

Abstract
The 12/15-lipoxygenase(s) (LOX), poly(ADP-ribose) polymerase (PARP-1) activity and mitochondrial apoptosis inducing factor (AIF) protein in the amyloid β (Aβ) toxicity were investigated in PC12 cells that express either wild-type (APPwt) or double Swedish mutation (APPsw) forms of human Aβ precursor protein. Different levels of Aβ secretion and free radicals formation characterize these cells. The results demonstrated a relationship between the Aβ levels and LOX protein expression and activity. High Aβ concentration in APPsw cells correlated with a significant increase in free radicals and LOX activation, which leads to translocation of p65/NF-κB into the nucleus. An increase in AIF expression in mitochondria was observed concurrently with inhibition of PARP-1 activity in the nuclear fraction of APPsw cells. We suggested that AIF accumulation in mitochondria may be involved in adaptive/protective processes. However, inhibition of PARP-1 may be responsible for the disturbances in transcription and DNA repair as well as the degeneration of APP cells. Under conditions of increased nitrosative stress, evoked by the nitric oxide donor, sodium nitroprusside (SNP, 0.5 mM), 70–80% of all cells types died after 24 h, significantly more in APPsw cells. There was no further significant change in mitochondrial AIF level and PARP-1 activity compared to corresponding non-treated cells. Only one exception was observed in PC12 control, where SNP significantly inhibits PARP-1 activity. Moreover, SNP significantly activated gene expression for 12/15-LOX in all types of investigated cells. Inhibitors of all LOX isoforms and specific inhibitor of 12-LOX enhanced the survival of cells that were subjected to SNP. We conclude that the LOX pathways may play a role in Aβ toxicity and in nitrosative-stress-induced cell death and that inhibition of these pathways offers novel protective strategies.
Keywords: Aβ toxicity, LOX, AIF, PARP, Free radicals, Stress, Cell death
Introduction
Alzheimer`s disease (AD) is a progressive neurodegenerative disorder characterized by the presence of amyloid β senile plaques. Other typical changes in AD include the following: tau protein-containing neurofibrillary tangles, degeneration of axons and dendrites, synaptic failure and neuronal cell death. The amyloid beta (Aβ) peptides are the crucial components responsible for AD pathology and are derived from amyloid precursor proteins (APPs) through an initial β-secretase degradation followed by a γ-secretase cleavage within the membrane-penetrating domain of APP [1]. Aβ peptides exist in several forms, depending on the milieu and the oligomerization/fibrillization stage. Aβ peptides have been implicated in free radical formation as well as alterations in several intracellular and intercellular signaling pathways [2–5]. Numerous hypotheses exist regarding the role of Aβ peptides in the pathogenesis of AD. Some data suggest a direct action of Aβ oligomers [6] whereas other studies provide evidence that the Aβ peptides activate cell surface receptors [7] or alter the immune response [8]. Cyclooxygenases (COXs) and lipoxygenases (LOXs) contribute to oxidative stress and AD pathogenesis. Moreover, it was observed that COX-2 expression could be one of the early events in Aβ toxicity occurring during the first stage of AD [8–10]. The Aβ-evoked free radical cascade involves several cellular pathways, including nitric oxide (NO)-mediated signaling. NO release leads to the activation of cytosolic phospholipase A2 (cPLA2) and the liberation of arachidonic acid (AA), which in turn, is metabolized by COX and LOX [2, 4, 11]. The LOX pathway produces leukotrienes, lipoxins, prostanes and other derivates. Several studies suggest that 12/15-LOX is a major source of oxidative stress [12]. In AD-affected frontal and temporal regions, 12/15-LOX protein levels were higher compared with controls. The metabolic products of 12/15-LOX, 12/15-hydroxyeicosatetraenoic acids, were also markedly elevated in AD brains [12, 13]. The increase in the activity of 12/15-LOX metabolic pathway was shown to correlate with an increasing of oxidative imbalance in the AD brain. The activation of 12-LOX plays also an important role in the generation of reactive oxygen species (ROS) and in the decrease of neurons viability in glucose deprivation/reload conditions [14]. The data from Lebeau et al. [15] showed that blockade of 12-LOX expression protected cortical neurons from Aβ- induced c-Jun-dependent apoptosis. That study suggested that 12-LOX might represent a new target for the treatment of AD. The Ikonomovic et al. [16] data indicated that intracellular 5-LOX level is the hallmark of AD pathological lesions. Firuzi et al. [17] reported that 5-lipoxygenase gene disruption reduces Aβ pathology in a model of AD. These findings suggested that LOXs might significantly contribute to the pathogenesis of the neurodegenerative processes in AD.
Consequently, Aβ-mediated oxidative stress alters the function of the DNA-bound enzyme, PARP-1, and several ion channel-coupled receptors, signaling pathways and mitochondrial processes [18–21].
Alteration of PARP influences the function of numerous transcription factors, including p53, NF-κB, AP-1 and more than 40 other nuclear proteins [22–26]. In addition to its basic role in local chromatin disassembly and the efficient positioning of DNA repair machinery, PARP over-activation can induce cell death [27, 28].
Moreover, the product of the enzyme poly (ADP-ribose) (PAR) can function as a death signaling molecule leading to liberation of apoptosis-inducing factor (AIF) from the mitochondria and its translocation to the nucleus [29–32]. Under resting conditions, AIF is anchored to the inner mitochondrial membrane where it acts as a complex I stabilizer and functions as an NADH oxidase [33]. Our previous results suggest a pro-survival role of AIF accumulation in mitochondria under conditions of nitrosative stress evoked by the NO donor, sodium nitroprusside (SNP) [34]. Based on our current knowledge, we suggest that PARP-1 and AIF can be considered as molecular targets for Aβ mediated signaling.
The aim of our study was to investigate the role of LOXs, AIF and PARP-1 in Aβ-evoked oxidative stress signaling in PC12 cells that were subjected to nitrosative stress; these cells were either transfected with wild type human amyloid precursor protein gene (APPwt) or carried a double Swedish mutation (APPsw).
Materials and Methods
Materials
The reagents and antibodies were obtained as follows: ECL reagents were purchased from GE Healthcare, Piscataway NJ, USA; anti-12/15 LOX antibody was obtained from Cayman Chemical, Ann Arbor MI, USA; anti-β-actin IgG was acquired from MP Biomedicals, Irvine CA, USA; anti-cytochrome c was purchased from BD Pharmingen, SD, USA anti-AIF and anti-p65 IgG were purchased from Santa Cruz Biotechnology, Santa Cruz CA, USA; anti-mouse IgG was obtained from GE Healthcare, Piscataway NJ, USA; and anti-PARP-1 antibody, anti-rabbit IgG and other biochemical and inorganic reagents were acquired from Sigma, St. Louis MO, USA.
Cell Culture
Rat pheochromocytoma PC12 cells were kindly provided by Prof. Walter E. Müller and Prof. A. Eckert from the Department of Pharmacology Biocenter (University of Frankfurt, Germany). The PC12 cells used for the experiments contained DNA expression constructs harboring human wild-type (APPwt) or mutant APP gene (APPsw, K670 M/N671L) inserted downstream of a cytomegalovirus promoter [4]. Cells transfected with an empty vector were used as controls. The cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 5% horse serum, 50 units/ml penicillin, 50 μg/ml streptomycin, 400 μg/ml G418 and 2 mM glutamine at 37°C in a humidified incubator containing 5% CO2. Cells were subcultured approximately twice a week after achieving 80% confluency. For experiments, confluent cells were subcultured into polyethyleneimine-coated 10-cm dishes or 24-well plates and were used at 75–90% confluence or 1 day after being plated into 24-well plates. Prior to treatment, the used media was replaced with fresh media containing 2% fetal bovine serum. PC12, APPwt, and APPsw cell lines secreted Aβ peptide in increasing amounts as previously described [2].
Cell Treatment Protocols
PC12 cells were treated with SNP at 0.5 mM for various times up to 24 h. Additionally LOX inhibitors [2.5 μM baicalein or 0.5 μM nordihydroguaiaretic acid (NDGA)] were used together with 0.5 mM SNP.
Isolation of Cytosolic, Mitochondrial and Nuclear Fractions
Fractinaction of the PC12 cells was made as previously described by Wang et al. [35]. Cells were washed, scraped into ice-cold PBS and pelleted at 1,000× rpm for 3 min at 4°C. The pellet was resuspended in hypotonic buffer (10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, 1 mM dithiotreitol, 1.5 mM MgCl2, 10 mM KCl and protease inhibitors cocktail); cell membranes were disrupted by 10 passes through a 26-gauge needle and pelleted at 500g for 10 min at 4°C. The pellet (P1, the crude nuclear fraction) was resuspended in 25 mM Tris, pH 7.4 with protease inhibitors and used for western blot analysis. The supernatant (S1) was centrifuged at 15,000g for 15 min at 4°C to obtain a cytosolic (S2) and a crude mitochondrial fraction (P2). The immunochemical determination of typical mitochondrial protein, cytochrome c, demonstrated that it is exclusively present in the mitochondria fraction, and indicated concomitantly that other subcellular fractions (nuclear and cytosolic) are not contaminated by mitochondria.
Then immunochemical detection of DNA bound enzyme PARP-1 was observed exclusively in nuclear fraction and has never been detected by us in mitochondrial and cytosolic fractions. It indicated that the other subcellular fractions are not contaminated.
Determination of Amyloid β1-40
Cells were plated at equal density in 6-well plates. After reaching confluence, 2 ml of conditioned media was collected. Media were centrifuged to remove cell fragments, and aliquots were then used to determine the Aβ1–40 level. For determination of secreted Aβ1–40, a specific sandwich enzyme linked immunosorbent assay (ELISA) employing monoclonal antibodies was used. The ELISA was performed according to the instructions given in the Aβ-ELISA kit by Genetics Company. The assay principle is that of a standard sandwich ELISA, which utilizes a site-specific rabbit anti-human N-terminal capture, a cleavage sitespecific rabbit anti-human Aβ1–40 terminal detection antibody, and anti-rabbit IgG peroxidase-conjugated secondary antibody.
Determination of Free Radicals Using DCF Probe
ROS production in PC12 cells was assessed by using the 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) probe. The cell media was changed to Phenol Red-free Hanks’ buffer and incubation was continued in the presence of 10 μM H2DCF-DA for 50 min at 37°C. Cells were washed three times in Hanks’ buffer and lysed with DMSO. The fluorescence (excitation 485 nm and emission 535 nm) was quantified in the lysate by using a LS-50B Spectrofluorimeter (Perkin Elmer).
MTT Reduction Assay
Cell viability and mitochondrial function were evaluated using 2-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). PC12 cells were seeded onto 24-well culture plates coated with 0.1% polyethyleneimine in 25 mM borate buffer and allowed to attach. Media containing 2% fetal bovine serum, 50 units/ml penicillin, 50 μg/ml streptomycin, and 0.5 mM SNP was added to the cells for a given period of time. MTT was then added to all wells and the cells were incubated at 37°C for 2 h, followed by lysis of the cells and spectrophotometric measurement at 595 nm.
Measurement of PARP-1 Activity
PARP activity was determined using 14C-labeled β-NAD+ as described previously Strosznajder et al. [27, 28]. The incubation mixture contained 200 μM [adenine-14C] β-NAD+ (4 × 105 d.p.m.), 100 mM Tris/HCl buffer (pH 8.0), 10 mM MgCl2, 5 mM DTT, 50 μM PMSF and 200 μg of protein in a final volume of 100 μl. The mixture was incubated for 1 min at 37°C and the reaction was stopped with 0.8 ml of ice-cold 25% trichloroacetic acid. Precipitates were collected on Whatman GF/B filters, washed three times with 5% trichloroacetic acid and left overnight for drying. The radioactivity was measured in Bray scintillation fluid using a LKB Wallac 1409 scintillator counter.
Measurement of Lipoxygenase (LOX) Activity
The activity of LOX was determined using the colorimetric method described by Waslidge and Hayes [36]. The cell lysate was diluted fourfold on ice with 50 mM Tris–HCl buffer, pH 7.4, transferred into an ice-cold 96-well plate. The assay was initiated by the addition of 50 μl arachidonic acid (final concentration, 70 μM) in 50 mM Tris–HCl buffer, pH 7.4, and incubated at 37°C for 10 min. The assay was terminated by the addition of 100 μl FOX reagent: sulfuric acid (25 mM), xylenol orange (100 μM), iron (II) sulfate (100 μM), methanol:water (9:1). Blanks contained enzyme during the incubation but substrate was added after the FOX reagent. The yellow color of the acidified xylenol orange was converted to a blue color by the lipid hydroperoxide-mediated oxidation of Fe2+ ions and interaction of the resulting Fe3+ ions with the dye. Absorbance of the ferrium ions complex at 620 nm was measured using Bio Rad plate reader.
Analysis of mRNA Levels (Real Time PCR for 12/15-LOX, AIF, PARP-1, β-Actin)
RNA was isolated using TRI-reagent from Sigma. The isolated RNA was dissolved in RNAse-free water (Promega Corporation). The amount and purity of RNA was determined using spectrophotometric measurement at 260 and 280 nm. The OD260/OD280 ratio of the RNA samples ranged from 1.6 to 1.9. Isolated RNA (5 μg) was used in RT–PCR. The reverse transcription was performed by using a High Capacity cDNA Reverse Transcription Kit according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA, USA). Quantitative PCR was performed on an ABI PRISM 7500 apparatus by using pre-developed TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions: Alox15 (15-LOX) Rn00696151_m1*, Aifm1 (AIF) m1-Rn00442540_m1, Adpr1- (PARP-1) Rn00565018, Actb (β-Actin) Rn01412977_g1. Actb was selected and used as a reference gene in all studies. The relative level of mRNA was calculated by the DDCt method.
Western Blot
After determination of the total protein content using the Lowry method, the cytosolic, mitochondrial, or nuclear fraction was mixed with 5× SDS-containing sample buffer and denatured for 5 min at 95°C. Protein (40 μg/lane) was loaded onto 10% denaturing polyacrylamide gels and resolved by SDS–PAGE. The proteins were transferred onto Polyvinylidene Fluoride membranes at 100 V. Membranes were incubated in 5% fatty acid free dry milk in TBS (Tris-buffered saline; if not otherwise indicated) with Tween-20 (TBS-T) for 1 h and were exposed overnight at 4°C to the following antibodies: anti-12/15-LOX 1:500; anti-AIF 1:750; anti-cytochrome c 1:500; anti p65 (in PBS-T) 1:100; anti-PARP-1 (in PBS-T) 1:500; or anti-actin 1:1,000. After treatment for 1 h with the appropriate horseradish peroxidase-coupled secondary antibodies (1:4,000, 1:8,000 anti-rabbit IgG, or 1:2,000, 1:4,000 anti-mouse IgG), the protein bands were visualized with enhanced chemiluminescence (GE Healthcare) and autoradiographed. The bands were analyzed using TotalLab™ densitometry package from Phoretix.
After stripping with a pH 2.5 solution of 50 mM glycine and 1% sodium dodecyl sulfate (SDS) for 30 min at room temperature, the membranes were re-used for subsequent western blot experiments.
Statistical Analysis
All experiments were repeated at least 3 times and were performed either in triplicate or duplicate. The presented data are the means ± SEM. For statistical comparison, Student’s t test or one-way ANOVA followed by Bonferroni post-hoc test were used; P values <0.05 were considered statistically significant.
Results
PC12 cells transfected with human wild-type APP (APPwt) or APP carrying the double Swedish mutation (APPsw) were used in this study and showed a 2.8 or 4.9-fold higher 1-40 Aβ production, respectively, than PC12 control cells transfected with an empty vector (Fig. 1a). APPwt and APPsw cells displayed significantly increased levels of free radicals as determined by DCF fluorescence (Fig. 1b). Moreover, the level of free radicals is significantly higher in APPsw cells comparing to APPwt. The protein immunoreactivity of 12/15-LOX was enhanced in both APP transfected cell types, but this enhancement was significant in the APPsw cells (Fig. 1c). LOX activity was also significantly higher in APPsw cells indicating a close relationship between these changes and Aβ concentration (Fig. 1d). We demonstrated previously that a γ-secretase inhibitor (DAPT) effectively decreased the Aβ-mediated release of arachidonic acid (AA), the substrate for LOXs, indicating a direct involvement of Aβ in this processes [2].
Fig. 1.
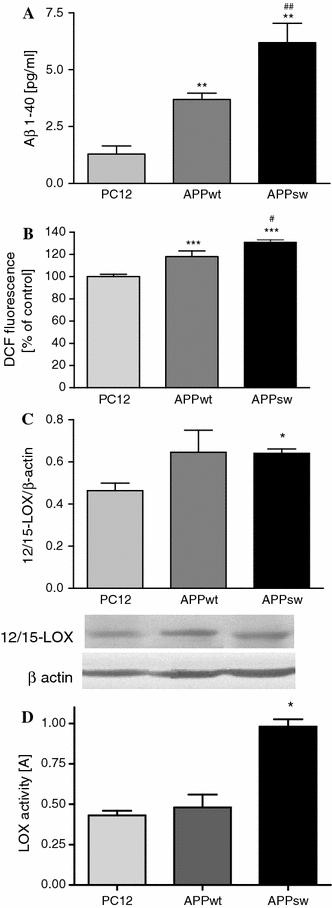
The level of amyloid β secretion, free radicals and LOX protein expression and activity in APP transfected PC12 cells. a The cells were transfected with an empty vector (control) or with an expression vector containing wt (APPwt) or mutated (APPsw) APP human gene. The amyloid β1-40 secretion was evaluated by Aβ-ELISA kit. b ROS generation in PC12, APPwt, APPsw cells was estimated using spectrofluorescence method (H2DCF-DA). c 12/15-LOX protein level in APP transfected PC12 cells was estimated using western blot analysis. A representative western blot from one typical experiment was selected from 3 independent experiments. d LOX activity in PC12, APPwt, APPsw cells was measured using the colorimetric method according to Waslidge and Hayes [36]. All data represent the mean value ± S.E.M for 3 independent experiments (in triplicate for LOX activity). *P < 0.05, **P < 0.01, ***P < 0.001 versus control cells # P < 0.05, ## P < 0.01 versus APPwt cells, ANOVA with Bonferroni post-hoc test
The oxidative stress evoked by Aβ may lead to the activation of NF-κB. APPsw cells characterized by high levels of Aβ secretion displayed significant enhancement of p65 (a subunit of the NF-κB) immunoreactivity in the nuclear fraction (Fig. 2a). Nordihydroguaiaretic acid (NDGA), an inhibitor of all lipoxygenase isoforms, significantly reduced the nuclear translocation of p65 in cells expressing APPsw (Fig. 2b). Baicalein, a 12/15-lipoxygenase inhibitor, also reduced the nuclear immunoreactivity of p65, although to a lesser degree (Fig. 2b).
Fig. 2.

Nuclear immunoreactivity of the p65 subunit of the NF-κB transcription factor in PC-12 cells and the effect of LOX inhibitors on the p65 level. a The effect of endogenous Amyloid β on p65 immunoreactivity. Nuclear p65 protein level in PC12, APPwt, and APPsw cells was estimated using western blot analysis. A representative western blot from one typical experiment is presented. b The effect of LOX inhibitors (Baicalein and NDGA) in APPsw cells on p65 immunoreactivity was estimated by western blot. Baicalein was used at 2.5 μM and NDGA at 0.5 μM. A representative western blot from one typical experiment is below the graph. All data represent the mean value ± S.E.M for 3 independent experiments normalized against the immunoreactivity of β-actin. *P < 0.05, ANOVA with Bonferroni post-hoc test
The Aβ peptides mediated oxidative stress also influenced mitochondrial function. Our previous data indicated that a γ-secretase inhibitor significantly reduced the level of Aβ and mitochondrial dysfunction by normalizing NO and ATP levels and the mitochondrial membrane potential in APPsw PC12 cells [4]. In the current study, a statistically significant increase in AIF levels in the mitochondria of APPsw cells was observed (Fig. 3a). However, AIF (Aifm1) mRNA level was not altered (Fig. 3a). The nuclear target for free radical cascade is DNA bound enzyme PARP-1. The activity of this enzyme was significantly decreased in APPsw cells but was not significantly increased in APPwt. (Fig. 3b). In both APP transfected cell lines, the PARP-1 (Adpr1) mRNA level was not changed (Fig. 3b). The statistically significant alterations in mitochondrial AIF levels and PARP-1 activity in the nuclear fraction were observed in APPsw cells, which are characterized by the highest Aβ production.
Fig. 3.
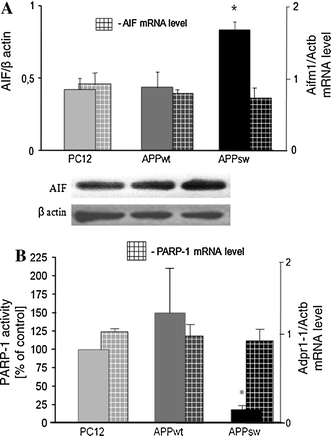
Mitochondrial AIF level and PARP-1 activity in PC12 cells transfected with human APP. a The mitochondrial AIF protein level and its gene expression (Aifm1) in PC12 control cells and APPwt or APPsw. Protein level was estimated by western blot. AIF mRNA was evaluated using Real Time PCR. The results are means ± S.E.M. of 4 experiments normalized against β-actin. b PARP-1 activity was measured radiochemically with [14C] NAD+ [27] in PC12 cell lines. PARP-1 (Adpr1) mRNA was evaluated using Real Time PCR. The data represent the mean value ± S.E.M for 3 independent experiments (for PARP-1 activity in triplicate). *P < 0.05, Student`s t test
To determine the susceptibility of the APP transfected cells to additional stress and to evaluate if the observed pronounced alterations of AIF and PARP-1 in APPsw cells will be futher changed, NO donor was used in further experiments.
In the following experiments, all cell lines were subjected to nitrosative stress evoked by sodium nitroprusside (SNP, 0.5 mM), an NO donor. The western blot signals for AIF in the mitochondrial fraction and PARP-1 activity were not changed by SNP as compared to corresponding SNP untreated controls. The significantly lower PARP-1 activity was observed only in SNP-treated PC12 control cells (data not shown) [34]. Moreover, SNP treatment significantly increased 12/15-LOX gene expression (determined by the level of Alox15 versus corresponding controls) in all examined cell lines. More pronounced activation of 12/15-LOX gene expression in APPsw cells versus APPwt was observed (Fig. 4a). Under these experimental conditions, SNP decreased the viability of all cells types by 70–80% after 24 h. APPsw cells displayed a higher susceptibility to SNP toxicity than PC12 control cells (Fig. 4b). The LOX inhibitors baicalein and NDGA significantly protected the cells against death caused by nitrosative stress (Fig. 4c).
Fig. 4.
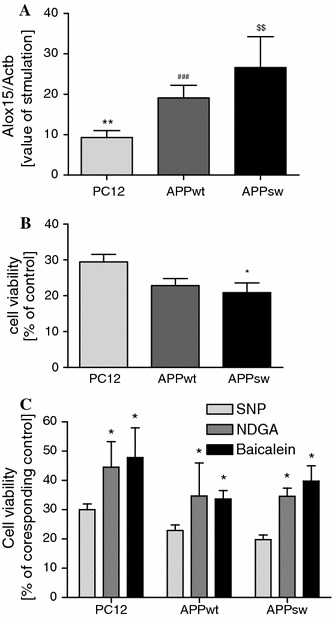
The role of LOX in SNP-induced PC12 cells death. a The effect of SNP treatment on 12/15-LOX expression (Alox15). PC12 cells and PC12 expressing wt (APPwt) or mutated APP human gene (APPsw) were treated with 0.5 mM SNP for 12 h. mRNA levels for 12/15-LOX was evaluated using Real Time PCR. The value expresses the fold of Alox15 gene stimulation normalized against Actb (β-actin) versus corresponding control (100%). b The effect of SNP treatment on cell viability. PC12, APPwt and APPsw cells were treated with 0.5 mM SNP for 24 h, and cell viability was determined by the MTT assay. c The effect of LOX inhibitors (Baicalein and NDGA) on the viability of cells treated with 0.5 mM SNP for 24 h was determined by MTT assay. Cells were incubated in the presence of LOX inhibitors, 2.5 μM Baicalein and 0.5 μM NDGA. All values were calculated as a percentage of control (without SNP) and are means ± S.E.M for 4 independent experiments carried out in triplicate or quadruplicate. ** P < 0.01 versus PC12 control (untreated), ### P < 0.001 versus APPwt control (untreated), $$ P < 0.01 versus APPsw control (untreated), *P < 0.05 versus SNP treated PC12 control, ANOVA with Bonferroni post-hoc test
Discussion
In our current work, we investigated the level of Aβ1-40 secretion, free radicals and the activity of LOX enzymes, AIF level and PARP-1 activity in Aβ-induced oxidative/nitrosative stress. The studies were performed using cell lines that secrete different amounts of Aβ similar to the concentrations observed previously [2, 3]. Our data demonstrate a close relationship between this molecular events in PC12 cells transfected with human APP gene bearing double Swedish mutation (APPsw), secreted 5 times more Aβ comparing to control PC12 and 1,7 times more comparing to APPwt. Moreover the level of free radicals was significantly higher in APPsw and also the liberation of NO was enhanced in this cells line [2].
Free radicals serve as important mediators of Aβ-induced toxicity. Aβ liberated nitric oxide (NO), mediated molecular processes described previously by us [3, 4]. It leads to liberation of arachidonic acid (AA), the LOXs substrate. The enhanced activity of LOXs is connected with the generation of the superoxide radical (O·−2). The interaction of O·−2 with NO· can form peroxynitrite (ONOO−), which leads to the activation of free radicals cascade and to oxidative stress. In this way NO, by regulation of AA level, may be involved in enhancement in LOX activity. On the basis of our results it is difficult to explain the mechanism of LOX activation. Several different covalent modification may occur. Until now, little is known about regulation of LOX activity. Some studies suggested, that dephosphorylation of LOX may by responsible for the stimulation of enzyme activity [50].
Our data presented for the first time the involvement of LOX in Aβ evoked free radicals signaling, that consequently may lead to the enhancement of p65 level in nuclear fraction, increase of AIF level in mitochondria and to inhibition of PARP activity. Despite of several biochemical and ultrastructural alterations described by us [2–5], the significant population of APPsw cells survive during 24 h culturing indicated, that concomitantly with degeneration processes several adaptive/protective mechanisms are activated. It seems that all these observations are very important and could help to better understand the mechanism of Aβ toxicity in Alzheimer Disease (AD). To this day, very few studies evaluated the sequence of events connected with Aβ mediated LOX pathway [37, 38]. Most studies were concentrated attention on cyclooxygenases (COX) the other family of enzymes involved in AA metabolism. However, increase of 12/15 LOX and the elevation of 12/15 LOX products were found in AD and in mild cognitive impairment by Practico et al. and Yao et al. [12, 51, 52]. Their results suggest that the activation of this enzymes occurs early in the course of AD and that the activation of LOXs mediated processes may play important role in the pathogenesis of this disease.
The precise biological role for LOXs in the brain under physiological condition and in AD has not yet be established. A few other studies indicated the involvement of 5-LOX,12-LOX in neuronal death [15, 49, 53]. Our data confirmed the involvement of LOX in oxidative stress. Transcription factor NF-κB may serve as an indicator of the Aβ/LOX-related free radical cascade. APPsw cells, which display enhanced 12/15-LOX level and activity, had higher nuclear levels of p65. This elevation of p65 immunoreactivity was suppressed by a specific inhibitor of all lipoxygenases, NDGA. Additionally, significantly increased mitochondrial AIF levels may suggest its protective role in APPsw cells. This protein possesses NADH reductase activity and has anti-oxidative properties [39–44]. Depending on the severity and duration of stress, AIF levels can be responsible for cell survival or death. In response to excessive stress, AIF may be translocated to the nucleus, where it induces apoptosis or other forms of programmed cell death [26, 31, 39]. It has been previously shown that exogenous Aβ activates the nuclear translocation of AIF [18, 45]. In this study, the endogenous pool of Aβ and its intracellular modification may be responsible for the increase in mitochondrial AIF level.
PARP-1 is another molecular target for stress signaling. Under mild stress, it is involved in DNA repair; however, massive oxidative stress could lead to PARP over-activation and cell death. In addition, under significant oxidative stress, PARP-1 could be poly(ADP-ribosylated) and inactivated [46, 47]. This covalent modification is probably responsible for the inhibition of this enzyme activity in APPsw cells. Based on our results, it is difficult to judge what type of mechanism is responsible for the significant suppression of PARP-1 activity. The NO-dependent mechanism of PARP-1 inhibition observed in our previous studies [34] may also be involved in these conditions. Moreover, Sidorkina et al. [47] showed that PARP-1 activity may be directly suppressed via NO action. In most studies, free radical-dependent DNA damage leads to the activation of PARP-1 [48], such as in APPwt cells; unfortunately the data are not significant in the current study. The suppression of PARP-1 activity in APPsw cells probably plays a dual role: it leads to the alteration of several processes and cell degeneration but may concurrently protect the cells against energy crises and allow most of them to survive.
Despite the significant enhancement of free radicals and the dramatic biochemical and morphological alterations, most of the APPwt and APPsw cells survived [2, 4, 5]. These data suggest that many different degenerative and adaptive/protective processes were concurrently activated in response to Aβ toxicity. It seems that similar changes may occur in brain cells during the early stages of AD. The additional stress evoked by SNP overcomes these adaptive/protective processes developed in the cells and leads to death, more significantly in the APPsw cells compared to PC12 control cells. Enhanced NO production by constitutive and inducible NO synthases was reported in PC12 cells transfected with the APP gene in our previous work [2, 4]. To better understand the role of excessive nitrosative stress in Aβ peptide toxicity, we investigated the effect of the NO donor, SNP.
Nitrosative stress caused by SNP led to significant 12/15-LOX gene expression. The data show that LOX-mediated death signaling occurs under nitrosative stress. The LOX inhibitor protected cells against SNP-induced death. The improvement in survival was greatest in the APPsw-expressing cell line, which exhibited the lowest survival rates without the inhibitors. Our current results add to the evidence that supports the role of AA metabolism by LOX in Aβ evoked oxidative/nitrosative stress and in the pathogenesis of AD.
Acknowledgments
We express our thanks to Anna Kazmierczak PhD, for the help in determination of Aβ level in APP transfected and control cell lines. This study was supported by scientific network 28/E-32/BWSN-0053/2008 and statutory themes NO 6, 7.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- AA
Arachidonic acid
- AIF
Apoptosis-inducing factor
- APP
Amyloid precursor Protein
- APPsw
PC12 cells transfected with an expression vector containing double Swedish-mutated APP gene
- APPwt
PC12 cells transfected with an expression vector containing wild—type APP gene
- DCF
2′,7-dichlorofluorescein
- DMSO
Dimethyl sulfoxide
- G418
An antibiotic used for selection of plasmid-bearing strains
- H2DCFDA
2′,7-dichlorofluorescein diacetate
- LOX
Lipoxygenase
- MTT
2-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- β- NDGA
Nordihydroguajaretic acid
- P1
The crude nuclear fraction
- P2
Mitochondrial fraction
- PAR
Poly(ADP-ribose)
- PARP-1
Poly(ADP-ribose) polymerase-1
- S2
Cytosolic fraction
- SNP
Sodium nitroprusside
Footnotes
Joanna B. Strosznajder and Magdalena Cieslik have contributed equally to this study.
References
- 1.Haass C. Take five-BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004;23:483–488. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chalimoniuk M, Stolecka A, Cakała M, Hauptmann S, Schulz K, Lipka U, Leuner K, Eckert A, Muller WE, Strosznajder JB. Amyloid beta enhances cytosolic phospholipase A2 level and arachidonic acid release via nitric oxide in APP-transfected PC12 cells. Acta Biochim Pol. 2007;54(3):611–623. [PubMed] [Google Scholar]
- 3.Kazmierczak A, Strosznajder JB, Adamczyk A. α-Synuclein enhances secretion and toxicity of amyloid beta peptides in PC12 cells. Neurochem Int. 2008;53:263–269. doi: 10.1016/j.neuint.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Muller-Spahn F, Haass C, Czech C, Pradier L, Muller WE. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004;279(48):50310–50320. doi: 10.1074/jbc.M405600200. [DOI] [PubMed] [Google Scholar]
- 5.Pajak B, Songin M, Strosznajder JB, Gajkowska B. Alzheimer’s disease genetic mutation evokes ultrastructural alterations: correlation to an intracellular Ab deposition and the level of GSK-3b-P (Y216) phosphorylated form. Neurotoxicology. 2009;30(4):581–588. doi: 10.1016/j.neuro.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. The ups and downs of Abeta. Nat Med. 2006;12(7):758–759. doi: 10.1038/nm0706-758. [DOI] [PubMed] [Google Scholar]
- 7.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23(7):2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoozemans JJ, Veerhuis R, Rozemuller JM, Eikelenboom P. Neuroinflammation and regeneration in the early stages of Alzheimer’s disease pathology. Int J Dev Neurosci. 2006;24(2–3):157–165. doi: 10.1016/j.ijdevneu.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Braak E. Neuropathological stageing of alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 10.Hoozemans JJ, Veerhuis R, Rozemuller AJ, Arendt T, Eikelenboom P. Neuronal COX-2 expression and phosphorylation of pRb precede p38 MAPK activation and neurofibrillary changes in AD temporal cortex. Neurobiol Dis. 2004;15:492–499. doi: 10.1016/j.nbd.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 11.Cakala M, Malik AR, Strosznajder JB. Inhibitor of cyclooxygenase-2 protects against amyloid beta peptide-evoked memory impairment in mice. Pharmacol Rep. 2007;59(2):164–172. [PubMed] [Google Scholar]
- 12.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee M-Y. 12/15-lipoxygenase is increased in alzheimer’s disease. Am J Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao L, Pratico D, Rader DJ, Funk CD. 12/15-Lipoxygenase gene disruption and vitamin E administration diminish atherosclerosis and oxidative stress in apolipoprotein E deficient mice through a final common pathway. Prostaglandins Other Lipid Mediat. 2005;78(1–4):185–193. doi: 10.1016/j.prostaglandins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Nagasawa K, Kakuda T, Higashi Y, Fujimoto S. Possible involvement of 12-lipoxygenase activation in glucose-deprivation/reload-treated neuron. Neurosci Lett. 2007;429:120–125. doi: 10.1016/j.neulet.2007.09.073. [DOI] [PubMed] [Google Scholar]
- 15.Lebeau A, Terro F, Rostene W, Pelaprat D. Blockade of 12-lipoxygenase expression protects cortical neurons from apoptosis induced by β-amyloid peptide. Cell Death Differ. 2004;11:875–884. doi: 10.1038/sj.cdd.4401395. [DOI] [PubMed] [Google Scholar]
- 16.Ikonomovic MD, Abrahamson EE, Uz T, Manev H, DeKosky ST. Increased 5-Lipoxygenase immunoreactivity in the hippocampus of patients with Alzheimer’s Diseas. J Histochem Cytochem. 2008;56(12):1065–1073. doi: 10.1369/jhc.2008.951855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Firuzi O, Zhuo J, Chinnici CM, Wisniewski T, Prawico D. 5-Lipoxygenase gene disruption reduces amyloid—pathology in a mouse model of Alzheimer’s disease. FASEB J. 2008;22:1169–1178. doi: 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adamczyk A, Czapski GA, Jesko H, Strosznajder RP. Non A beta component of alzheimer’s disease amyloid and amyloid beta peptides evoked poly(ADP-ribose) polymerase-dependent release of apoptosis-inducing factor from rat brain mitochondria. J Physiol Pharmacol. 2005;56(Suppl 2):5–13. [PubMed] [Google Scholar]
- 19.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPs, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 20.Leuner K, Hauptmann S, Abdel-Kader R, Scherping I, Keil U, Strosznajder JB, Eckert A, Muller WE. Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease? Antioxid Redox Signal. 2007;9(10):1659–1675. doi: 10.1089/ars.2007.1763. [DOI] [PubMed] [Google Scholar]
- 21.Strosznajder RP, Banasik M. Amyloid beta protein affects poly (ADP-ribose) polymerase activity in PC-12 cells in culture. Acta Neurobiol Exp (Wars) 2000;60(2):215. doi: 10.55782/ane-2000-1340. [DOI] [PubMed] [Google Scholar]
- 22.Altmeyer M, Hottiger MO. Poly (ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging. 2009;1:458–469. doi: 10.18632/aging.100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, Gersbach M, Imhof R, Hottiger MO. Acetylation of poly (ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. 2005;280(49):40450–40464. doi: 10.1074/jbc.M507553200. [DOI] [PubMed] [Google Scholar]
- 24.Hassa PO, Hottiger MO. The functional role of poly (ADP-ribose) polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol Life Sci. 2002;59(9):1534–1553. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strosznajder RP, Jesko H, Zambrzycka A. Poly (ADP-ribose) polymerase: the nuclear target in signal transduction and its role in brain ischemia-reperfusion injury. Mol Neurobiol. 2005;31(1–3):149–167. doi: 10.1385/MN:31:1-3:149. [DOI] [PubMed] [Google Scholar]
- 26.Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB. Poly (ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol. 2010;41(2–3):187–196. doi: 10.1007/s12035-010-8124-6. [DOI] [PubMed] [Google Scholar]
- 27.Strosznajder RP, Gadamski R, Czapski GA, Jesko H, Strosznajder JB. Poly (ADP-ribose) polymerase during reperfusion after transient forebrain ischemia: its role in brain edema and cell death. J Mol Neurosci. 2003;20(1):61–72. doi: 10.1385/JMN:20:1:61. [DOI] [PubMed] [Google Scholar]
- 28.Strosznajder RP, Jesko H, Adamczyk A. Effect of aging and oxidative/genotoxic stress on poly(ADP-ribose) polymerase-1 activity in rat brain. Acta Biochim Pol. 2005;52(4):909–914. [PubMed] [Google Scholar]
- 29.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dowson VL, Dowson TM. Poly (ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, Susin SA. Sequential activation of poly (ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strosznajder R, Gajkowska B. Effect of 3-aminobenzamide on Bcl-2, Bax and AIF localization in hippocampal neurons altered by ischemia-reperfusion injury. The immunocytochemical study. Acta Neurobiol Exp (Wars) 2006;66(1):15–22. doi: 10.55782/ane-2006-1583. [DOI] [PubMed] [Google Scholar]
- 32.Yu S-W, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly (ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heeres JT, Hergenrother PJ. Poly (ADP-ribose) makes a date with death. Curr Opin Chem Biol. 2007;11(6):644–653. doi: 10.1016/j.cbpa.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 34.Pytlowany M, Strosznajder JB, Jesko H, Cakala M, Strosznajder RP. Molecular mechanism of PC12 cell death evoked by sodium nitroprusside, a nitric oxide donor. Acta Biochim Pol. 2008;55(2):339–347. [PubMed] [Google Scholar]
- 35.Wang H, Yu S-W, Koh DW, Lew J, Coombos C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Apoptosis-Inducing Factor Substitutes for Caspase Executioners in NMDA-Triggered Excitotoxic Neuronal Death. J Neurosci. 2004;24(48):10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waslidge NB, Hayes DJ. A colorimetric method for the determination of lipoxygenase activity suitable for use in a high throughput assay format. Anal Biochem. 1995;231:354–358. doi: 10.1006/abio.1995.0063. [DOI] [PubMed] [Google Scholar]
- 37.Chinnici CM, Yao Y, Ding T, Funk CD, Praticò D. Absence of 12/15 lipoxygenase reduces brain oxidative stress in apolipoprotein E-deficient mice. Am J Pathol. 2005;167(5):1371–1377. doi: 10.1016/S0002-9440(10)61224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nivsarkar M, Banerjee A, Padh H. Cyclooxygenase inhibitors: a novel direction for Alzheimer’s management. Pharmacol Rep. 2008;60(5):692–698. [PubMed] [Google Scholar]
- 39.Brown D, Yu BD, Joza N, Bénit P, Meneses J, Firpo M, Rustin P, Penninger JM, Martin GR. Loss of Aif function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc Natl Acad Sci USA. 2006;103(26):9918–9923. doi: 10.1073/pnas.0603950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joza N, Oudit GY, Brown D, Bénit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction. skeletal muscle atrophy and dilated cardiomyopathy. Mol Cell Biol. 2005;25(23):10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Tekahido S, Elia AJ, Cheng H-YM, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong Y-Y, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410(6828):549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 42.Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 43.Urbano A, Lakshmanan U, Choo PH, Kwan JC, Ng PY, Guo K, Dhakshinamoorthy S, Porter A. AIF suppresses chemical stress-induced apoptosis and maintains the transformed state of tumor cells. EMBO J. 2005;24(15):2815–2826. doi: 10.1038/sj.emboj.7600746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schägger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23(23):4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Movsesyan VA, Stoica BA, Faden AI. MGLuR5 activation reduces beta-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J Neurochem. 2004;89(6):1528–1536. doi: 10.1111/j.1471-4159.2004.02451.x. [DOI] [PubMed] [Google Scholar]
- 46.Ueda K, Hayashi O (1982) ADP- ribosylation reactions. Biology and Medicine. Molecular biology an international series of monographs and textbooks. Academic Press, New York
- 47.Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Inhibition of poly (ADP-ribose) polymerase (PARP) by nitric oxide and reactive nitrogen oxide species. Free Radic Biol Med. 2003;35(11):1431–1438. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Dawson VL, Dawson TM, Synder SH. Nitric oxide activation of poly (ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y, Wei EQ, Fang SH, Chu LS, Wang ML, Zhang WP, Yu GL, Ye YL, Lin SC, Chen Z. Spatio-temporal properties of 5-lipoxygenase expression and activation in the brain after focal cerebral ischemia in rats. Life Sci 20. 2006;79(17):1645–1656. doi: 10.1016/j.lfs.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 50.Thornber K, Colomba A, Ceccato L, Delsol G, Payrastre B, Gaits-Iacovoni F. Reactive oxygen species and lipoxygenases regulate the oncogenicity of NPM-ALK-positive anaplastic large cell lymphomasROS regulate NPM–ALK in ALCLs. Oncogene. 2009;28:2690–2696. doi: 10.1038/onc.2009.125. [DOI] [PubMed] [Google Scholar]
- 51.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VM-Y. 12/15-Lipoxygenases is increased in alzheimer’s disease. Am J Pathol. 2004;164(5):1655–1661. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao Y, Clark CM, Trojanowski MD, Lee VM-Y, Pratico MD. Elevation of 12/15 Lipoxygenase products in AD and mild cognitive impairment. Ann Neurol. 2005;58:623–626. doi: 10.1002/ana.20558. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/S0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]