

Abstract
The levels of selected neuroregulatory proteins that inhibit or promote apoptotic cell death were measured in the striatum of piglets subjected to precisely controlled 1 h hypoxic insult followed by 0, 2 and 4 h recovery and compared to sham operated animals. The anti-apoptotic proteins: there were increases in Survivin at 0 (157%, P = 0.031) and 4 h (171%, P = 0.033), in Bcl-XL at 0 (138%, P = 0.028) and 4 h (143%, P = 0.007), in VEGF at 4 h (185%, P = 0.019) and Hsp27 at 2 h (144%, P = 0.05) and 4 h (143%, P = 0.05). The pro-apoptotic proteins: caspases-1 and 7 increased at 4 h (135%, P = 0.05) and (129%, P = 0.038), respectively. Bim increased after 4 h (115%, P = 0.028), Apoptosis Inducing Factor after 2 h (127%, P = 0.048) and Calpain after 4 h (143% of control, P = 0.04). Hypoxia causes increase in levels of both anti- and pro-apoptotic proteins. Their relative activity determines the outcome in terms of cell damage and neuronal deficit.
Keywords: Newborn brain, Hypoxic injury, Apoptotic neuronal injury, Striatal injury
Introduction
Neonatal brain injury resulting from hypoxia–ischemia is a most common form of brain damage and can lead to various neurological disorders [1–3]. Hypoxia is etiologically linked to cerebral palsy, hearing and vision loss, mental retardation, learning disabilities, attention deficit hyperactivity disorder, schizophrenia and neuronal migration disorders.
The mechanisms underlying hypoxic-ischemic brain damage are only partially understood but it is well established that following hypoxia–ischemia of neonatal brain, neuronal death can occurs by both apoptosis and necrosis [4–6]. However, the relative involvement of necrosis and apoptosis to the injury that develops after cerebral hypoxia–ischemia is still not clear [7]. The occurrence of these processes has been reported to be dependent on the severity and duration of insult and also on the region and degree of maturation of the brain [8–10]. Numerous studies have reported that, following hypoxia–ischemia in the neonatal brain, there is greater apoptotic than necrotic cell death and that, in neonates, hypoxia can activate several pro-apoptotic pathways [9–11]. Some of the pro-apoptotic regulatory proteins involved in this process include caspases, members of the Bcl-2 protein family such as Bax, Bad, Bid or Bim, apoptosis inducing factor (AIF), and calpain-calcium binding protease.
Over-expression of other proteins that can occur following hypoxia–ischemia, such as anti-apoptotic proteins of the Bcl-2 protein family- Bcl-2 or Bcl-XL, Survivin, VEGF, hypoxia inducing factor -27 (Hsp-27) are known to suppress apoptotic cell death.
Our previous studies revealed significant regional differences in expression of Bcl-2 and Bax (the Bcl-2/Bax ratio is considered an indicator of apoptotic pathway activation) in the brain of newborn piglet following 1 h hypoxia. The Bcl-2/Bax ratio was lower in the striatum than in other brain regions, suggesting that the striatum may be particularly susceptible to apoptotic injury.
The purpose of the present investigation was to assess the responses of other pro- and anti-apoptotic proteins in the striatum of newborn piglets in the same hypoxia model.
Experimental Procedure
Animal Preparation
Newborn piglets, age 3–5 days, were used for this study. Anesthesia was induced with 4% isoflurane (Novaplus, Hospira Inc., Lake Forest, IL). Pulse oximetry, ECG and temperature measurements begun immediately after induction of anesthesia. A 1.0% lidocaine-HCl (Abbott Laboratories, North Chicago, IL) was used as a local anesthetic for tracheotomy and insertion of femoral arterial and venous lines. A bolus of fentanyl citrate (30 μg/kg) was administered intravenously (IV). Monitoring of systemic blood pressure and heart rate began immediately following insertion of the arterial line and blood gas measurements (pO2, pCO2, pH, glucose, and ions) were made at regular intervals throughout the procedure. Isoflurane was reduced to 1% after tracheotomy and pancuronium (Apothecon, Bristol-Meyers Squibb, Princeton, NJ) was injected (0.3 mg/kg, IV) to help maintain a balanced anesthesia and to induce respiratory paralysis. The piglets were placed on mechanical ventilation (Sechrist Infant Ventilator, model IV-100 B), isoflurane was withdrawn entirely and piglets were ventilated with a mixture of oxygen and nitrous oxide (at control condition with 21–22% oxygen and 79–78% nitrous oxide). Anesthesia was maintained during the experiments using nitrous oxide, fentanyl (10 μg/kg/h, IV) and pancuronium (0.1 mg/kg, IV). This model provides a balanced anesthesia using an inhalation agent (nitrous oxide) for amnesia and analgesia, a potent fast acting narcotic agent (fentanyl) for primary analgesia, and a neuromuscular blocking agent (pancuronium) to provide surgical and experimental stillness and control of ventilation. The piglets were monitored closely and any increase in heart rate or blood pressure of more than 10% was treated with supplemental fentanyl at a dose sufficient to return the values of control levels.
Blood pressure, body temperature, heart rate and end-tidal CO2 were continuously monitored. Arterial blood samples were taken every 15 min and blood pH, arterial CO2 pressure (PaCO2) and arterial oxygen pressure (PaO2) were measured using a Rapid Lab 850 Analyzer (Bayer). Hydration was maintained by iv injection of physiological saline as needed. At the conclusion of the experiments, the anesthetized animals were euthanized by intravenous administration of saturated KCl solution. After euthanasia the brain was quickly removed and placed in 0–4°C saline solution, a medial cut used to separate it into the left and right halves and the all of the striatum, putamen and caudate nucleus, dissected out. The striatal tissue was rapidly frozen by placing it on aluminum foil lying in a bed of powdered dry ice. The frozen tissue was stored at −80°C until use.
All animal use procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the local Animal Care Committee.
Experimental Model
Hypoxia was induced by changing the FiO2 from 21 to 6%, lowering the mean cortical oxygen pressure to 10 mm Hg over 20 min and then holding a level of 8 mm Hg for 40 min followed by reoxygenation with 21% FiO2 for 0, 2 or 4 h.
Protein Array (Full Moon Bioscience)
Protein Labeling
Labeling reagent was prepared by adding 100 μl of DMF to 1 mg of biotin reagent was added to a final concentration of 10 μg/μl. Labeling buffer was added to a homogenate of the striatal tissue containing 145 μg protein to bring the volume to 50 μl and then 2 μl of the biotin/DMF was added to the protein sample with labeling buffer. The solution was mixed and incubated at room temperature (RT) for 2 h with mixing. Twenty five micro litre of stop reagent was added and the mixture incubated for 30 min at RT with mixing.
Coupling
The Antibody Microarrays were submerged in blocking buffer and shaken for 40 min at RT. The arrays were rinsed with Milli-Q grade deionized (DI) water and incubated in the coupling chamber with 145 μg of labeled protein sample in 6 ml coupling solution on an orbital shaker for 2 h at RT. The slides were removed from the coupling chamber, washed three times with fresh wash buffer, and rinsed extensively with DI water.
Detection
Thirty micro litre of Cy3-Streptavidin (1 mg/ml) was added to a 60-ml bottle containing detection buffer. The arrays were submerged in 30 ml of Cy3-Streptavidin solution and incubated on an orbital shaker for 45 min at RT in the dark. The slides were washed three times with fresh wash buffer and rinsed extensively with DI water. They were then dried with compressed nitrogen and scanned on an Axon GenePix Array Scanner.
Data Analysis
Scanned images of microarrays were analyzed using Scion Image software (NIH). The data are presented as the mean ± SEM for the density of bands for six independent experiments in each group. This is an exploratory study. We did not adjust for type 1 errors and statistical significance of differences were assessed using a two-tailed t test with P < 0.05 considered significant. Our hypothesis, that hypoxia would result in significant increase in pro and anti apoptotic signaling in the striatum, predicts a direction for the changes in protein levels and a one tailed t test would be appropriate. However, multiple pro and anti apoptotic proteins were measured in the same samples and a correction for multiple comparisons would be appropriate. Using both the one tailed t test and Bonferroni’s correction for multiple comparisons, which is generally believed to underestimate the significance, significance at the 95% for any one of the proteins being different from control would be equivalent to P < 0.03 calculated by the two tailed t test.
Results
Effect of Hypoxia on Early Response of Selected Anti-Apoptotic Proteins in the Striatum of Newborn Piglets
There were no significant differences as compared to sham operated animals in expression of Bcl-2, Bcl-2a, Bcl-6 and Bcl-XL. There were, however, significant increases in the levels of several other proteins believed to have significant roles in protection of the brain from hypoxic injury (Fig. 1a–d). As can be seen in Fig. 1a, at 0 h level of Survivin increased from 2,749 ± 475 to 4,326 ± 414 arbitrary units (157% of control, P = 0.031) and by 4 h to 4,705 ± 631 arbitrary units (171% of control, P = 0.033). Similarly, Bcl-XL increased significantly as compared to control at 0 h from 12,807 ± 1,162 to 17,666 ± 1,485 arbitrary units (138% of control, P = 0.028) and 4 h of recovery to 18,323 ± 1,115 arbitrary units (143% of control, P = 0.007; Fig. 1b). VEGF was also increased, from 1,382 ± 276 to 2,560 ± 317 arbitrary units (185% of control, P = 0.019) after 4 h of recovery (Fig. 1c) and Hsp27 increased from 5,137 ± 859 arbitrary units to 7,379 ± 528 arbitrary units after 2 h of recovery (144% of control, P = 0.05) and to 7,343 ± 524 arbitrary units after 4 h (143% of control, P = 0.05; Fig. 1d).
Fig. 1.
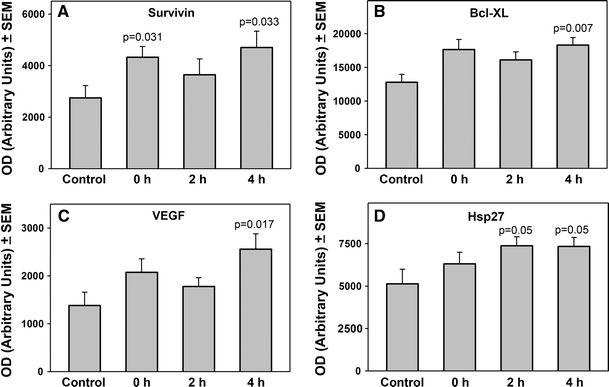
Expression of Survivin (a), Bcl-XL (b), VEGF (c) and Hsp27 (d) in the striatum of newborn piglets at 0, 2 and 4 h of post-hypoxic recovery. Analysis was of homogenates of striatal tissue. The data are presented as the mean ± SEM for the density of bands for six independent experiments. The statistical significance of differences from control were assessed using a two-tailed t test with P < 0.05 considered significant
Effect of Hypoxia on the Early Response of Selected Pro-Apoptotic Proteins in the Striatum of Newborn Piglets
There were no significant differences between sham operated animals and post-hypoxic groups in levels of Caspase-2, -3, -5, -6, -8 and -9. Increase in expression of Caspase-7 from 1,947 ± 192 arbitrary units to 2,508 ± 136 arbitrary units was observed at 4 h of post-hypoxic recovery (129% of control, P = 0.038; Fig. 2a). Similarly, Caspase-1 was increased significantly from 2,878 ± 303 to 3,884 ± 377 arbitrary units after 4 h of recovery (135% of control, P = 0.05; Fig. 2b). Bim expression was increased after 4 h from 539 ± 21 to 621 ± 24 arbitrary units (115% of control, P = 0.028; Fig. 3a). Apoptosis Inducing Factor was increased after 2 h of recovery from 5,636 ± 485 to 7,130 ± 452 arbitrary units (127% of control, P = 0.048 and at 4 h to 7,643 ± 512 arbitrary units (136% of control, P = 0.017; Fig. 3b). Calpain was increased from 5,004 ± 691 to 7,181 ± 609 arbitrary units (143% of control, P = 0.04) after 4 h of recovery (Fig. 3c).
Fig. 2.
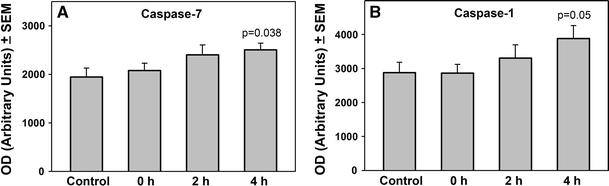
Expression of Caspase-7 (a) and Caspase-1 (b) in the striatum of newborn piglets at 0, 2 and 4 h of post-hypoxic recovery. The data are presented as the mean ± SEM for the density of bands for six independent experiments
Fig. 3.
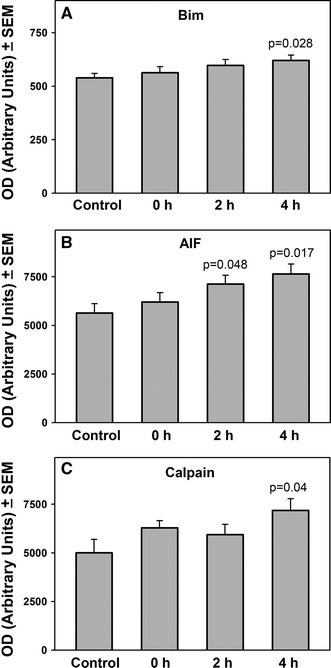
Expression of Bim (a), AIF (b) and Calpain (c) in the striatum of newborn piglets at 0, 2 and 4 h of post-hypoxic recovery. The data are presented as the mean ± SEM for the density of bands for six independent experiments
Discussion
Hypoxic/ischemic insult to the brain activates a number of pathways, some of which, if unopposed, lead to apoptotic or necrotic neuronal damage while others have a protective role, in part by countering the former. The data presented in this paper show that, in the striatum of newborn piglets, hypoxic insult results in rapid changes in expression of many proteins believed to regulate the processes that both promote and prevent apoptotic cell death. Striatum was chosen for this study because we have shown earlier that this region is particularly sensitive to hypoxia [12]. This was manifested by significant decrease in Bcl-2/Bax ratio, an indicator of an increased susceptibility of cells to apoptosis.
Each protein, reported in this study to undergo significant changes in response to hypoxic insult to the brain, is considered likely to have a significant role in determining the outcome of the apoptotic process. Survivin, VEGF, Bcl-XL and Hsp27 have been shown to be elevated following the hypoxic episode and all these proteins have been reported to help protect cells by inhibiting the processes leading to apoptotic death.
Survivin is a member of the inhibitor of apoptosis protein family (IAP family) and is reported to diminish apoptosis by interfering with the activity of caspase-3, caspase-7, and caspase-9 [13–16]. Additionally, Survivin can increase cell survival through its effects on mitosis and cell cycle progression [17–21]. Conway et al. [22] reported that hypoxic/ischemic insult in a mouse stroke model resulted in increased expression of Survivin in the brain after 2 days of recovery. Our study shows that increase in expression of this protein occurs immediately after hypoxia, suggesting that Survivin can act quickly after a hypoxic insult as well as during prolonged recovery.
Survivin can be upregulated by angiogenic factors, including vascular endothelial cell growth factor (VEGF; [23, 24]). Our data show that VEGF expression increased significantly only after 4 h of recovery whereas increase in expression of Survivin was observed already at the end of the hypoxic insult (0 h of recovery). Therefore, it can be suggested that, at least at 0 h of recovery, the increase of Survivin level occurred independently of VEGF. This is consistent with the report by Conway et al. [22] that hypoxia alone can induce Survivin expression in the brain, and this response is at least partially independent of VEGF.
The increase in VEGF observed after hypoxic insult in our study is consistent with reports of other investigators [25–29]. However, the role of this increase on apoptotic pathways in the newborn brain will require future investigation. VEGF has been reported to have two opposing roles in the brain. It can have pro-inflammatory properties and increases brain edema and tissue damage following ischemia [30]. On the other hand, it can also induce angiogenesis and increase vascular permeability [31, 32], and these would be expected to be neuro-protective by inducing proliferation of astrocytes and preventing death of dopaminergic neurons [33]. As mentioned earlier, VEGF has been reported to have anti-apoptotic properties by up-regulation of Survivin, Akt and Bcl-2, key anti-apoptotic proteins [24, 34–36].
Another anti-apoptotic protein which was increased during post-hypoxic recovery is Bcl-XL. Bcl-XL has been proposed as a strong endogenous neuronal survival factor against neonatal hypoxic-ischemic injury [37]. Bcl-XL is an integral protein localized primarily in the mitochondrial membrane and had been shown to diminish cell death by suppressing release of apoptogenic proteins [38–40].
It was also observed increase in expression of Heat-shock protein 27 (Hsp27) during post-hypoxic recovery. Hsp27 has several functions; one of them is regulation of apoptosis. It has been reported that Hsp27 interacts with outer mitochondrial membranes and, by interfering with the activation of cytochrome c-Apaf1-dATP complex, inhibits activation of procaspase-9 [41]. Hsp27 can also inhibit Daxx apoptotic protein and thus prevent association of Daxx with Fas and Ask1 [42].
It appears, not surprisingly, that cells stressed by hypoxic/ischemic episode respond by increasing the amount of many proteins that help protect them from serious long term injury and/or death. These proteins form a coordinated stress response system designed to counter the deleterious effects of stressful episode on the cell, and affect almost every facet of the cell structure and metabolism. The question, therefore, arises: what is the response of some pro-apoptotic proteins in the striatum of the newborn to hypoxic insult?
In the present study, the levels of caspase-2, -5, -6, -8 and -9 did not change in response to the hypoxic episode when compared to sham operated animals. In contrast, caspase-1 and -7, as well as Bim, Apoptosis Inducing Factor (AIF), and calpain, were all significantly increased in the striatum after hypoxic-ischemic insult. Caspase-3 was not measured in the present study, but we have previously reported no significant changes in expression of this enzyme following hypoxic insult [12].
The caspase cascade plays a vital role in the induction, transduction, amplification and execution of apoptotic signals within the cell. Activation of caspases (cysteine proteases) is an essential component of the process of apoptosis [43]. Caspases have been reported to be activated after hypoxia–ischemia [6, 44] and inhibition of caspase activity has been reported to have neuroprotective effects in the immature brain [45, 46]. Upon their activation, through the intrinsic and/or extrinsic pathways, caspases can destroy essential cellular proteins, promoting controlled cell death. There are two tiers of caspase activation during apoptosis. Initiator caspases (-2, -8, -9 and -10) are activated through the apoptosis-signaling pathways and activate the effector caspases (-3, -6 and -7) which, in an expanding cascade, carry out the apoptosis. In addition, caspase-1 can activate inflammatory processes. The present study shows that hypoxic insult activates caspase-1 and -7. The increased expression of caspase-1 is in agreement with other studies showing an increase of caspase-1 level in brain neurons in response to hypoxic insults both in vivo and in vitro [47, 48]. Caspase-1 catalyzes cleavage of inactive precursor of IL-1b to generate mature cytokine. IL-1b acts to initiate inflammation, and this contributes to neuronal injury [49]. Caspase-7 had been generally characterized as an executioner of apoptosis [50]. However, Lakhani et al. [51] showed that this caspase plays also key role in the initial phase of apoptosis such as Bax translocation and cytochrome c release. Increase in activation of caspase-7 is in agreement with Yin et al. [52] who reported that in a rat pup model of hypoxia–ischemia caused activation of caspase 7 and also other caspases such as -3, -8 and -9. Lack of increase in expression of caspases-3, -8 and -9 in our study could be due to differences in severity of the hypoxic insult, animal age, species of experimental animals, and anatomical region examined.
When caspase activation is inhibited, neurons can still be injured through caspase-independent pathways [53, 54]. A major regulatory protein in caspase-independent cell death pathway is the apoptosis-inducing factor (AIF; [40, 55, 56]). AIF has been shown to increase apoptotic pathway induced by glutamate or oxidative stress [57, 58], trauma and ischemia [59, 60]. The present study shown increase in expression of AIF during post-hypoxic recovery in striatum of newborn piglets. Data from literature indicated that after hypoxic-ischemic insult to the developing brain, AIF and caspases could act through parallel pathways that can lead to neuronal death [61, 62]. Observed in our model, increase in expressions of caspase-1 and -7 and AIF are consistent with the mechanism(s) of apoptotic injury proposed above by other investigators [61, 62]. Bim, a pro-apoptotic member of Bcl-2 family, is another protein up-regulated following hypoxia. Bim has been reported to play an important role in neuronal death following neonatal hypoxia–ischemia [63]. The authors reported that deficiency of the pro-apoptotic BH3-only molecules Bad or Bim is protective in the Rice–Vannucci model of neonatal hypoxia–ischemia. The increased level of Bim observed in our study is in agreement with the reports of other investigators showing that Bim increased following hypoxic-ischemic insults [64, 65]. Another pro-apoptotic protein that increased following hypoxia was calpain, calcium binding protease. This is consistent with the report of Carloni et al. [66] showing significant increase in calpain 30 min after hypoxia–ischemia in a neonatal rat model. As response to hypoxia, there is increase in cytoplasmic free calcium derived from the combined influx of extracellular calcium and the release of calcium present in the endoplasmic reticulum (ER; [67]). This can activate calcium dependent proteins, such as calpain, related to cell death [68, 69].
In conclusion, this study was designed to gain understanding of the metabolic signaling changes that are initiated by hypoxic insult and can lead to cell loss through apoptosis. Through understanding the signaling processes associated with progression toward cellular injury and apoptotic cell death, it should be possible to design rational protective interventions. Such protective interventions would be designed to suppress pro-apoptotic and/or promote anti-apoptotic signaling. Our results show that 1 h of hypoxic insult to newborn brain causes increases in the levels of several proteins which, if unopposed, can promote apoptotic or necrotic cell death. However, it also leads to increased levels of regulatory proteins that have protective roles, in part by countering the effects of the pro-apoptotic proteins. The final outcome, as measured in loss of brain cells and neuronal function, is determined by the relative activity of these pro- and anti- apoptotic regulatory processes. Treatments that preferentially favor one or the other process would be expected to markedly affect the final outcome.
Acknowledgments
Supported in part by Grants NS-31465 and HL058669 from the US National Institutes of Health.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Bracci R, Perrone S, Buonocore G. The timing of neonatal brain damage. Biol Neonate. 2006;90:145–1552. doi: 10.1159/000092517. [DOI] [PubMed] [Google Scholar]
- 2.Kadhim H, Sebire G, Kahn A, et al. Causal mechanisms underlying periventricular leukomalacia and cerebral palsy. Curr Pediatr Rev. 2005;1:1–63. doi: 10.2174/1573396052953471. [DOI] [Google Scholar]
- 3.Vexler ZS, Ferriero DM. Molecular and biochemical mechanisms of perinatal brain injury. Semin Neonatol. 2001;6:99–108. doi: 10.1053/siny.2001.0041. [DOI] [PubMed] [Google Scholar]
- 4.Blomgren K, Zhu C, Wang X, et al. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of ‘pathological apoptosis’? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- 5.Hu BR, Liu CL, Ouyang Y, et al. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab. 2000;20:1294–1300. doi: 10.1097/00004647-200009000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Zhu C, Wang X, Xu F, et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005;12:162–176. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]
- 7.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 8.Puka-Sundvall M, Gajkowska B, Cholewinski M, et al. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia ischemia in immature rats. Brain Res Dev Brain Res. 2000;125:31–41. doi: 10.1016/S0165-3806(00)00110-3. [DOI] [PubMed] [Google Scholar]
- 9.Northington FJ, Ferriero DM, Flock DL, et al. Delayed neurodegeneration in neonatal rat thalamus after hypoxia–ischemia is apoptosis. J Neurosci. 2001;21:1931–1938. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Northington FJ, Ferriero DM, Martin LJ. Neurodegeneration in the thalamus following neonatal hypoxia–ischemia is programmed cell death. Dev Neurosci. 2001;23:186–191. doi: 10.1159/000046141. [DOI] [PubMed] [Google Scholar]
- 11.Felderhoff-Mueser U, Taylor DL, Greenwood K, et al. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic–ischemic injury to the developing rat brain. Brain Pathol. 2000;10:17–29. doi: 10.1111/j.1750-3639.2000.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pirzadeh A, Mammen A, Kubin J, et al. Early regional response of apoptotic activity in newborn piglets brain following hypoxia and ischemia. Neurochem Res. 2010;36(1):83–92. doi: 10.1007/s11064-010-0267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamm I, Wang Y, Sausville E, et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58:5315–5320. [PubMed] [Google Scholar]
- 14.Shin S, Sung BJ, Cho YS, et al. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry. 2001;40:1117–1123. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 15.Altieri DC. Survivin in apoptosis control and cell cycle regulation in cancer. Prog Cell Cycle Res. 2003;5:447–452. [PubMed] [Google Scholar]
- 16.Kobayashi K, Hatano M, Otaki M, et al. Expression of a murine homologue of the inhibitor of apoptosis protein is related to cell proliferation. Proc Natl Acad Sci USA. 1999;96:1457–1462. doi: 10.1073/pnas.96.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ambrosini G, Adida C, Altieri D. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 18.Skoufias DA, Mollinari C, Lacroix FB, et al. Human survivin is a kinetochore-associated passenger protein. J Cell Biol. 2000;51:1575–1582. doi: 10.1083/jcb.151.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, Ackermann EJ, Bennett CF, et al. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461–466. doi: 10.1038/70242. [DOI] [PubMed] [Google Scholar]
- 20.Uren AG, Wong L, Pakusch M, et al. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319–1328. doi: 10.1016/S0960-9822(00)00769-7. [DOI] [PubMed] [Google Scholar]
- 21.Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 22.Conway EM, Femke Zwerts F, Van Eygen V, et al. Survivin-dependent angiogenesis in ischemic brain. Molecular mechanisms of hypoxia-induced up-regulation. Am J Pathol. 2003;163:935–946. doi: 10.1016/S0002-9440(10)63453-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran J, Rak J, Sheehan C, et al. Marked induction of the IAP family antiapoptotic proteins surviving and XIAP by VEGF in vascular endothelial cells. Biochem Biophys Res Comm. 1999;264:781–788. doi: 10.1006/bbrc.1999.1589. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor DS, Schechner JS, Adida C, et al. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol. 2000;156:393–398. doi: 10.1016/S0002-9440(10)64742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aly H, Hassanein S, Nada A, et al. Vascular endothelial growth factor in neonates with perinatal asphyxia. Brain Dev. 2009;31:600–604. doi: 10.1016/j.braindev.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 27.Cheung CY. Vascular endothelial growth factor: possible role in fetal development and placental function. J Soc Gynecol Investig. 1997;4:169–177. doi: 10.1016/S1071-5576(97)00025-7. [DOI] [PubMed] [Google Scholar]
- 28.Marti HJ, Bernaudin M, Bellail A, et al. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. doi: 10.1016/S0002-9440(10)64964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lennmyr F, Ata KA, Funa K, et al. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–882. doi: 10.1097/00005072-199809000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Croll SD, Goodman JH, Scharfman HE. Vascular endothelial growth factor (VEGF) in seizures: a double-edged sword. Adv Exp Med Biol. 2004;548:57–68. doi: 10.1007/978-1-4757-6376-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gluzman-Poltorak Z, Cohen T, Herzog Y, et al. Nuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165. J Biol Chem. 2000;275:18040–18045. doi: 10.1074/jbc.M909259199. [DOI] [PubMed] [Google Scholar]
- 32.Saito Y, Uppal A, Byfield G, et al. Activated NAD(P)H oxidase from supplemental oxygen induces neovascularization independent of VEGF in retinopathy of prematurity model. Invest Ophthalm Vis Sci. 2008;49:1591–1598. doi: 10.1167/iovs.07-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Temburni MK, Jacob MH. New functions for glia in the brain. Proc Natl Acad Sci USA. 2001;98:3631–3632. doi: 10.1073/pnas.081073198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 35.Tran J, Rak J, Sheehan C, et al. Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem Biophys Res Comm. 1999;264:781–788. doi: 10.1006/bbrc.1999.1589. [DOI] [PubMed] [Google Scholar]
- 36.Zhu WH, MacIntyre A, Nicosia RF. Regulation of angiogenesis by vascular endothelial growth factor and angiopoietin-1 in the rat aorta model: distinct temporal patterns of intracellular signaling correlate with induction of angiogenic sprouting. Am J Pathol. 2002;161:823–830. doi: 10.1016/S0002-9440(10)64242-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsadanian AS, Cheng Y, Keller-Peck CR, et al. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci. 1998;18:1009–1019. doi: 10.1523/JNEUROSCI.18-03-01009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kluck RM, Bossy-Wetzel E, Green DR, et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 40.Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 41.Sarto C, Binz PA, Mocarelli P. Heat shock proteins in human cancer. Electrophoresis. 2000;21:1218–1226. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1218::AID-ELPS1218>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 42.Charette SJ, Lavoie JN, Lambert H, et al. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol Cell Biol. 2000;20:7602–7612. doi: 10.1128/MCB.20.20.7602-7612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 44.Hu BR, Liu CL, Ouyang Y, et al. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab. 2000;20:1294–1300. doi: 10.1097/00004647-200009000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Zhu C, Hagberg H, et al. X-linked inhibitor of apoptosis (XIAP) protein protects against caspase activation and tissue loss after neonatal hypoxia-ischemia. Neurobiol Dis. 2004;16:179–189. doi: 10.1016/j.nbd.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 46.Cheng Y, Deshmukh M, D’Costa A, et al. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J, Nagayama T, Jin K, et al. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamatani M, Mitsuda N, Matsuzaki H, et al. A pathway of neuronal apoptosis induced by hypoxia/reoxygenation: roles of nuclear factor-UB and Bcl-2. J Neurochem. 2000;75:683–693. doi: 10.1046/j.1471-4159.2000.0750683.x. [DOI] [PubMed] [Google Scholar]
- 49.Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog Neurobiol. 2002;67:161–172. doi: 10.1016/S0301-0082(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 50.Kuribayashi K, Mayes PA, El-Deiry WS. What are caspases 3 and 7 doing upstream of the mitochondria? Cancer Biol Ther. 2006;5:763–765. doi: 10.4161/cbt.5.7.3228. [DOI] [PubMed] [Google Scholar]
- 51.Lakhani SA, Masud A, Kuida K, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin W, Cao G, Johnnides MJ, et al. TAT-mediated delivery of Bcl-XL protein is neuroprotective against neonatal hypoxic–ischemic brain injury via inhibition of caspases and AIF. Neurobiol Dis. 2006;21:358–371. doi: 10.1016/j.nbd.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 53.Susin SA, Daugas E, Ravagnan L, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida H, Kong YY, Yoshida R, et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/S0092-8674(00)81733-X. [DOI] [PubMed] [Google Scholar]
- 55.Cheung EC, Melanson-Drapeau L, Cregan SP, et al. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX dependent and BAX-independent mechanisms. J Neurosci. 2005;25:1324–1334. doi: 10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 57.Dumont C, Durrbach A, Bidere N, et al. Caspase-independent commitment phase to apoptosis in activated blood T lymphocytes: reversibility at low apoptotic insult. Blood. 2000;96:1030–1038. [PubMed] [Google Scholar]
- 58.Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 59.Cao G, Pei W, Ge H, et al. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X, Chen J, Graham SH, et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–191. doi: 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]
- 61.Zhu C, Wang X, Huang Z, et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007;14:775–784. doi: 10.1038/sj.cdd.4402053. [DOI] [PubMed] [Google Scholar]
- 62.Zhu C, Qiu L, Wang X, et al. Involvement of apoptosis inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem. 2003;86:306–317. doi: 10.1046/j.1471-4159.2003.01832.x. [DOI] [PubMed] [Google Scholar]
- 63.Ness JM, Harvey CA, Strasser A, et al. Selective involvement of BH3-only Bcl-2 family members Bim and Bad in neonatal hypoxia–ischemia. Brain Res. 2006;1099:150–159. doi: 10.1016/j.brainres.2006.04.132. [DOI] [PubMed] [Google Scholar]
- 64.Kuan CY, Whitmarsh AJ, Yang DD, et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Nat Acad Sci USA. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shibata M, Hattori H, Sasaki T, et al. Temporal profiles of the subcellular localization of Bim, a BH3-only protein, during middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2002;22:810–820. doi: 10.1097/00004647-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 66.Carloni S, Carnevali A, Cimino M, et al. Extended role of necrotic cell death after hypoxia–ischemia-induced neurodegeneration in the neonatal rat. Neurobiol Dis. 2007;27:354–361. doi: 10.1016/j.nbd.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 67.Seta KA, Yuan Y, Spicer Z, et al. The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium. 2004;36:331–340. doi: 10.1016/j.ceca.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 68.Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 69.Liu J, Liu MC, Wang KK (2008) Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal 1:re1 [DOI] [PubMed]