

Abstract
MauG is a diheme enzyme responsible for the post-translational formation of the catalytic tryptophan tryptophylquinone (TTQ) cofactor in methylamine dehydrogenase (MADH). MauG can utilize hydrogen peroxide, or molecular oxygen and reducing equivalents, to complete this reaction via a catalytic bis-Fe(IV) intermediate. Crystal structures of diferrous, Fe(II)-CO and Fe(II)-NO forms of MauG in complex with its preMADH substrate have been determined and compared to one another as well as to the structure of the resting diferric MauG-preMADH complex. CO and NO each bind exclusively to the 5-coordinate high-spin heme with no change in ligation of the 6-coordinate low-spin heme. These structures reveal likely roles for amino acid residues in the distal pocket of the high-spin heme in oxygen binding and activation. Glu113 is implicated in the protonation of heme-bound diatomic oxygen intermediates in promoting cleavage of the O-O bond. Pro107 is shown to change conformation on binding of each ligand and may play a steric role in oxygen activation by positioning the distal oxygen near Glu113. Gln103 is in a position to provide a hydrogen bond to the Fe(IV)=O moiety that may account for the unusual stability of this species in MauG.
MauG is a novel c-type diheme enzyme responsible for the post-translational modification of methylamine dehydrogenase (MADH) to generate a catalytic tryptophan tryptophylquinone cofactor (TTQ) from two Trp residues (βTrp57 and βTrp108 in Paracoccus denitrificans MADH) (1). MADH is a 119 kDa α2β2 heterotetrameric enzyme with two independent active sites each containing a TTQ cofactor. The substrate for this reaction is a monohydroxylated precursor (preMADH) where one oxygen atom has been inserted into each of the βTrp57 indole rings in the two active sites (2, 3). Maturation of the TTQ cofactor is a 6-electron oxidation process comprised of three 2-electron oxidations: i) insertion of the second -OH group into βTrp57, ii) formation of the crosslink between βTrp57 and βTrp108, and iii) oxidation of the quinol to quinone (Figure 1). The order of these modifications is currently unknown.
Figure 1.

MauG-dependent TTQ formation.
MauG can catalyze the 6-electron oxidation of preMADH using three equivalents of H2O2 or, O2 plus reducing equivalents (4). This catalytic activity distinguishes MauG from cytochrome c peroxidases (CCPs)1, as does its ability to utilize O2 as well as H2O2, although it is currently unclear which is the physiologically relevant substrate. The low micromolar affinity of diferric MauG for H2O2 (5) and the sensitivity of diferrous MauG to oxidation in air (1, 6) suggests that either may be physiologically relevant. Regardless of the oxidant source, an unprecedented bis-Fe(IV) intermediate is the high valent species that is generated and required for each of the three 2-electron oxidation reactions that complete TTQ synthesis (7). Based on Mössbauer data, the bis-Fe(IV) intermediate hemes are described as a ferryl (Fe(IV)=O) heme, with the other being a 6-coordinate Fe(IV) heme with two amino acid ligands (7). Thus, MauG stores one oxidizing equivalent on each of its c-type hemes generating an Fe(V) formal oxidation state equivalent to compound I: an Fe(IV)=O heme plus porphyrin/amino acid cation radical that is the more common high valent intermediate formed by heme oxygenases and peroxidases.
The redox cooperativity of the hemes enable them to act as a single 2-electron cofactor with no observation of a mixed-valence state (8). This is in contrast to most bacterial diheme CCPs (BCCPs) with which MauG is ~30% homologous, that require formation of a Fe(II)/Fe(III) state for reactivity with H2O2 (9). The crystal structure of the MauG-preMADH complex revealed a 5-coordinate heme with a 6-coordinate heme ligated by His205 and Tyr294 (10). The observation of catalytic activity in these crystals upon addition of H2O2 demonstrated conclusively that MauG oxidizes the TTQ site by long-range electron transfer from preMADH to the Fe(IV) hemes of MauG rather than by direct O-atom insertion by the Fe(IV)=O heme (10). The appearance of positive difference electron density upon H2O2 treatment distal to the 5-coordinate heme indicated this as the high-spin heme of MauG and the site of oxygen binding and activation (10). This heme is furthest from the MauG-preMADH interface, and more than 40 Ǻ from the site of TTQ synthesis in preMADH.
In addition to binding and activating O2 to form the high-valent species used in TTQ maturation, fully reduced (diferrous) MauG is also capable of binding CO defined by UV-visible spectroscopy (1, 11) and NO defined by EPR (12) to form stable complexes. This property is highly unusual among c-type hemes of which the large majority are 6-coordinate with two amino acid residues (13) and principally involved in electron transfer reactions. Thus, structures of CO and NO complexes with c-type heme proteins in the literature are quite rare, one example being cytochrome c′ of Alcaligenes xylosoxidans for which structures of 6-coordinate CO and 5-coordinate NO complexes have been determined (14). Here we describe crystal structures of the diferrous, Fe(II)-CO and Fe(II)-NO MauG in complex with preMADH. These structures provide an opportunity to analyze interactions engaged by diatomic ligands analogous to O2 in the distal pocket of MauG, and illustrate the potential importance of several conserved distal residues in the positioning of O2 and H2O2 and subsequent O-O bond cleavage.
EXPERIMENTAL PROCEDURES
Protein expression, purification and crystallization
The expression and purification of Paracoccus denitrificans MauG (1) and preMADH (2) were carried out as described previously. Crystallization was performed as described previously (10). Briefly, crystals were grown by hanging drop vapor diffusion at 20 °C in drops containing 1 μL of preformed protein complex (100 μM MauG and 50 μM preMADH in 10 mM potassium phosphate, pH 7.5) and 3 μL of reservoir solution (23–25 % w/v PEG 8000, 0.1 M sodium acetate, 0.1 M MES pH 6.4).
Formation of diferrous, Fe(II)-CO and Fe(II)-NO MauG-preMADH Crystals
Crystal trays were brought into an anaerobic chamber (Belle Technologies, UK) and incubated for 24 h. All procedures up to and including flash freezing of the crystals were carried out in the anaerobic chamber. For crystal soaking a degassed artificial mother liquor solution was used (24 % w/v PEG 8000, 0.1 M sodium acetate, 0.1 M MES pH 6.4) that additionally contained 2 mM sodium dithionite (Sigma) to reduce MauG to the diferrous form, and 10% PEG 400 as a cryoprotectant. Diferrous MauG-preMADH crystals were formed by soaking the crystals for 2 min in the artificial mother liquor prior to flash freezing in liquid nitrogen. CO-bound crystals were generated by 2 min soaks in the mother liquor pre-saturated with CO by purging with CO gas for ~ 10 min, and then flash frozen in liquid nitrogen. NO-bound crystals were formed by taking diferrous MauG-preMADH crystals generated as above, and transferring these for 2 min to artificial mother liquor without reductant, but containing ~2 mM NO, which were then flash frozen in liquid nitrogen. The NO solution was formed by diluting a concentrated stock solution of the NO donor DEA-NONOate (Sigma) in 10 mM NaOH into anaerobic cryoprotectant and incubating for 1 h in a sealed container. The formation of the desired oxidation and ligation states as well as stability during X-ray data collection were confirmed by single crystal UV-visible spectroscopy (4DX Systems AB, Sweden) (Figure S1).
X-ray data collection, processing and refinement
X-ray diffraction data were collected at 100 K on GM/CA-CAT beamline 23-ID-D of the Advanced Photon Source (APS), Argonne National Laboratory, Argonne IL using a MARmosaic 300 CCD. The beam size was adjusted to match the crystal size and orientation, and attenuated by 200–250-fold. 180° of diffraction data were collected on a single crystal, during which time no significant degradation of diffraction quality was observed. Data collection statistics are given in Table 1. The treated crystals described here were isomorphous to those of the untreated protein crystals, and were in space group P1 with one complex in the asymmetric unit (2 MauG + 1 preMADH; preMADH has two sites of TTQ synthesis) (10). Structure solution was by direct Fourier synthesis using the 2.1 Å resolution resting state MauG-preMADH crystal structure without ligands and waters (Protein Data Bank (PDB) ID, 3L4M) to generate initial phases, and also as the starting model in refinement. The reflection equivalents used to calculate Rfree in the refinement of the untreated crystals were used to calculate Rfree during refinement of the datasets described here. Cycles of model building with COOT (15) and restrained refinement using REFMAC5.5 with TLS were performed until all interpretable regions of the 2Fo-Fc and Fo-Fc electron density maps, excepting density representing ligands bound to the high-spin heme, were explained (16). The final step involved building the CO and NO ligands into the models and refining their positions. The high occupancy and differing geometries of the two ligands were confirmed by Fo-Fo electron density maps, and corroborated by the final atomic B-factors being similar to those of the heme. In the case of CO, the Fe-C bond had to be restrained to 1.9 Å in order to obtain satisfactory fits to the electron density, whereas no restraints were placed on the geometry of the NO ligand. The data refinement statistics are given in Table 1.
Table 1.
Data collection and refinement statisticsa.
| Diferrous MauG-preMADH | Fe(II)-CO MauG-preMADH | Fe(II)-NO MauG-preMADH | |
|---|---|---|---|
| Data Collection | |||
| Wavelength (Å) | 1.03317 | 1.03317 | 1.03316 |
| Resolution (Å) | 50.00 - 2.22 (2.28 - 2.22) | 50.00 - 2.16 (2.22 - 2.16) | 50.00 - 2.11 (2.17 - 2.11) |
| Space group | P1 | P1 | P1 |
| Unit cell lengths (Å) | 55.6 × 83.6 × 107.9 | 55.4 × 83.5 × 108.1 | 55.5 × 83.6 × 107.9 |
| Unit cell angles (°) | 110.0, 91.7, 105.5 | 110.1, 91.8, 105.6 | 110.0, 91.6, 105.7 |
| Measured reflections | 160,038 | 174,415 | 180,141 |
| Unique reflections | 83,223 | 90,587 | 95,254 |
| Completeness (%) | 96.5 (89.8) | 97.2 (88.1) | 95.3 (84.2) |
| Rmerge (%)b | 8.5 (34.3) | 8.3 (38.0) | 8.8 (36.2) |
| I/σI | 8.9 (2.1) | 9.3 (2.0) | 8.2 (2.2) |
| Multiplicity | 1.9 (1.8) | 1.9 (1.9) | 1.9 (1.9) |
| Refinement | |||
| Resolution (Å) | 44.49 - 2.22 (2.28 - 2.22) | 44.49 - 2.16 (2.22 - 2.16) | 44.49 - 2.11 (2.17 - 2.11) |
| No. reflections; working/test | 78,610/4,161 | 85995/4,534 | 90,417/4,810 |
| Rwork (%)c | 17.7 | 17.7 | 18.9 |
| Rfree (%)d | 22.9 | 22.6 | 24.2 |
| Protein atoms | 13,299 | 13,264 | 13,264 |
| Other atoms | 1,272 | 1,295 | 1,438 |
| Ramachandran statisticse | |||
| Allowed (%) | 99.9 | 100.0 | 100.0 |
| Outliers (%) | 0.1 | 0.0 | 0.0 |
| RMS Deviation | |||
| Bond lengths (Å) | 0.008 | 0.007 | 0.008 |
| Bond angles (°) | 1.329 | 1.323 | 1.328 |
| Average B-factor (Å2) | 27.15 | 27.81 | 25.50 |
| ESU (Å)f; Rwork/Rfree | 0.343/0.226 | 0.285/0.206 | 0.281/0.213 |
| Protein Data Bank ID | 3PXS | 3PXT | 3PXW |
Data in parentheses are for the highest resolution shell.
Rmerge = Σi|Ihkl,i - 〈Ihkl〉|/Σhkl Σi Ihkl,i, where I is the observed intensity and 〈Ihkl〉 is the average intensity of multiple measurements.
Rwork = Σ||Fo|-|Fc||/Σ|Fo|, where |Fo| is the observed structure factor amplitude, and |Fc| is the calculated structure factor amplitude.
Rfree is the R factor based on 5% of the data excluded from refinement.
Based on values obtained from PROCHECK (38).
Estimated standard uncertainties generated for Rwork and Rfree in REFMAC5.5 (16).
RESULTS
Overall structure and diferrous MauG-preMADH crystal structure
The crystal structures of diferrous, Fe(II)-CO and Fe(II)-NO MauG-preMADH were determined to 2.2, 2.2 and 2.1 Å resolution, respectively (Table 1). The overall structures of these forms and the resting state structure previously characterized (10) are superimposable with rmsd ≤ 0.17 Å, indicating that no major structural rearrangements accompany heme reduction or ligand binding. This similarity extends to the local environment of the high-spin heme of the diferrous form (Figure 2). Like the resting state MauG, the high-spin ferrous heme iron is proximally coordinated by His35, with no evidence of an ordered distal ligand making this heme 5-coordinate. A conserved pattern of ordered waters from the protein surface is stabilized by hydrogen bonds to Glu113, culminating in a water molecule (W0) at ~ 3.7 Å from the heme iron on the distal side (Figure 2 and 3A). No structural differences are observed between the low-spin hemes in any of the above four structures.
Figure 2.
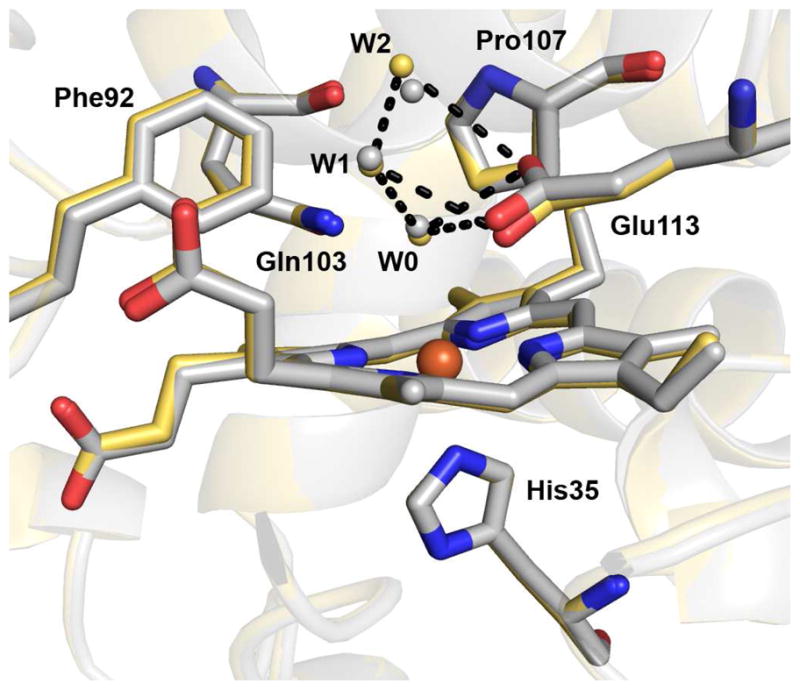
Superimposition of diferrous MauG and resting state MauG structures (PDB ID, 3L4M) within the context of MauG-preMADH complex crystals (10). The high-spin hemes, proximal ligand, and distal residues are shown in stick form colored according to element (carbon color: gold, diferrous; gray, resting state). Water molecules are shown as gold gray spheres for the diferrous and resting state MauG structures, respectively. Iron is drawn as an orange sphere. The remainder of MauG is shown as a secondary structure cartoon (gold diferrous; gray, resting state). Dotted lines represent hydrogen bonds involving three waters (W0, W1, W2) drawn between atoms in the diferrous MauG structure. The figure was produced using PyMOL (http://www.pymol.org/).
Figure 3.

The high-spin heme of (A) diferrous, (B) Fe(II)-CO and (C) Fe(II)-NO MauG within the context of MauG-preMADH complex crystals. 2Fo-Fc electron densities are contoured to 1.5σ. The proximal His35 ligand and distal residues are shown as sticks and waters as spheres colored by atom. Iron is drawn as an orange sphere. The remainder of MauG is shown as a secondary structure cartoon (gold, diferrous; cyan, CO; pink, NO). The figure was produced using PyMOL (http://www.pymol.org/).
Fe(II)-CO and Fe(II)-NO MauG-preMADH ligand geometries
For the Fe(II)-CO and Fe(II)-NO structures, modeling CO and NO ligands bound to the distal side of the high-spin heme in the final step of refinement results in a good fit to the electron density (Figure 3B and C). The positions of the terminal oxygen atoms of the CO and NO ligands are occupied by the water, W0, in the diferrous and resting state enzyme. Fo-Fo difference maps of Fe(II)-CO and Fe(II)-NO minus diferrous MauG-preMADH show the strongest positive density above the formerly 5-coordinate heme consistent with diatomics bound at high occupancy (Figure 4A and B). As ligand binding displaces the W0 water molecule in diferrous MauG, this is reflected in the weaker Fo-Fo electron density for the distal atoms. These difference maps in conjunction with a lack of residual Fo-Fc difference density with final models containing 100% ligand occupancy support complete ligation of the heme by CO and NO. This is further validated by the temperature factors for the terminal oxygen atoms of the CO and NO ligands in the final model being 22.4 and 22.2 Å2 and 25.1 and 25.9 Å2 for the two independent copies of MauG in the asymmetric unit (A and B chains, respectively). These values are similar to those of the heme iron and pyrrole nitrogen atoms which range from 20.5 to 23.7 Å2.
Figure 4.

Fo-Fo difference electron density maps. (A) Fe(II)-CO minus diferrous and (B) Fe(II)-NO minus diferrous contoured to 5.0σ around heme, ligand, and proximal and distal residues. (C) Fe(II)-NO minus Fe(II)-CO contoured to 3.0σ around bound NO. Positive and negative Fo-Fo are colored green and red respectively. Panel A shows the Fe(II)-CO model, and panels B and C the Fe(II)-NO model. The heme and proximal and distal residues are drawn as sticks colored by atom. Iron is drawn as an orange sphere. The remainder of MauG is shown as a secondary structure cartoon (cyan, CO; pink, NO). The figure was produced using PyMOL (http://www.pymol.org/).
CO is bound nearly perpendicular to the heme with Fe-C-O angles of 157° and 171° for the A and B chains, respectively, while both Fe-C distances refine to 2.0 Å. NO adopts a bent geometry with FeN-O angles of 118° and 122° for the A and B chains, respectively, while both Fe-N distances refine to 1.9 Å. An Fe(II)-NO minus Fe(II)-CO MauG-preMADH Fo-Fo difference map is consistent with the decreased bonding angle of NO relative to CO (Figure 4C). The geometries of these complexes are typical for what is observed in heme proteins and similar to those of myoglobin (Fe-CO = 1.8 Å, Fe-C-O = 171° (17), Fe-NO = 1.9 Å, Fe-N-O = 112° (18)).
Distal pocket changes upon CO and NO binding
The distal pocket of the high-spin heme of MauG is lined with the residues Phe92, Gln103, Pro107, and Glu113 (Figure 2). Only Pro107 and Glu113 show any evidence of movement going from diferrous to NO- and CO-bound states (Figure 5A). Interestingly, the Pro107 pucker changes upon CO and NO binding. The proline ring can adopt two different conformation states known as upPro and downPro that differ by the sign of their dihedral angles (19). Upon binding CO or NO, Pro107 converts from the upPro to the downPro form (Figure 5). For trans isomers of Pro (which is the case for MauG Pro107) there seems to be little preference for one form over the other (20). Although this change is subtle, attempts to manually position and then refine an upPro conformation in the Fe(II)-CO and Fe(II)-NO MauG-preMADH structures or a downPro in the diferrous MauG-preMADH structure resulted in conformer switching during refinement. Further, the electron density clearly supports the conformational assignments (Figure 5B). Thus, the conversion from upPro to downPro upon ligand binding seems to represent a mechanism for relieving steric crowding by Pro107 through movement of the β carbon 0.6 and 1.0 Å away from the NO and CO ligands, respectively (Figure 5A). In addition to Pro107, Glu113 is sterically perturbed by ligand binding (Figure 5A). Both of these perturbations are more pronounced in the CO-bound versus the NO-bound structure, likely as a consequence of their different binding geometries. Within co-ordinate error Phe92 and Gln103 are unaffected by ligand binding (Figure 5A).
Figure 5.

(A) Superimposition of the high-spin heme and distal residues for the diferrous (gold), Fe(II)-CO (cyan), and Fe(II)-NO (pink) complexes. (B) 2Fo-Fc electron density contoured at 1.5σ for Pro107 of diferrous (gold carbons and gold mesh) and Fe(II)-CO (cyan carbons and blue mesh) MauG. The figure was produced using PyMOL (http://www.pymol.org/).
Intriguingly, the steric restraint imposed by Pro107 orients the terminal oxygen atoms of both CO and NO toward the distal Glu113 residue. The terminal oxygen atoms of both CO and NO ligands occupy a similar position as the water molecule, W0, observed in resting state and diferrous structures (Figure 5A). This places the terminal oxygen atoms of CO and NO ligands at 3.5 and 3.1 Å respectively from the nearest carboxyl group oxygen of Glu113. As it is unlikely that this solvent accessible residue is protonated at pH 6.4, this observation is best described as an electrostatic interaction between the negatively charged carboxylate and the partial positive charge carried on the terminal oxygen atoms as has been described for the CO and NO complexes of chloroperoxidase (CPO) (21).
As NO binds in a bent geometry similar to that expected for O2, this ligand is a particularly good probe for interactions engaged by potential diatomic oxygen intermediates such as Fe(II)-O2 and Fe(III)-OOH- in the MauG catalytic cycle. Interactions between the NO ligand and distal residues are shown in detail in Figure 6. The closest contact to the bound NO in MauG is from the side-chain amide of Gln103, which is 2.9 Å from the proximal nitrogen atom of NO, consistent with a hydrogen bond. After conversion from upPro to downPro, the β and γ-carbon atoms of Pro107 are 3.4 Å from the distal oxygen and proximal nitrogen atoms of bound NO, respectively. As mentioned above, Glu113 is also nearby with its carboxyl oxygen atoms at 3.1 and 3.3 Å from the NO distal oxygen atom. In addition to its interaction with bound NO, this residue appears to stabilize a well-conserved network of water molecules to the protein surface, positioning the two nearest (W1 and W2) at 3.4 and 3.5 Å, respectively, from the terminal oxygen atom of NO. The potential role of this residue in O-O bond cleavage is discussed below.
Figure 6.
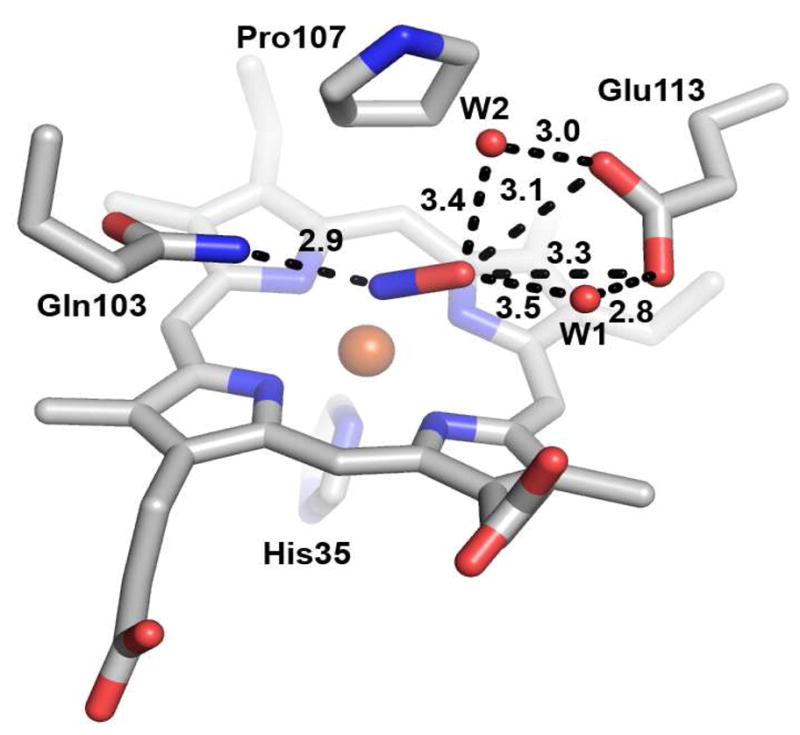
Distal environment of the high-spin heme in Fe(II)-NO MauG. Dotted lines indicate interaction distances in angstroms. The figure was produced using PyMOL (http://www.pymol.org/).
DISCUSSION
CO and NO have been used extensively as stable oxygen analogues in heme systems. Although c-type hemes do not typically bind these ligands, binding is observed for diferrous MauG and enabled the structural characterization of CO and NO adducts of MauG in complex with its preMADH substrate. These structures allowed the identification of residues which are likely vital to the catalytic functions of MauG, namely Gln103, Pro107, and Glu113. Consistent with this is the observation that these residues are absolutely conserved among known MauG orthologs (Figure S2). The Fe(II)-CO and Fe(II)-NO structures show that these ligands bind exclusively at the high-spin heme, causing no change to the overall structure of MauG or to the low-spin heme. However, perturbations in the positions of two residues in the distal pocket of the high-spin heme, Pro107 and Glu113, are observed. The previously reported structure of H2O2-treated MauG shows some positive electron density over the heme iron suggesting partial occupancy at this site, likely attributable to a mixture of oxygen species as previously discussed (10). Intriguingly, there is evidence within this mixture for similar motions of Pro107 and Glu113 (Figure S3). This observation provides some evidence that the movement of Pro107 and Glu113 observed upon CO and NO binding is also relevant to the binding and activation of H2O2.
The fact that even the high-spin heme distal residues show little perturbation upon ligand binding is consistent with the MauG catalyzed reaction being random order and the high-spin heme being constitutively “active” for dioxygen activation, which contrasts with the ordered mechanism of cytochromes P450 where binding of organic substrate is a prerequisite (22). The Pro107 ring in MauG and the substrate camphene in the ES complex of cytochrome P450cam (23) are at comparable distances to the heme irons in these two structures (4.3 Ǻ). This led to the proposal that Pro107 in MauG may fulfill the structural role of substrate in cytochrome P450cam, providing a stable O2-binding site (10). The structures reported herein indicate a more dynamic role for Pro107 in O2 activation, as it is able to switch conformation from upPro to downPro in response to ligand binding. This change relieves steric crowding with the ligand and may be instrumental in positioning bound O2 in close contact with Glu113, the likely active site acid/base. Proline conformational switching in response to ligand binding is an unusual observation, and may represent a previously unrecognized mechanism for ligand sensing in signalling proteins.
The hydrogen bond network between distal waters, Glu113, and NO suggests that Glu113 plays a key role in proton donation to and/or Fe(III)-OOH- intermediates either directly or by Fe(II)-O2 stabilizing a network of catalytic water molecules. The latter mechanism has been implicated in oxygen activation by cytochrome P450cam where well-ordered waters are observed in the active site of the Fe(II)-O2 form of the enzyme (24, 25). The homologous diheme BCCP of Nitrosomonas europea (NeBCCP; 28% sequence identity) also employs a glutamate residue in the distal pocket to potentially serve as a catalytic acid/base promoting cleavage of the O-O bond (26). In fact, the entire set of MauG high-spin heme distal residues are conserved in NeBCCP (Phe81, Gln92, Pro96, Glu102). Generally the diheme BCCPs require reduction of the second, low-spin (high-potential) heme to cause a distal His ligand to dissociate from the low-potential heme, allowing H2O2 to bind. However, NeBCCP differs in that its high-spin (low-potential) heme is five-coordinate regardless of the oxidation state of the enzyme. At least for Pseudomonas aeruginosa BCCP, this dissociation event is proposed to correlate with a conformational change allowing proper positioning of a fully conserved catalytic Glu residue leading to a distal environment similar to that of NeBCCP (26, 27). In support of this, mutants of Glu117 in Rhodobacter capsulatus BCCP show no peroxidase activity (28).
Caldariomyces fiimago choloroperoxidase (CPO), an enzyme capable of peroxidase as well as P450-type chemistry, also contains an active site glutamate residue (Glu183) thought to fulfill a role in acid/base catalysis (29). Structures of CPO bound to a number of exogenous heme ligands including NO and CO have been solved and demonstrate that the carboxyl group of Glu183 forms a direct electrostatic interaction with the terminal oxygen atoms of NO and CO ligands at 2.88 and 3.18 Å, respectively (21). An intermediate in the peroxidase mechanism known as compound 0 (Fe(III)-OOH) was trapped in CPO and the terminal oxygen atom was observed at 2.7 Å from the carboxyl group of Glu183, indicating that this residue likely participates in proton transfer to the terminal oxygen atom of bound reaction intermediates and demonstrating the utility of NO and CO ligands as stable analogues of diatomic oxygen species (30). Although potentially mediated by water molecules in MauG, this demonstrates precedence for the use of a catalytic glutamate residue in oxygen activation by heme enzymes.
The Fe(II)-NO structure of MauG suggests a critical role for Gln103 in MauG catalysis as well. This residue interacts with the proximal nitrogen of bound NO by donation of a hydrogen bond. Gln103 could also hydrogen bond to bound oxygen species during the catalytic cycle, including the Fe(IV)=O intermediate. Such a hydrogen bond interaction was recently suggested on the basis of DFT calculations of the bis-Fe(IV) MauG hemes (31). A distal Arg residue has been proposed to fulfill an analogous role in horseradish peroxidase (HPO), donating hydrogen bonds to both Fe(III)-hydroperoxo and Fe(IV)=O intermediates during formation of compound I (32).
The key difference between MauG and the above mentioned enzymes lies in how oxidizing equivalents are stored. CPO and HPO, having only a single heme, store one oxidizing equivalent as Fe(IV)=O and the other as a porphyrin cation radical (compound I) (33). Most eukaryotic monoheme CCPs utilize this same intermediate (34), except the yeast enzyme which stores the second oxidizing equivalent as a Trp cation radical (35). The bacterial diheme CCPs, which are active in a mixed-valance (Fe(III)/Fe(II)) state, store the second oxidizing equivalent as Fe(III) at the second heme (9), although NeBCCP can also utilize compound I if the second heme is ferric at the start of catalysis (36). MauG is the only enzyme known to form a bis-Fe(IV) heme two-electron oxidant (7). This remarkable property must have little to do with the environment of the high-spin heme, which is very similar to that of NeBCCP. The unusual His-Tyr ligation of the low-spin heme of MauG (10) appears key to stabilizing this intermediate. In this regard, it should be noted that the Tyr ligand to the low-spin heme is also fully conserved among MauG proteins (Figure S2). When the Tyr294 is mutated to His, which acts as a ligand to give a six-coordinate His-His heme, the mutant no longer supports formation of the bis-Fe(IV) (37). Although it can form a high-valent intermediate (probably compound I), it is inactive in TTQ biosynthesis. The hemes of the Tyr294His mutant still retain the redox co-operativity of wild-type (8, 12), although the linked oxidation-reduction midpoint potentials are altered (37). As such CO binding at the high-spin heme is affected by the change of ligation at the low-spin heme, leading to a significant increase in CO affinity over that of wild-type.
Our results indicate that Glu113 of MauG likely plays a key role in catalysis, donating one or more protons, either directly or by stabilizing a network of catalytic waters, to bound O2 intermediates leading to O-O bond cleavage and formation of the bis-Fe(IV) catalytic intermediate. In addition to its proposed role in maintaining an activated high-spin heme environment (10), Pro107 may play a steric role in O2-activation, positioning the distal oxygen atom near the carboxylate of Glu113. Once the O-O bond has been cleaved, Gln103 may be in a position to hydrogen bond with the Fe(IV)=O moiety. This, coupled with charge stabilization provided by the negatively charged Glu113, may account for the unusual stability of this species in MauG in the absence of substrate (7). Structural characterization and activity measurements of distal residue mutants should help to refine our understanding of the mechanism of oxygen activation and high-valent heme stabilization in this unusual enzyme.
Supplementary Material
Acknowledgments
We thank staff at sector 23, GM/CA-CAT, of the Advanced Photon Source, Argonne National Laboratory, Argonne, IL, USA, for assistance. GM/CA-CAT is funded by National Cancer Institute Grant Y1-CO-1020 and National Institute of General Medical Sciences Grant Y1-GM-1104. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract DE-AC02-06CH11357. Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute, and we thank Can Ergenekan for support. We also thank Ed Hoeffner at the Kahlert Structural Biology Laboratory (KSBL) at the University of Minnesota.
ABBREVIATIONS
- MADH
methylamine dehydrogenase
- TTQ
tryptophan tryptophylquinone
- preMADH
the biosynthetic precursor protein of MADH with incompletely synthesized TTQ
- CCP
cytochrome c peroxidase
- BCCP
bacterial diheme cytochrome c peroxidase
- bis-Fe(IV) MauG
redox state of MauG with one heme as Fe(IV)=O and the other as Fe(IV)
- PDB
Protein Data Bank
- CPO
chloroperoxidase
- HPO
horseradish peroxidase
- NeBCCP
Nitrosomonas europea diheme cytochrome c peroxidase
Footnotes
This work was supported by NIH grants GM-66569 (CMW), GM-41574 (VLD) and Minnesota Partnership for Biotechnology and Medical Genomics Grant SPAP-05-0013-P-FY06 (CMW)
Co-ordinates and structure factors have been deposited in the Protein Data Bank as entries 3PXS (diferrous MauG-preMADH), 3PXT (Fe(II)-CO MauG-preMADH) and 3PXW (Fe(II)-NO MauG-preMADH)
SUPPORTING INFORMATION AVAILABLE
Single crystal UV-visble spectra of diferric, diferrous, Fe(II)-CO and Fe(II)-NO MauG-preMADH crystals before and after X-ray data collection; multiple sequence alignment of MauG orthologs; comparison of the high-spin heme structures of diferrous and Fe(II)-CO MauG with that of MauG treated with H2O2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wang Y, Graichen ME, Liu A, Pearson AR, Wilmot CM, Davidson VL. MauG, a novel diheme protein required for tryptophan tryptophylquinone biogenesis. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 2.Pearson AR, De La Mora-Rey T, Graichen ME, Wang Y, Jones LH, Marimanikkupam S, Agger SA, Grimsrud PA, Davidson VL, Wilmot CM. Further insights into quinone cofactor biogenesis: probing the role of mauG in methylamine dehydrogenase tryptophan tryptophylquinone formation. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. MauG-dependent in vitro biosynthesis of tryptophan tryptophylquinone in methylamine dehydrogenase. J Am Chem Soc. 2005;127:8258–8259. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 4.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Mechanistic possibilities in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:13276–13283. doi: 10.1021/bi061497d. [DOI] [PubMed] [Google Scholar]
- 5.Shin S, Abu Tarboush N, Davidson VL. Long-range electron transfer reactions between hemes of MauG and different forms of tryptophan tryptophylquinone of methylamine dehydrogenase. Biochemistry. 2010;49:5810–5816. doi: 10.1021/bi1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin S, Lee S, Davidson VL. Suicide inactivation of MauG during reaction with O2 or H2O2 in the absence of its natural protein substrate. Biochemistry. 2009;48:10106–10112. doi: 10.1021/bi901284e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Fu R, Lee S, Krebs C, Davidson VL, Liu A. A catalytic di-heme bis-Fe(IV) intermediate, alternative to an Fe(IV)=O porphyrin radical. Proc Natl Acad Sci U S A. 2008;105:8597–8600. doi: 10.1073/pnas.0801643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Feng M, Wang Y, Tachikawa H, Davidson VL. Evidence for redox cooperativity between c-type hemes of MauG which is likely coupled to oxygen activation during tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:821–828. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pettigrew GW, Echalier A, Pauleta SR. Structure and mechanism in the bacterial dihaem cytochrome c peroxidases. J Inorg Biochem. 2006;100:551–567. doi: 10.1016/j.jinorgbio.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Jensen LM, Sanishvili R, Davidson VL, Wilmot CM. In crystallo posttranslational modification within a MauG/pre-methylamine dehydrogenase complex. Science. 327:1392–1394. doi: 10.1126/science.1182492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S, Shin S, Li X, Davidson V. Kinetic mechanism for the initial steps in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2009;48:2442–2447. doi: 10.1021/bi802166c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu R, Liu F, Davidson VL, Liu A. Heme iron nitrosyl complex of MauG reveals an efficient redox equilibrium between hemes with only one heme exclusively binding exogenous ligands. Biochemistry. 2009;48:11603–11605. doi: 10.1021/bi9017544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fufezan C, Zhang J, Gunner MR. Ligand preference and orientation in b- and c-type heme-binding proteins. Proteins. 2008;73:690–704. doi: 10.1002/prot.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawson DM, Stevenson CE, Andrew CR, Eady RR. Unprecedented proximal binding of nitric oxide to heme: implications for guanylate cyclase. EMBO J. 2000;19:5661–5671. doi: 10.1093/emboj/19.21.5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 17.Vojtechovsky J, Chu K, Berendzen J, Sweet RM, Schlichting I. Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brucker EA, Olson JS, Ikeda-Saito M, Phillips GN., Jr Nitric oxide myoglobin: crystal structure and analysis of ligand geometry. Proteins. 1998;30:352–356. [PubMed] [Google Scholar]
- 19.Nemethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, Zagari A, Rumsey S, Scheraga HA. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. The Journal of Physical Chemistry. 1992;96:6472–6484. [Google Scholar]
- 20.Vitagliano L, Berisio R, Mastrangelo A, Mazzarella L, Zagari A. Preferred proline puckerings in cis and trans peptide groups: implications for collagen stability. Protein Sci. 2001;10:2627–2632. doi: 10.1110/ps.ps.26601a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundaramoorthy M, Terner J, Poulos TL. Stereochemistry of the chloroperoxidase active site: crystallographic and molecular-modeling studies. Chem Biol. 1998;5:461–473. doi: 10.1016/s1074-5521(98)90003-5. [DOI] [PubMed] [Google Scholar]
- 22.Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 23.Raag R, Poulos TL. Crystal structures of cytochrome P-450CAM complexed with camphane, thiocamphor, and adamantane: factors controlling P-450 substrate hydroxylation. Biochemistry. 1991;30:2674–2684. doi: 10.1021/bi00224a016. [DOI] [PubMed] [Google Scholar]
- 24.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 25.Nagano S, Poulos TL. Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J Biol Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu H, Schuller DJ, Lanzilotta WN, Sundaramoorthy M, Arciero DM, Hooper AB, Poulos TL. Crystal structure of Nitrosomonas europaea cytochrome c peroxidase and the structural basis for ligand switching in bacterial di-heme peroxidases. Biochemistry. 2001;40:13483–13490. doi: 10.1021/bi011481h. [DOI] [PubMed] [Google Scholar]
- 27.Fulop V, Ridout CJ, Greenwood C, Hajdu J. Crystal structure of the di-haem cytochrome c peroxidase from Pseudomonas aeruginosa. Structure. 1995;3:1225–1233. doi: 10.1016/s0969-2126(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 28.De Smet L, Savvides SN, Van Horen E, Pettigrew G, Van Beeumen JJ. Structural and mutagenesis studies on the cytochrome c peroxidase from Rhodobacter capsulatus provide new insights into structure-function relationships of bacterial di-heme peroxidases. J Biol Chem. 2006;281:4371–4379. doi: 10.1074/jbc.M509582200. [DOI] [PubMed] [Google Scholar]
- 29.Sundaramoorthy M, Terner J, Poulos TL. The crystal structure of chloroperoxidase: a heme peroxidase--cytochrome P450 functional hybrid. Structure. 1995;3:1367–1377. doi: 10.1016/s0969-2126(01)00274-x. [DOI] [PubMed] [Google Scholar]
- 30.Kuhnel K, Derat E, Terner J, Shaik S, Schlichting I. Structure and quantum chemical characterization of chloroperoxidase compound 0, a common reaction intermediate of diverse heme enzymes. Proc Natl Acad Sci U S A. 2007;104:99–104. doi: 10.1073/pnas.0606285103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ling Y, Davidson VL, Zhang Y. Unprecedented Fe(IV) species in a diheme protein MauG: A quantum chemical investigation on the unusual Mössbauer spectroscopic properties. J Phys Chem Lett. 1:2936–2939. doi: 10.1021/jz101159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlsson GH, Nicholls P, Svistunenko D, Berglund GI, Hajdu J. Complexes of horseradish peroxidase with formate, acetate, and carbon monoxide. Biochemistry. 2005;44:635–642. doi: 10.1021/bi0483211. [DOI] [PubMed] [Google Scholar]
- 33.Rutter R, Hager LP, Dhonau H, Hendrich M, Valentine M, Debrunner P. Chloroperoxidase compound I: Electron paramagnetic resonance and Mössbauer studies. Biochemistry. 1984;23:6809–6816. doi: 10.1021/bi00321a082. [DOI] [PubMed] [Google Scholar]
- 34.Sivaraja M, Goodin DB, Smith M, Hoffman BM. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science. 1989;245:738–740. doi: 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- 35.Dolphin D, Forman A, Borg DC, Fajer J, Felton RH. Compounds I of catalase and horse radish peroxidase: pi-cation radicals. Proc Natl Acad Sci U S A. 1971;68:614–618. doi: 10.1073/pnas.68.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arciero DM, Hooper AB. A di-heme cytochrome c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced states. J Biol Chem. 1994;269:11878–11886. [PubMed] [Google Scholar]
- 37.Abu Tarboush N, Jensen LM, Feng M, Tachikawa H, Wilmot CM, Davidson VL. Functional importance of tyrosine 294 and the catalytic selectivity for the bis-Fe(IV) state of MauG revealed by replacement of this axial heme ligand with histidine. Biochemistry. 49:9783–9791. doi: 10.1021/bi101254p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.