to further corroborate that downregulation of Ral signaling is indeed an underlying cause of the phenotypic changes in pancreatic cancer cells observed upon inhibition of endogenous CDK5 function, we attempted to rescue the malignant phenotype by constitutively activating RalA and RalB in pancreatic cancer cells expressing dnCDK5. Therefore, we generated dnCDK5 MIAPaca-2 clones with enforced expression of a constitutively active form (Rlf-CAAX) of the Ral guanine exchange factor (RalGEF), Rgl2 (25). This RalGEF can activate RalA and RalB, independent of signaling from oncogenic Ras. Rlf-CAAX expression restored RalA-GTP and RalB-GTP levels in multiple independent dnCDK5 MIAPaCa-2 clones (Figure 6A), and significantly “rescued” the phenotype of anchorage independent growth, compared to vector transfected dnCDK5 clones (Figure 6B and C). Of note, in vivo tumorigenicity was also partially rescued, as demonstrated using subcutaneous xenografts. While vector-transfected CDK5dn-expressing MIAPaCa-2 cells (‘MIAPaCa-2-CDK5dn1-control”) formed xenografts in only 2/7 (29%) mice, MIAPaCa-2-CDK5dn1-Rgl2-CAAX cells formed xenografts in 7/7 (100%) mice (Figure 6). Thus, our results place CDK5 as an essential signaling intermediate in the Ral effector pathway, downstream of oncogenic Ras. The precise mechanism by which CDK5 modulates Ral activation remains a matter of investigation; nonetheless, this result indicates that CDK5 inhibition blocks the malignant properties of pancreatic cancer cells by disrupting an obligate Ras-CDK5-Ral signaling axis.
Figure 6. Overexpression of Rgl2-CAAX partially rescues the phenotype of anchorage independent growth and tumorigenecity in CDK5-dn MIAPaCa-2 cells.
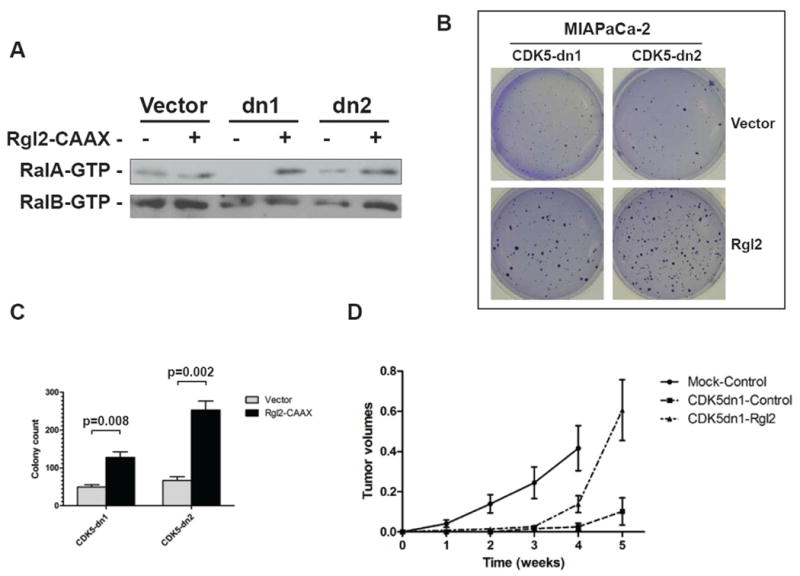
(A) Enforced expression of Rgl2-CAAX restituted GTP-bound RalA protein levels in MIAPaCa-2 clones with blockade of endogenous CDK5 function (dn1 and dn2). (B, C) Restoration of RalA activity was associated with rescue of the anchorage independent growth phenotype in both dnCDK5 clones, compared to vector-transfected cells. (D) In vivo tumorigenecity was also partially rescued as observed in murine xenograft experiments.
Blockade of Ral effector function through CDK5 inhibition cooperates with inhibition of RAF/MEK/ERK and PI3K/Akt pathways in limiting pancreatic cancer growth
While the Ral signaling pathway has been shown to be critical for Ras-driven human tumorigenesis, other Ras effector pathways, including the RAF/MEK/ERK and PI3K/Akt pathways, also are important in human cancer ((26); Supplementary Figure 7A). In pancreatic cancer cells, it has been shown that inhibitors of the Ras effector pathways RAF/MEK/ERK and PI3K/Akt could induce G0/G1 cell cycle arrest, and, in some instances, apoptosis (27, 28). Therefore, we reasoned that inhibition of the Ral pathway, via blockage of CDK5 activity, in combination with inhibition of either the RAF/MEK/ERK or PI3K/Akt pathway, might result in a more pronounced effect on the transformed properties of Ras-driven pancreatic cancer cells. To test this possibility, we compared the effect of the MEK inhibitor U0126 and the PI3K inhibitor LY294002 in MIAPaCa-2 cells containing the previously described dnCDK5 for inhibition of CDK5, or vector-only control cells. The combination of either small molecule antagonist at 10 μM concentration with CDK5 inhibition resulted in marked decrease in anchorage independent growth compared to blockade of any one Ras effector pathway alone, and this effect was even more accentuated when all three major downstream effectors of Ras were inhibited simultaneously, with essentially no visible colony formation in soft agar (Supplementary Figure 7B and C). Similar results were obtained when the Raf inhibitor sorafenib was used instead of U0126 to block signaling through the RAF/MEK/ERK axis (Supplementary Figure 8). Thus, Ral inhibition mediated by CDK5 blockage appears to cooperate with blockade of other Ras effector pathways, further reiterating its potential as a bona fide therapeutic target in pancreatic cancer.
Discussion
In the current study, we show that inhibition of CDK5 results in suppression of cancer growth and metastatic progression in preclinical models of pancreatic cancer. It is noteworthy that the levels of p35 in pancreatic cancer are not uniformly higher than those in non-neoplastic pancreatic epithelial cells. In addition, enforced overexpression of p35 above the observed endogenous levels in MIAPaCa-2 cells led to slightly increased in vitro cell migration but did not further enhance colony formation in soft agar of xenograft growth in athymic mice (data not shown). This suggests that the relative level of CDK5 activity, over some threshold, may not further increase tumorigenicity, and that CDK5 activity is necessary, although not sufficient, for pancreatic tumor growth and progression.
These results have significant therapeutic implications for this almost uniformly lethal disease. The potential for therapeutic application is especially relevant to pancreatic cancer, since our data indicate that CDK5 signaling is critical for Ras signaling through the Ral pathway, which has emerged as an important determinant of the malignant phenotype in pancreatic cancer cells (12, 13). The Ras pathway is activated by KRAS2 gene mutations in greater than 90% of cases of pancreatic cancer (14), and KRAS2 mutations are among the earliest genetic aberrations observed in low-grade PanIN lesions during the multistep progression model leading to fully invasive pancreatic cancer (29). The striking frequency and early onset of this mutation strongly indicate that Ras signaling is central to the malignant phenotype in pancreatic cancer, and identify Ras as a prime therapeutic target in this disease. Indeed, in preclinical studies, disruption of KRAS2 function via RNA interference, antisense DNA, or expression of dominant negative KRASN17 attenuates the tumorigenicity of PDAC cell lines (30-32). Unfortunately, strategies to inhibit the Ras pathway directly in patients have been largely unsuccessful, as exemplified by the lack of clinical activity of farnesyltransferase inhibitors (FTIs), which interfere with critical post-translational modification of RAS proteins (33). Similarly, targeting downstream effectors of the Ras pathway has not yet been therapeutically effective [reviewed by (34)]. In this context, one may speculate that inhibition of specific Ras effector pathways may be ineffective because either a) the specific targeted pathways are not critical for pancreatic cancer, or b) the multiple signal transduction pathways activated by Ras provide some redundancy, necessitating simultaneous inhibition of more than one effector pathway.
Given the impediments in therapeutic targeting of Ras effector pathways, our finding that CDK5 activity is required for Ral activation may be quite significant for pancreatic cancer. In prior reports by Counter and colleagues (12, 13), inhibition of RalA signaling resulted in abrogation of the malignant phenotype in KRAS2-mutant pancreatic cancer cell lines, while downregulation of RalB inhibited experimental (tail vein injection) metastasis. These findings underscore the Ral pathway as an important therapeutic target for pancreatic cancer. Nevertheless, like other GTP-binding proteins in the Ras family, Ral has been difficult to inhibit, although there has been recent progress in use of a geranylgeranyltransferase inhibitor in vitro, for blocking Ral membrane localization (35). Our data, demonstrating that CDK5 inactivation results in inhibition of Ral activation, identify a facile, druggable kinase target to access this important effector arm of the Ras signal transduction pathway. Inhibition of CDK5 function alone in pancreatic cancer cells is sufficient to abrogate the activation of both Ral proteins, which may account for the observed phenotype of decreased tumorigenicity (through RalA inhibition) as well as reduced systemic metastases (through the RalB effector). Further, our experiments employing CDK5 inhibition in combination with inhibition of other Ras effector pathways (Supplementary Figures 7 and 8) suggest that such a combination strategy may be especially effective in pancreatic cancer. It is significant that CDK5 inhibitors are already in pharmaceutical development (36, 37), in part since CDK5 has been considered a potential therapeutic target for neurodegenerative diseases (reviewed by (38, 39)); therefore, development of CDK5 as a therapeutic target for pancreatic cancer could be relatively rapid.
The molecular mechanism by which CDK5 activity regulates Ral activation will be a fascinating focus of future study. As is the case for other GTP-binding proteins in the Ras superfamily, the level of Ral activation corresponds to the level of GTP occupancy of the Ral proteins, which is proximally controlled by the action of guanine nucleotide exchange factors (RalGEFs, which load GTP) and GTPase activating proteins (RalGAPs, which promote GTP hydrolysis to GDP). There are five known RalGEFs; in contrast, while RalGAP activity has been detected in cells (40, 41), a specific RalGAP has only recently been identified (42). Activated Ras can activate Ral by recruiting RalGEFs to the cell membrane, where their nucleotide exchange function is activated allosterically (43). One may envision, for example, that CDK5 may participate in the activation of Ral by direct phosphorylation of Ral or a RalGEF, or indirectly, by phosphorylating an interacting protein, thus promoting guanine nucleotide exchange or inhibiting GTPase activity. Alternatively, CDK5 may phosphorylate or otherwise deactivate a RalGAP or interacting protein, inhibiting Ral GTPase activity and increasing GTP occupancy. Control of other GEFs and GAPs by such phosphorylation events has been extensively documented (reviewed by (44)). CDK5 itself has been reported to regulate the activity of modulating factors for other GTP binding proteins, by direct phosphorylation of the GEF RasGRF2 (45), release from sequestration of RhoGDI (46), or increased expression of RhoGAP (47), further suggesting that such mechanisms may underlie CDK5 regulation of Ral.
In summary, we show that inhibition of CDK5 activity antagonizes tumorigenic and metastatic properties of pancreatic cancer cells. CDK5 appears to inhibit Ras signaling in these cells, blocking the critical Ral effector arm of the Ras signal transduction pathway. Mutated Ras is central to tumorigenesis in the overwhelming majority of cases of pancreatic cancer, and therefore has been considered a prime therapeutic target for this disease. Unfortunately, both Ras and Ral have been virtually intractable therapeutically. Our demonstration that CDK5 activity is necessary for Ras pathway signaling and tumorigenic behavior in pancreatic cancer provides optimism that selective kinase inhibitors of CDK5 may finally allow us to successfully address Ras as a therapeutic target in pancreatic cancer.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by NIH R01CA134767 to BDN and AM. BDN was supported by NIH R01 CA085567, and grants from the Flight Attendant Medical Research Foundation (FAMRI) and the National Pancreas Foundation. AM was supported by the Sol Goldman Pancreatic Cancer Research Center, the Michael Rolfe Foundation for Pancreatic Cancer Research, and NIH R01CA113669. GF was supported in part by BONFOR grant number O-111.0001.2.
Footnotes
Conflicts of Interest: Nothing to declare.
Additional, more detailed descriptions of experimental procedures used in this work are provided as supporting online material.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA: a cancer journal for clinicians. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–59. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 3.Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–23. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 4.Lew J, Huang QQ, Qi Z, et al. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–6. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 5.Ohshima T, Ward JM, Huh CG, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:11173–8. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noble W, Olm V, Takata K, et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–65. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 7.Tsai LH, Lee MS, Cruz J. Cdk5, a therapeutic target for Alzheimer's disease? Biochimica et biophysica acta. 2004;1697:137–42. doi: 10.1016/j.bbapap.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Thiery JP, Boyer B, Tucker G, Gavrilovic J, Valles AM. Adhesion mechanisms in embryogenesis and in cancer invasion and metastasis. Ciba Found Symp. 1988;141:48–74. doi: 10.1002/9780470513736.ch4. [DOI] [PubMed] [Google Scholar]
- 9.Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007;7:246–55. doi: 10.1038/nrc2108. [DOI] [PubMed] [Google Scholar]
- 10.Rosales JL, Lee KY. Extraneuronal roles of cyclin-dependent kinase 5. Bioessays. 2006;28:1023–34. doi: 10.1002/bies.20473. [DOI] [PubMed] [Google Scholar]
- 11.Strock CJ, Park JI, Nakakura EK, et al. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer research. 2006;66:7509–15. doi: 10.1158/0008-5472.CAN-05-3048. [DOI] [PubMed] [Google Scholar]
- 12.Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer cell. 2005;7:533–45. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 13.Lim KH, O'Hayer K, Adam SJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385–94. doi: 10.1016/j.cub.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 14.Jones S, Zhang X, Parsons DW, et al. Science (New York, NY. 2008. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–54. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 16.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–82. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calhoun ES, Hucl T, Gallmeier E, et al. Identifying Allelic Loss and Homozygous Deletions in Pancreatic Cancer without Matched Normals Using High-Density Single-Nucleotide Polymorphism Arrays. Cancer research. 2006;66:7920–8. doi: 10.1158/0008-5472.CAN-06-0721. [DOI] [PubMed] [Google Scholar]
- 18.Lee KM, Yasuda H, Hollingsworth MA, Ouellette MM. Notch 2-positive progenitors with the intrinsic ability to give rise to pancreatic ductal cells. Lab Invest. 2005;85:1003–12. doi: 10.1038/labinvest.3700298. [DOI] [PubMed] [Google Scholar]
- 19.Feldmann G, Dhara S, Fendrich V, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer research. 2007;67:2187–96. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fendrich V, Waldmann J, Esni F, et al. Snail and Sonic Hedgehog activation in neuroendocrine tumors of the ileum. Endocr Relat Cancer. 2007;14:865–74. doi: 10.1677/ERC-07-0108. [DOI] [PubMed] [Google Scholar]
- 21.Dong J, Feldmann G, Huang J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–33. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feldmann G, Fendrich V, McGovern K, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7:2725–35. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mullendore ME, Koorstra JB, Li YM, et al. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res. 2009;15:2291–301. doi: 10.1158/1078-0432.CCR-08-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bisht S, Feldmann G, Koorstra JB, et al. In vivo characterization of a polymeric nanoparticle platform with potential oral drug delivery capabilities. Mol Cancer Ther. 2008;7:3878–88. doi: 10.1158/1535-7163.MCT-08-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–25. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 26.Hamad NM, Elconin JH, Karnoub AE, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–57. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer research. 2005;65:4870–80. doi: 10.1158/0008-5472.CAN-04-2848. [DOI] [PubMed] [Google Scholar]
- 28.Mirza AM, Gysin S, Malek N, Nakayama K, Roberts JM, McMahon M. Cooperative regulation of the cell division cycle by the protein kinases RAF and AKT. Molecular and cellular biology. 2004;24:10868–81. doi: 10.1128/MCB.24.24.10868-10881.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–88. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aoki K, Yoshida T, Sugimura T, Terada M. Liposome-mediated in vivo gene transfer of antisense K-ras construct inhibits pancreatic tumor dissemination in the murine peritoneal cavity. Cancer research. 1995;55:3810–6. [PubMed] [Google Scholar]
- 31.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer cell. 2002;2:243–7. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 32.Hirano T, Shino Y, Saito T, et al. Dominant negative MEKK1 inhibits survival of pancreatic cancer cells. Oncogene. 2002;21:5923–8. doi: 10.1038/sj.onc.1205643. [DOI] [PubMed] [Google Scholar]
- 33.Van Cutsem E, van de Velde H, Karasek P, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–8. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 34.Yeh JJ, Der CJ. Targeting signal transduction in pancreatic cancer treatment. Expert Opin Ther Targets. 2007;11:673–94. doi: 10.1517/14728222.11.5.673. [DOI] [PubMed] [Google Scholar]
- 35.Falsetti SC, Wang DA, Peng H, et al. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Molecular and cellular biology. 2007;27:8003–14. doi: 10.1128/MCB.00057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wan Y, Hur W, Cho CY, et al. Synthesis and target identification of hymenialdisine analogs. Chem Biol. 2004;11:247–59. doi: 10.1016/j.chembiol.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 37.Rzasa RM, Kaller MR, Liu G, et al. Structure-activity relationships of 3,4-dihydro-1H-quinazolin-2-one derivatives as potential CDK5 inhibitors. Bioorganic & medicinal chemistry. 2007;15:6574–95. doi: 10.1016/j.bmc.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends in molecular medicine. 2004;10:452–8. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nature reviews. 2007;6:464–79. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 40.Emkey R, Freedman S, Feig LA. Characterization of a GTPase-activating protein for the Ras-related Ral protein. The Journal of biological chemistry. 1991;266:9703–6. [PubMed] [Google Scholar]
- 41.Bhullar RP, Seneviratne HD. Characterization of human platelet GTPase activating protein for the Ral GTP-binding protein. Biochimica et biophysica acta. 1996;1311:181–8. doi: 10.1016/0167-4889(96)00002-x. [DOI] [PubMed] [Google Scholar]
- 42.Shirakawa R, Fukai S, Kawato M, et al. Tuberous sclerosis tumor suppressor complex-like complexes act as GTPase-activating proteins for Ral GTPases. J Biol Chem. 2009;284:21580–8. doi: 10.1074/jbc.M109.012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian X, Rusanescu G, Hou W, Schaffhausen B, Feig LA. PDK1 mediates growth factor-induced Ral-GEF activation by a kinase-independent mechanism. The EMBO journal. 2002;21:1327–38. doi: 10.1093/emboj/21.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29:356–70. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kesavapany S, Amin N, Zheng YL, et al. p35/cyclin-dependent kinase 5 phosphorylation of ras guanine nucleotide releasing factor 2 (RasGRF2) mediates Rac-dependent Extracellular Signal-regulated kinase 1/2 activity, altering RasGRF2 and microtubule-associated protein 1b distribution in neurons. J Neurosci. 2004;24:4421–31. doi: 10.1523/JNEUROSCI.0690-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang HS, Hinds PW. Phosphorylation of ezrin by cyclin-dependent kinase 5 induces the release of Rho GDP dissociation inhibitor to inhibit Rac1 activity in senescent cells. Cancer research. 2006;66:2708–15. doi: 10.1158/0008-5472.CAN-05-3141. [DOI] [PubMed] [Google Scholar]
- 47.Gillardon F, Steinlein P, Burger E, Hildebrandt T, Gerner C. Phosphoproteome and transcriptome analysis of the neuronal response to a CDK5 inhibitor. Proteomics. 2005;5:1299–307. doi: 10.1002/pmic.200400992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.