

Abstract
Background
We studied the safety and efficacy of lithium in combination with riluzole in ALS. Recently, a pilot study demonstrated a dramatic effect of lithium in slowing ALS progression. To confirm or refute these findings, United States and Canadian funding organizations and investigators collaborated to design and execute a multicenter, double-blind placebo controlled trial in a rapid and efficient manner.
Methods
Eligible participants had familial or sporadic ALS diagnosed as clinically possible, laboratory supported probable, probable, or definite ALS according to El Escorial criteria and were taking a stable dose of riluzole for at least 30 days. Subjects were equally randomized by a centralized computer to receive either lithium (serum levels maintained between 0.4-0.8 mEq/L) or placebo. Subjects, caregivers and investigators were blinded to treatment assignment throughout the study. The study used a ‘time to an event’ design, novel to ALS trials. An event was defined as ≥ 6 points drop in the ALS Functional Rating Scale-Revised (ALSFRS-R) score or death. The primary efficacy analysis used a log-rank test to compare the distributions of the time to an event between the lithium and placebo groups. The first interim analysis occurred after 84 of 250 participants were randomized. The stopping boundary for futility at first interim analysis was a p-value ≥ 0.68.
Findings
The study was terminated early at the first intent-to-treat interim analysis as criterion for futility was met. A log-rank statistical analysis testing the superiority of lithium favored placebo (p-value = 0.78). In the final dataset, 22/40 subjects experienced an event in the lithium group compared to 20/44 subjects in the placebo group (p= 0.51). The point estimate (95% CI) for the hazard ratio of reaching the primary endpoint was 1.126 (0.6116 to 2.073). There were no major safety concerns. Fall (p=0.04) and back pain (p=0.05) were significantly more common in the lithium group.
Interpretation
Based on the standard error around the observed point estimate of effect, lithium in combination with riluzole did not reach the pre-specified threshold of a 43% or greater slowing in ALS disease progression which contradicts the pilot study. The ‘time to an event’ design enhanced enrollment and expeditiously answered an important clinical question while optimizing patient resources and funds.
Keywords: Amyotrophic lateral sclerosis, time-to-event, lithium carbonate, riluzole, clinical trial
Introduction
Amyotrophic lateral sclerosis (ALS) is a rare, devastating neurological disease with disproportionate socioeconomic impact due to the age at which the disease strikes, the extent and duration of disability, and the cost of long term care for patients with extensive medical needs. Recently, a small pilot study performed in Italy reported dramatic slowing of neurological decline in patients with ALS treated with lithium carbonate and riluzole as measured by the ALS Functional Rating Scale-Revised (ALSFRS-R), decline in forced vital capacity, and survival 1. Fornai et al. evaluated lithium as an agent to induce autophagy and maintained a targeted serum range of 0.4-0.8 mEq/L 1,2. Both autophagy and the proteasome are important for the clearance of aggregate-prone proteins like mutant SOD1, mutant huntingtin and alpha-synucleins 3,4. Fornai and colleagues observed increase in autophagic vacuoles in spinal cord sections from wild type and G93A mutant mice treated with lithium 1. Filimonenko et al. found that TDP-43 clearance is dependent on autophagy activation and that the mutation responsible for disease produces defective autophagy 5.
While the effect of lithium in the pilot study was dramatic, the number of treated subjects was very small (n=16), the participants were not blinded to treatment assignment, and at study entry the cohort treated with lithium appeared to have more slowly progressive disease than the general population of ALS patients. Despite these caveats, the promising results in this pilot study combined with preclinical data supporting the use of lithium as a potential therapeutic in ALS mandated further investigation. Because lithium is readily available by prescription and the results of the pilot study generated intense interest in lithium treatment, a high level of off-label use in patients with ALS has been noted in many ALS clinical centers. To maximize subject participation, we designed a double-blind, placebo controlled trial that allowed subjects to transition to lithium from placebo after they progressed to a predefined level. Granting agencies in the United States [the National Institute of Neurological Disorders and Stroke (NINDS) and the ALS Association] and Canada [the ALS Society of Canada] provided funds outside of the normal grant cycle following peer review from ALS Association while awaiting NIH peer review, and a novel trial design was employed to test the hypothesis that lithium carbonate and riluzole slowed ALS progression more than riluzole alone. Rapid enrollment of participants, the use of a time to event endpoint, and a pre-specified interim analysis with stopping rules allowed a clear result to be obtained less than 1 year after study initiation.
Methods
Study Design
A randomized, double-blind, placebo controlled trial of 250 subjects with ALS was planned. Enrollment occurred in a staged manner. The FDA exempted the study and Health Canada approval was obtained (9427-S2019-42C). All enrolling sites had institutional regulatory board (IRB) / Research Ethics Board (REB) approval and all research participants provided written informed consent prior to initiation of study procedures. Randomization was 1:1 placebo to lithium. The randomization scheme was independently developed by the Biostatistics Center at Massachusetts General Hospital (MGH), and it determined the treatment assignment for each participant number. The duration of treatment was up to 52 weeks. From January - June 2009, 84 participants were enrolled at 21 clinical sites (11 US and 10 Canadian centers) from the Northeast ALS (NEALS) and Canadian ALS (CALS) Consortia.
The primary outcome measure was the time to an event, where an event was defined as ≥ 6-point drop in the overall ALSFRS-R score from the baseline value or death. Participants assigned to placebo were switched to lithium in a blinded fashion after the time to event endpoint was reached. Those assigned originally to lithium remained on lithium after the time to event endpoint was reached. Secondary efficacy variables included rate of decline in the ALSFRS-R 6-8 and slow vital capacity (SVC), tracheostomy-free survival, along with analysis of the ALS-Specific Quality of Life (ALSSQOL) 9, and the Quick Inventory of Depressive Symptomatology Self Report Questionnaires (QIDS-SR16) 10,11. Lithium blood levels and safety and tolerability of lithium were also measured. Data were entered into a web-based electronic data capture (EDC) system. On-site monitoring was performed with reconciliation of source to electronic data.
Organization
The trial was managed through the MGH Neurology Clinical Trials Unit (NCTU) Project Coordination Center and individual Canadian (Sunnybrook Health Sciences Centre, University of Toronto) and United States (Columbia University) Site Management Centers that worked closely for seamless project management. Outcome measure training, compliance, and study monitoring for the trial were provided by the NEALS Outcome Measures and Monitoring Center at the State University of New York (SUNY) Upstate Medical University in Syracuse, NY. The MGH Biostatistics Department provided the randomization, statistical plans and analyses for the study. The Steering Committee and an independent Data and Safety Monitoring Board (DSMB) appointed by NINDS monitored the safety, data integrity, and conduct of the trial. The trial was registered at www.clinicaltrials.gov, identifier NCT00818389.
Study Drug
Lithium carbonate was purchased from Apotex, Inc. (Toronto, Canada) and dispensed as 150 mg capsules. Apotex Inc. provided matching placebo capsules in the same shell. Research participants initiated the study drug at 3 capsules per day: 1 capsule in the morning and 2 in the evening. The dosage was titrated based on drug levels obtained at each in-person visit to achieve a serum lithium concentration of 0.4 to 0.8 mEq/L; this concentration range matched that targeted in the Italian pilot study. Lithium levels were adjusted centrally by an unblinded drug monitor (JS), and levels were not available to site personnel. For levels below 0.4 mEq/L, the dosage was adjusted up by 1 capsule per day. Research participants taking an odd number of capsules, the odd capsule will have been taken at night; in this situation, the research participant was instructed to increase the AM dose by 1 capsule. For research participants taking an even number of capsules, the dose was increased by adding the extra capsule to the PM dose. For levels between 0.8-1.2 mEq/L, the dosage was adjusted down by 1 capsule in a similar manner based on the odd (reduce from PM dose) or even (reduce from AM dose) number of capsules taken. For levels above 1.2 mEq/L, dosage suspension occurred for 3-5 days and rechallenge at half the previous dosage occurred if a repeat level was less than 0.4 mEq/L. To maintain subject and investigator blinding throughout the study, sham dosage modifications were made for participants assigned to placebo.
Participant Selection Criteria
Eligible participants had familial or sporadic ALS diagnosed as clinically possible, laboratory supported probable, probable, or definite ALS according to the World Federation of Neurology El Escorial criteria 12. Inclusion criteria included age 18 years or older, ability to provide informed consent and comply with study procedures; disease duration less than 36 months from symptom onset; SVC greater than 60% predicted value for sex, height, and age; serum creatinine less than 1.5 mg/ dl (133 umol/L); euthyroid for at least 3 months, absence of or inactive psoriasis for at least 30 days prior to the screening visit, geographic accessibility to the study site, and fluency in English, Spanish or French (Canadian). All participants were required to be on a stable dose of riluzole for at least 30 days prior to the screening visit. Women of childbearing potential could be included if using adequate birth control and a screening pregnancy test was negative. Exclusion criteria included known sensitivity or intolerability to lithium; prior exposure to lithium within 90 days, exposure to any investigational agent within 30 days, use of digoxin or iodide salts, and malnourishment, dehydration or sodium free diet. Patients were excluded if they had substance abuse within the past year, active significant medical (cardiac, pulmonary, renal, hepatic, endocrine, hematologic, active malignancy or infectious disease) or psychiatric disease (psychosis or untreated major depression with in 90 days of screening visit), AIDS or AIDS related complex, pregnant or breast-feeding (female volunteers), clinically significant abnormal laboratory values (TSH > 20% above the upper limit), or significant cardiac conduction abnormality identified on screening EKG.
Training and Study Monitoring
Each site investigator, clinical evaluator, and research coordinator were trained on study procedures. Site evaluators were NEALS certified in the administration of the ALSFRS-R and SVC, and met reliability criteria established by NEALS. The ALSFRS-R is the summed score of 12 functional and respiratory items rated on a scale of 0 to 4 7. The amount of decline on ALSFRS-R that defines the time to event (≥ 6-point drop) was not known to either participants or clinical evaluators. The ALSFRS-R questionnaire was administered by the clinical evaluators, but the total score was not tallied and the scores from previous visits were not available in the EDC. The local study site staff and all personnel at the Coordination Center and Site Management Centers were blinded to total ALSFRS-R scores and date of transition to active study drug for participants initially assigned to placebo.
The site personnel were instructed to enter information within 48 hours of the visit. An edit checking and data clarification process was in place to ensure accuracy and completeness of the database. Logic and range checks as well as more sophisticated rules were built into the Web-based electronic Case Report Forms (eCRFs) to provide immediate error checking of the data entered. The system automatically created electronic queries on behalf of the Data Manager if saved eCRFs contained data that were out of range, out of window, missing or not calculated correctly. The Data Manager identified the errors in the EDC system by using electronic logic checks and the Study Monitors identified errors by direct visualization and comparison of data entered into the system with the source documents. In addition, a Medical Reviewer was appointed and was responsible for reviewing all adverse events and corresponding concomitant study data remotely via EDC for accuracy and consistency. Any inconsistent or questionable data points were queried to the sites and followed up on by the Study Monitors, Data Managers and Project Manager as needed.
Study Procedures
Participants were assessed for eligibility at the screening visit. After eligibility confirmation by the site Principal Investigator (PI) via an electronic signature in the Electronic Data Capture (EDC) system, the site randomized the subject electronically in the EDC. The Biostatistics Center at MGH used statistical software package R to generate the randomization sequence. Randomization was stratified by site to ensure a balanced number of lithium and placebo participants within each site. Their method was to generate a list of three digit subject numbers for each site. The Subjects were randomly allocated to one of the two treatment groups in blocks of two. The three-digit patient IDs were listed alongside the list of randomly allocated pairs of treatment/placebo codes. This algorithm generated a randomized block randomization sheet unique for each site that was then sent to each site Pharmacist (unblinded). The lists of subject numbers for each site were also available in the EDC. Once the site hit the randomization button in EDC, the EDC then generated a 3-digit randomization code. The 3-digit site number followed by the 3-digit randomization number then became the subject's 6-digit Subject ID number. The unblinded site Pharmacist assigned the treatment by matching the randomization number to a corresponding treatment from the randomization code sheet. The site Pharmacist then dispensed either lithium carbonate or placebo according to the site-specific randomization sheet to be initiated at the baseline visit. Accrued participants returned for the baseline visit (week 0) within 21 days of the screening visit. The following measures were obtained at the baseline visit: vital signs, weight, ALSFRS-R, ALSSQOL, and QIDS-SR16 questionnaires, and slow vital capacity. In-person visits were to occur at weeks 4, 8, 12, 20, 28, 36, 44, and 52; at these visits, vitals signs, weight, ALSFRS-R, slow vital capacity, a review of adverse events and concomitant medications, and assessment of drug accountability were completed. Trough lithium levels were drawn at in-person visits. Telephone visits were to occur at weeks 16, 24, 32, 40, and 48. The ALSFRS-R was obtained at each telephone visit and adverse events were assessed.
Statistical Analysis
The trial was intent-to-treat. All 84 participants who were randomized and received at least one dose of study drug were considered for the primary efficacy analysis. If a participant was lost to follow-up, they were censored in the primary analysis. The primary efficacy analysis used a log-rank test to compare the distributions of the time to an event between the lithium/riluzole and placebo/riluzole groups 13. This measure accounts for the variable length of time that each subject remained in the study while employing all available data for each participant. From the NEALS database encompassing placebo groups of prior clinical trials, ALSFRS-R was calculated to decline by an average of 1 point per month. Therefore, the time to event endpoint in this study was expected to occur after approximately 6 months in the placebo treated subjects. A 6-month period of active treatment was thought to be the minimum duration for which we could reasonably expect to see a therapeutic response.
Random effects models were used to examine the rates of decline over time for the secondary efficacy variables (ALSFRS-R, SVC, and QIDS-SR16) in each treatment group. Each subject was included in these analyses from baseline until the time that they experienced an event, dropped out, or until their final visit after the study was terminated following the first interim analysis. A group sequential design was employed to establish the time points for the interim analyses according to a mathematical function that is proportional to the number of events predicted to have occurred 14. A total of 167 events were expected in the study. Stopping rules were developed to determine at each interim analysis whether the study should be stopped for either efficacy or futility (e-data Figure1). The first interim analysis was planned after 84 subjects had been accrued to the trial. At that time, 1 of 4 decisions could be made: the trial could stop for efficacy, for futility, continue accrual, or stop accrual and continue to follow the 84 participants and reanalyze the data after 6 months. The second interim analysis was planned six months after the 84th participant has accrued or after there were 55 events. The third or the last interim analysis was planned after there were 100 events. At each interim analysis, the stopping boundaries for futility and efficacy were pre-specified based on the monitoring method described in “Group Sequential Methods with Applications to Clinical Trials” 14, 15. The log-rank statistic was calculated based upon the number of events that had occurred. The futility stopping boundary was defined for a one-sided p-value testing the superiority of lithium. A p-value < 0.50 would favor lithium, while a p-value > 0.50 would favor placebo. At the first interim analysis, the stopping boundary for futility was calculated as a p-value of 0.68. The trial was designed to have greater than 80% power to detect a 40% decrease in the rate of decline in the treatment group.
Tolerability was defined as the ability to complete 52 weeks of treatment on study drug. Distributions of baseline characteristics, adverse events and laboratory abnormalities were compared using Fisher's exact tests and t-tests.
Results
Enrollment, Baseline Characteristics, and Study Compliance
Between January and June 2009, 97 total subjects with ALS were screened at 21 participating sites across the US and Canada. Eighty-four eligible subjects consented to the study and were randomized to receive either lithium/riluzole (n = 40) or placebo/riluzole (n = 44) treatments (Figure 1). Subjects were followed for a mean period of 5.4 (SD 1.5) months (range 0.8 to 8.3 months) until study was terminated for futility in September 2009.
Figure 1. Participant flow chart.

The disposition of subjects in study, including the number screened, randomized, failed, early terminated, and completed.
Baseline characteristics are shown in Table 1. There were no significant differences in demographic features, clinical variables, and values of primary and secondary outcome variables between the 2 treatment groups at baseline.
Table 1. Baseline Characteristics.
| Variable | Lithium/Riluzole (n=40) | Placebo/Riluzole (n=44) |
|---|---|---|
| Mean age, yrs (SD) | 58.3 (10.2) | 55.5 (11.9) |
| Male Gender (%) | 30 (75) | 24 (55) |
| Race, % white | 39 (98) | 42 (95) |
| Family history of ALS, % | 3 (8%) | 1 (2%) |
| Mean time since symptom onset to diagnosis (SD) | 1.1 (0.6) | 1.0 (0.5) |
| Mean time from symptom onset to baseline, yr (SD) | 1.6 (0.6) | 1.7 (0.7) |
| Limb onset, % | 34 (85%) | 33 (75%) |
| Mean BMI | 26.6 (3.7) | 26.2 (4.6) |
| Mean VCmax % predicted (SD) | 94.0 (18.1) | 86.9 (16.9) |
| Mean ALSFRS-R score (SD) | 38.4 (4.6) | 36.5 (5.7) |
| Mean ALS-Specific QOL score (SD) | 417.8 (70.7) | 425.2 (81.4) |
| Mean QIDS-SR16 score (SD) | 6.6 (2.7) | 6.3 (4.2) |
Efficacy of Lithium/Riluzole
Interim Analysis Results
The first interim analysis was conducted after 84 subjects were enrolled. Due to the pre-planned nature of the interim analysis, we were able to institute methods for ensuring quick data entry and data validation, monitoring and cleaning. At the interim analysis, a log-rank statistical analysis testing the superiority of lithium favored placebo, p = 0.78. The one-sided p-value of 0.68 to stop for futility was exceeded (the p-value for stopping for efficacy was 0.001). The results were reviewed by the DSMB. The DSMB recommended, with NINDS concurrence, that the trial be terminated for futility. All participants were asked to stop study drug and were instructed to schedule a final safety visit with the site study staff.
Final Analysis Results
In the final dataset, in the lithium/riluzole group, 22/40 subjects (55.0%) experienced an event (defined as ≥ 6-point drop in the ALSFRS-R score from the baseline value or death) compared with 20/44 subjects (45.5%) in the placebo/riluzole group (p= 0.51) (Figure 2). The point estimate for the hazard ratio of reaching the primary endpoint was 1.126 (95% CI 0.6116 to 2.073). There were no significant differences in the rates of decline for placebo and lithium treated subjects in the ALSFRS-R (p = 0.61; 95% CI -0.43 to 0.73) (Figure 3a), SVC (p = 0.08; 95% CI -2.58 to 0.13) (Figure 3b), and the QIDS-SR16 (p = 0.83; 95% CI -0.37 to 0.30). Subjects were included up to the time that they experienced an event and crossed over (placebo subjects blindly switched to lithium treatment, while lithium subjects remained on lithium), or until their final visit after the trial was halted, whichever occurred first. In the final analysis, the 95% confidence interval around the difference in rates of decline of the ALSFRS-R total score was [-0.43, 0.73], suggesting that while a modest benefit of lithium was not ruled out by this study, an effect of 43% or more could be eliminated.
Figure 2. Survival curve for time to failure.
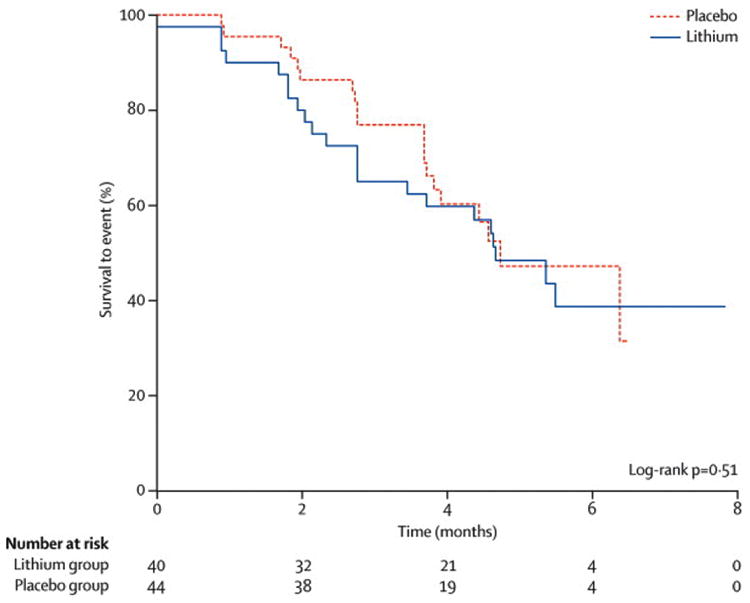
Kaplan-Meier survival curve of survival to event (defined as ≥ 6 points drops in ALSFRS-R score or death) of lithium (solid line) versus placebo (dashed line).
Figure 3.
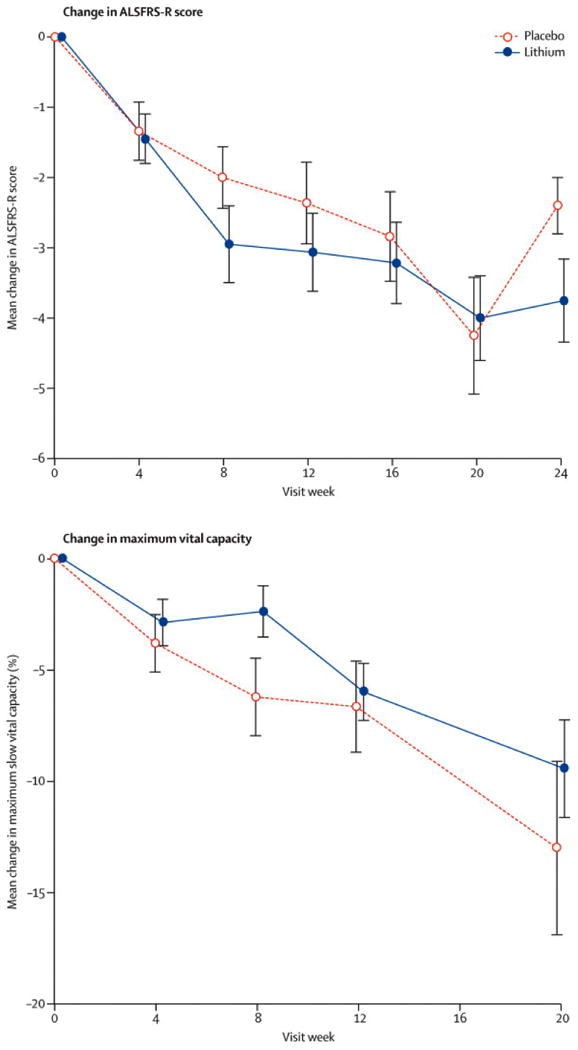
Figure 3a: ALSFRS-R Total Change. The ALSFRS-R total change from baseline by visit (in weeks) in the lithium treated subjects (solid line) vs. the placebo group (dashed line). The values plotted are mean difference in scores from baseline value. The bars indicate 1 standard error of the mean.
Figure 3b: Change in Vital Capacity. The Vital Capacity change from baseline by visit (in weeks) in the lithium treated subjects (solid line) vs. the placebo group (dashed line). The values plotted are mean difference in scores from baseline value. The bars indicate 1 standard error of the mean.
Lithium Levels
Lithium levels were drawn for all research participants (whether assigned to study drug or placebo) at each visit 8-12 hours +/- 1 hour post the last dose. Subjects were informed by site staff regarding holding the dose to time the draw, and this information was provided in writing in the consent form. Any levels drawn outside the window were redrawn. The lithium doses in the study ranged from 150 mg – 1050 mg per day (1 capsule per day so 150 mg to 7 capsules per day, 3 am and 4 pm so 450 mg am + 600 mg pm). At the week 4 visit, 36.84% of subjects (14/38) in the lithium treated group had serum lithium levels in the target range of 0.4-0.8 mEq/liter. The mean lithium level at week 4 was 0.31 mEq/liter. By week 8, 46.37% of lithium-treated subjects (18/38) were in target range (mean 0.36 mEq/liter), and at week 12, the majority of subjects 73.53% (25/34) had therapeutic serum lithium levels (mean 0.40 mEq/liter). However, given the pharmacokinetics of lithium, the above data suggest that subjects who were in the therapeutic range at week 12 reached that level by week 9. Of the 40 subjects initially randomized to lithium, 6 (15%) never attained a lithium level in the therapeutic range 0.4-0.8 mEq/liter; 2 of these subjects discontinued study drug early at 1 dose and 2 weeks after drug initiation. The boxplots of the lithium levels over the course of the study for subjects in the lithium/riluzole group are displayed in Figure 4. Two subjects assigned to the placebo group (5.6%) had a detectable lithium level at their week 12 visit. One additional placebo subject with a detectable lithium level at week 12 had previously discontinued study drug and initiated lithium treatment outside the study.
Figure 4. Lithium levels.

Box plots of plasma lithium levels by visit (in weeks). The band represents the median lithium levels; the bottom and the top of the boxes represent the 25th and 75th percentiles respectively. The whiskers represent 1.5 times the lower and upper interquartile range.
Tolerability
Overall, lithium in combination with riluzole was well-tolerated. Sixty-eight percent (27/40) of subjects in the lithium/riluzole treatment group completed the study up to the final analysis without any dosage reductions, suspensions or permanent discontinuations due to adverse events compared with 86% (38/44) in the placebo group (p-value = 0.07).
There were 12 permanent study drug discontinuations (excluding deaths) reported in the first interim analysis; 7 in the placebo group and 5 in the lithium group. Time to study drug discontinuation did not differ significantly between the treatment groups (log rank, p = 0.55). The reasons for permanent study drug discontinuation in the placebo group included: disease progression, elevated TSH and constitutional symptoms such as fatigue, anorexia and nausea. In the lithium group study drug was discontinued secondary to an episode of delirium (deemed unrelated to study drug), tremor and dizziness, depression, consent withdrawal and disease progression. One participant in the lithium group stopped study drug due to perceived lack of efficacy and study drug was discontinued in another subject on the recommendation of the drug monitor due to critical lithium levels (≥ 1.2 mEq/liter).
Safety
The most common adverse events included muscle weakness in the lower or upper extremities, fatigue, nausea, fall, dyspnea, limb edema, fasciculations, dysphagia, headache and whole body/generalized muscle weakness (Table 2). Fall (p=0.04) and back pain (p=0.05) were significantly more common in the lithium group. Six participants died during the study; one prior to randomization, 2 in the lithium group (1 greater than 30 days after stopping study drug) and 3 in the placebo group. All deaths were deemed unlikely or unrelated to the study drug. Thirty serious adverse events (SAEs) occurred in 19 participants during the course of the study; 1 prior to randomization, 14 events in 10 subjects in the lithium/riluzole group and 15 events in 8 subjects in the placebo/riluzole group. All SAEs were unexpected with respect to the study drug with the exception of five: 2 in the placebo group (nausea and somnolence/depressed level of consciousness due to over-sedation from another drug) and 3 in the lithium group (syncope, encephalopathy/delirium due to anticholinergic reaction and dysphagia secondary to a traumatic seizure after a motor vehicle accident). One SAE (depression with suicide attempt) in a lithium treated participant was unexpected and thought to be possibly related to study drug. There was no significant difference in the occurrence of serious adverse events between the two groups.
Table 2. Adverse Events.
| CTCAE Term | Lithium/Riluzole | Placebo/Riluzole | P-value |
|---|---|---|---|
| % (# of subjects with the event in each group/total # of subjects with the event) | |||
| Muscle weakness: Extremity-lower | 48.5 (16/33) | 51.5 (17/33) | 1.00 |
| Muscle weakness: Extremity-upper | 54.1 (20/37) | 45.9 (17/37) | 0.38 |
| Fatigue | 60.9 (14/23) | 39.1 (9/23) | 0.15 |
| Nausea | 52.6 (10/19) | 47.4 (9/19) | 0.79 |
| Fall | 80.0 (8/10) | 20.0 (2/10) | 0.04† |
| Dyspnea | 57.1 (8/14) | 42.9 (6/14) | 0.56 |
| Edema: Limb | 46.2 (6/13) | 53.8 (7/13) | 1.00 |
| Fasciculations | 63.6 (7/11) | 36.4 (4/11) | 0.34 |
| Dysphagia | 40.0 (6/15) | 60.0 (9/15) | 0.58 |
| Headache | 46.7 (7/15) | 53.3 (8/15) | 1.00 |
| Muscle weakness: Whole body/generalized | 36.4 (4/11) | 63.6 (7/11) | 0.53 |
| Pain: Back | 85.7 (6/7) | 14.3 (1/7) | 0.05† |
Discussion
Lithium was initially tested in animal models and humans with ALS because of its demonstrated effect of inducing autophagy 1,2. Lithium pretreatment protected cultured brain neurons from glutamate-induced, N-methyl-D-aspartate (NMDA) receptor-mediated apoptosis. Administration of lithium to G93A mice prolonged survival along with recovery of the cell pathology 1,16,17. In rats with thoracic spinal cord transection or contusion injuries, GSK-3 inactivation with lithium enhances descending corticospinal and serotonergic axon sprouting in caudal spinal cord and promotes locomotor functional recovery 18. In a more recent study, lithium enhanced the neuronal differentiation of neural progenitor cells (NPCs) in vitro and after transplantation into the avulsed ventral horn of adult rats through the secretion of brain-derived neurotrophic factor 19. Our results provide evidence that lithium in combination with riluzole does not have the dramatic effect of reducing disease progression in participants with ALS previously reported in a pilot study 1. We did not eliminate the possibility that lithium has a small positive effect or whether other serum concentration ranges might be beneficial.
Our goal was to test the validity of the results published by Fornai and colleagues; therefore we targeted the same plasma range used in their study. Moreover, there are preclinical data that suggest a potential limited range of efficacy for lithium and a level above which there may be activation of different pathways and potential for toxicity. In a recent study, lithium-induced neural progenitor cell differentiation towards neurons reached a plateau at 1mM in the dose-response curve 19,20.
In this study, approximately half of the subjects on active compound were in the therapeutic range at week 8 and 73.53% of subjects were therapeutic at week 12. We recognize that the target lithium levels took several months to achieve. However, it should be noted that we only checked lithium levels at monthly interval for the first three months to avoid inconvenience to the subjects although stable lithium levels are reached within a week of dosage adjustment. Thus subjects therapeutic at week 8 or 12 visit had been in the therapeutic range since week 9 and week 13 respectively. The trial was terminated for futility based on the strength of the evidence, which was sufficient to support that a large effect of lithium would not be seen by enrolling additional participants or following subjects already enrolled in the trial to increase the percentage of subjects with lithium levels in the target range. A smaller but meaningful effect of lithium may not be ruled out by this study.
It is possible that if a greater portion of subjects were treated for longer periods of time at therapeutic levels a smaller benefit of lithium may have been demonstrated. It is unknown from the pilot Italian study how quickly and what portion of subjects achieved therapeutic lithium levels, however, in the pilot study significant benefits were observed in the Norris scale, the ALSFRS-R and FVC in lithium treated subjects after only 3 months. Insights into the long-term effects of lithium on ALS progression may be determined from other ongoing controlled studies.
The feasibility of our novel time-to-event design for ALS trials has been demonstrated. Our study includes 3 innovative features: 1) the use of a time-to-event endpoint as the primary outcome measure rather than a conventional random effects model evaluating slope of ALSFRS-R decline; 2) blinded crossover to the active compound for participants initially assigned to placebo once they reach the pre-specified event; 3) multiple planned interim analyses for futility and efficacy. One reason for a time to failure design is to make a clinical trial more attractive to patients who may not want to take a placebo for a long period of time when the active drug is readily available and is thought by patients to be effective. Time to event endpoints essentially convert an interval scaling measure into a binary measure. Binary measures have the attribute that clinical relevance is incorporated into the endpoint. If the study were positive, conversion of the results into a “number needed to treat” estimate is also straightforward. This design can be analyzed by survival analysis, as we did with our primary analysis, or it can be analyzed using a random effects model as we did in our secondary analysis. A conventional fixed time design could be analyzed in both these ways as well. A detailed comparison of the four possible combinations of designs and analysis plans is beyond the scope of this paper. We studied the four possibilities by simulation and found that in most cases there is a small power loss in using a time to failure design but more power loss using survival analysis rather than the random effects model.
There is also a small power loss when a futility stopping rule is employed. The stopping rule depends on the alternative hypothesis. You specify the probability of stopping early given that the alternative is true. This trial was designed with an alternative that specified that lithium was much better than placebo, and the chance of stopping for futility if this had been the case was very small. However, if the benefit of lithium was moderate, say 30%, as we assumed for Celebrex 21, then our chance of stopping for futility would be much higher and there would be a larger loss of power as compared to a trial without futility stopping. It should be noted that this was not a futility design; the study was designed to accrue 250 patients unless early results showed that continuation of the trial was futile based on predefined stopping rules.
Given the clear lack of signal in this study after 84 subjects, it is unlikely that enrolling more patients would have changed the ultimate result. It is unclear, however, whether the time to failure endpoint was the major determinant in reaching futility. More studies will be needed to determine whether time to failure endpoint improves the sensitivity of futility analyses.
This design was chosen in part to enhance patient acceptance of a placebo controlled trial in the setting of intense patient interest and easy availability of lithium outside the trial. The inclusion of a placebo group and blinding in a randomized clinical trial testing novel therapeutics is necessary to control for the placebo effect in this heterogeneous disease and is required to detect mild to moderate treatment effects. The time to an event design was thought to be attractive to potential subjects in that participants would only be exposed for an average of 7 months to placebo and participants with a rapid progression in the placebo group would receive active compound earlier in the course of their disease. This trial design provides a good compromise between the need to effectively determine therapeutic efficacy and the desire to limit the period of time participants are on placebo. It should be considered in the design of future ALS trials, especially when the active compound is easily accessible.
In conclusion, this randomized, double-blinded clinical trial failed to demonstrate a significant slowing of disease progression or vital capacity decline in ALS patients treated with lithium. Whether smaller beneficial effects of lithium in ALS are still possible and compounds targeting autophagy induction have therapeutic potential, needs to be demonstrated in future studies. At this time, there remains no convincing evidence for the use of lithium as a treatment for patients with ALS.
Supplementary Material
Figure 1: Interim Analysis Flow Chart
Figure provides the details of the planned interim analysis for the trial.
Acknowledgments
We thank Drs. Robin A Conwit and Claudia Moy and Louise Ritz of the National Institute of Neurological Disorders and Stroke (NINDS), Lucie Bruijn of the ALS Association, and Denise Figlewicz of the ALS Society of Canada for their support. We thank the members of the DSMB for their guidance, Drs. Carl Leventhal, James Russell, Judy Hung, and Francine Vriesendorp and the patients and caregivers for their commitment and time. We would also like to thank Apotex Inc. for providing the lithium and matching placebo at a discounted rate.
Role of the funding Sources: This trial was initiated and undertaken by the Northeast and Canadian ALS Consortia. The funding sources for this trial approved the design and the protocol, but they had no involvement in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
Members of the Northeast ALS and Canadian ALS Consortia include (affiliation at time of study): Participating Site Investigators and Site Staff
Lorne Zinman, MD, MSc, FRCPC, Jane McKinley, RN, Hanika Pinto, Yael Friedman, MD, MSc, FRCPC, John Iazzetta, PharmD, Betty Chrichton, BSc Pharm, Sunnybrook Health Sciences Centre/University of Toronto;
Monique D'Amour, MD, Frederique Souchon, MD, Claire Lefebvre, RN, BSc(N), Louise Blais INH, Eric Cardin, INH, Lise Allard, BSc Pharm, Georges Quessy, BSc Pharm, Antonietta Limbo, MSc Pharm, Kenny Cabral, BScN, Nadia Spinelli, BScN, Gail Robertson, BSc, Phillip Lewis, CHUM Notre-Dame Hospital/University of Montreal;
Ezgi Tiryaki, MD, Scott Bundlie, MD, Tzivia Leviton, RPh, Cindy Rohde, RN, Sandra Swanson, PT; Hennepin County Medical Center/Berman Center for Outcomes and Clinical Research;
Kevin Boylan, MD, Kathleen Kennelly, PhD, MD, Amelia Johnston, BA, Pamela DeSaro, BA, Paula Fuqua, PharmD, Tammie Wright, CPhT, Cynthia Ward, RPh, Sandy Schoenberger, RPh, Amy Swan, PharmD, Gleydiane DeOliveira, PT, Angela Huser, RN, BSN, Mayo Clinic Jacksonville;
William S. David, MD, PhD, Anne-Marie Wills, MD, Nazem Atassi, MD, Alison R. Goldenberg, BS, Darlene E. Pulley, RN, Jennifer L. Berndt, BS, Maria Del Carmen Castrillo-Viguera, MD, Matthew Bellanich, NP, Matthew N. Jaffa, BS, Cherylann Reilly-Tremblay, RPh, Massachusetts General Hospital;
Angela Genge, MD, FRCPC, Danielle Lavoie, MD, FAANC, Jo-WEn Wang, MSc, Dolores Bertone, RRT, Kathleen Normandin, BPharm, MSc, Montreal Neurological Institute / McGill University;
Kevin Scott, MD, Zachary Simmons, MD, Stacey L. Clardy, MD, PhD, Allyson Brothers, MA, Chrsitine Schaeffer, RN, CCRC, Helen Elizabeth Stephens, MA, CCRC, Heather Heisey, RPh, Shirlynn Mottilla, RN, GCRC Grants NIH M01-RR10732, C06 RR016499 and Pennsylvania Department of Health Tobacco Settlement Funds, Pennsylvania State University;
Daragh Heitzman, MD, Brent Spears, BS, Shari L. Hand, BA, CCRC, Lori White, RN, Texas Neurology;
James B. Caress MD, Michael S. Cartwright, MD, Mozhdeh Marandi, MD, CCRC, Christine O'Neil, CCRC, GCTRC Grant #: MO1RR07122, Wake Forest University;
Rodney Li Pi Shan, MD, FRCPC, Walter Hader, MD FRCP(C), Scott Ridley, BSP, Layne Bruce, (Phar. Tech), Jeanine Munchinsky, B Mus, ARCT, Vanina Dal Bello-Haas, BSc (PT), M Ed, PhD, University of Saskatchewan;
Richard Bedlack, MD, PHD, Karen Harward-Grace, RN, BSN, Candace Boyette MSN, FNPC, Duke University;
Jean-Pierre L. Bouchard, MD, FRCPC, Annie Dionne, MD, FRCPC, Nicolas Dupre, MD, FRCPC, Lucie Morel, RN, BAA, Genevieve Roy, RN, MSN, Tuong-Vi Tran, B. Pharm, Laval University;
Christopher White, MD, FRCPC, Lawrence Korngut, MD, FRCPC, Shefina Mawani, RN, BN, CDE, Susan Munro, B.Sc. OT, Candice Cameron, BA, BSP, ACPR, University of Calgary;
I. A. Grant, MD, FRCP(C), T. J. Benstead, MD, FRCP(C), S. L. Reidy, BSc, CCRP, S.L. Hebert, Pharmacy Research Technician, Dalhousie University, Halifax;
Terry Heiman-Patterson, MD; Anahita Deboo, MD; Luisa Rojas, MD; Sara Feldman, MA, PT, ATP; Monica Mazurek, RN; Christine Barr, RN; Jeffrey Deitch, PhD, Drexel University;
J. Turnbull MD, PhD, FRCP(C), Joan Martin MSc; McMaster University;
Laura Simionescu, MD, Deborah Bradshaw, MD, Mary Lou Watson, BA, RRT, Megan Grosso, PA, Travis Boevin, RPh, SUNY Upstate Medical University;
Christen Shoesmith, MD, FRCP(C), Michael Strong MD, FRCP(C), Anne Row, RN, Karen Findlater, PT, Jennifer Verheyden, BMSc, University of Western Ontario/London Health Sciences Centre;
Brett M. Morrison, MD, PhD, Jeff Rothstein, MD, PhD, Richard M. Kimball, MSN, MPH, APRN-BC, PhD, Lora L. Clawson, MSN, CRNP, Kristen M. Riley, PhD, Johns Hopkins University;
Alan Pestronk, MD, Matthew Harms, MD, Julaine Florence, PT, DPT, Pamela Townsend RN, BSN, MPH, Kathryn Vehe, PharmD, Elizabeth Malkus, PT, MHS, Washington University St. Louis;
Colleen O'Connell, MD, FRCPC, Scott Worley, MD, BScPT, FRCPC, Susan Brophy-LeBlanc, RN/BN, CRNC, Stan Cassidy Centre for Rehabilitation;
Petra Kaufmann, MD, MSc, Jinsy Andrews, MD, MSc, Darleen Vecchio, MS, Columbia University;
John T. Kissel, MD, Steven M. Nash, MD, Stephen J. Kolb, MD, PhD, Adam D. Quick, MD, Sharon A. Chelnick, MHSA, Wendy M. King, PT, Robert P. Fudge, RPh, Jerold Reynold, PhD, Amy Bartlett, Ohio State University;
Abirami Muthukumaran, MD, Eugene Tsimerinov MD PhD, Hope Gruendler, MSN, CNS, Cedars-Sinai Medical Center;
Robert M. Pascuzzi, MD, John C. Kincaid, MD, Riley J. Snook, MD, Cynthia L. Bodkin, MD, Sandra Guingrich, LPN, CCRC, Angela Micheels, PT, Karl Gregory Humma, RPh, Indiana University;
David Saperstein MD, Todd Levine, MD, Nicole Hank, MHSM, Kathie Clarke, RN, MS, CVNS, CCRN, Phoenix Neurological Associates;
Michel Melanson, MD, FRCP, Evelyn Vee McBride, RN, BScN, Queen's University, Kingston;
Wendy Johnston, MD, FRCPC, Sanjay Kalra, MD, FRCPC, Ramnik Sekhon, BSc, University of Alberta;
Hannah Briemberg, MD, FRCP(C), Charles Krieger, MD, FRCP(C), Marife Fabros, RN, Brigitte Poirier, MPO, SLP(C); University of British Columbia;
Lomen-Hoerth, Catherine, MD, PhD, Ahmed, Fizaa, BS, University of California San Francisco;
Edward Kasarskis, MD, PhD, Kathie Vanderpool, Debbie Taylor, R. Stephen Wells, Stephen C. Sitzlar, University of Kentucky VA Medical Center, Lexington;
Alan Casey, MD, FRCPC, Karen Ethans, MD, FRCPC, Tracey Olafson, RN, University of Manitoba, Winnipeg;
Khema R. Sharma, MD, Donald Koggan, MD, Sherley Valdez, MD, Gina Gonzalez, ARNP, Mario Perez, RN, Maru Palomeque, RN, Emilio Weiss, PhD, University of Miami;
Rup Tandan, MD, FRCP, Paramjit Singh, MD, Christine Potter, BA, GCRC Grant # MO1-RR00109, University of Vermont;
Lawrence H. Phillips, MD, Ted M. Burns, MD, Guillermo Solorzano, MD, Kristen, Keller, BA, Judy Warder, BS, University of Virginia;
Agnes Jani-Acsadi, MD, Richard A. Lewis, MD, Stacey Masse, RN, Marissa Mariani, PharmD., Joanne, L. MacDonald, PharmD, Wayne State University;
NEALS & CALS Steering Committee—Swati Aggarwal, MD, Massachusetts General Hospital, Lorne Zinman, MD, MSc, FRCPC, Sunnybrook Health Sciences Centre/University of Toronto, Merit Cudkowicz, MD, MSc, Massachusetts General Hospital; Jeremy Shefner, MD, PhD, SUNY Upstate Medical University; David Schoenfeld (Biostatistician), PhD, Massachusetts General Hospital; Petra Kaufmann, MD, MSc, Columbia University (This work was done while Dr. Kaufmann was at Columbia University and is not related to NINDS/NIH); Kevin B. Boylan, MD, Mayo Clinic, Jacksonville; Robert Brown, MD, DPhil, UMass Medical Center Worcester; Robin Conwit, MD, NINDS/NIH; Angela Genge, MD, McGill University; Edward Kasarskis, MD, University of Kentucky VA Medical Centers, Lexington; Jeffrey Rothstein, MD, PhD, Johns Hopkins University; Christen Shoesmith, MD, University of Western Ontario; John Turnbull, MD, McMaster University;
NEALS Massachusetts General Hospital Coordination Center Staff: Swati Aggarwal, MD, Merit Cudkowicz, MD, MSc, Elizabeth Simpson, Katherine E. Jackson, Alex Sherman, MS, Hong Yu, MSc, Veena Lanka, MBBS, Lauren Schnupp, Jing Deng, Oscar Padilla, Irina Badayan, Padmaja Yerramilli-Rao, MBBS, Erin Rosenbaum, Marianne Kearney, Maria Maloutas.
CALS Sunnybrook Health Sciences Centre Coordination Staff: Lorne Zinman, MD, MSc, FRCPC, Jane McKinley, RN, Hanika Pinto, Canadian Coordination Centre, University of Toronto.
NEALS SUNY Upstate Medical University Outcomes Measures and Monitoring Center: Jeremy Shefner, MD, PhD, Mary Lou Watson RRT, Katherine Tindall, RN, BSN, Kristina Money, Betsy Varghese, Brittany Lew, Katie Markis, Holly Hand.
NEALS University of Columbia Site Management Center Staff: Petra Kaufman, MD, MSc, Jinsy Andrews, MD, MSc, Darleen Vecchio, MS.
Footnotes
Contributors: SPA, LZ, RAC, DS, JS, and MC participated in conception and design of the study. ES, JM, KEJ, & HP helped with timely data collection and cleaning. SPA, LZ, ES, DS, JS, and MC had full access to all the data in the study and contributed to analysis and interpretation of data, writing of the paper, and have final responsibility for the decision to submit for publication. All authors have seen and approved the final version of the manuscript.
Data and safety monitoring board: Drs. R Holloway (Chair); C Coffey, M Benatar, M Trivedi, S Wisniewski and C Joyce
Medical Monitors: Drs. Carl Leventhal and James Russell
Conflicts of interest: SPA, LZ, ES, JM, KEJ, HP, RAC, DS, JS, and MC have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences. 2008;105(6):2052–2057. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fornai F, Longone P, Ferrucci M, et al. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy. 2008;4(4):527–30. doi: 10.4161/auto.5923. [DOI] [PubMed] [Google Scholar]
- 3.Sarkar S, Floto R, Berger Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. Journal of Cell Biology. 2005;170(7):1101–11. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarkar S, Rubinsztein DC. Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy. 2006;2(2):132–4. doi: 10.4161/auto.2387. [DOI] [PubMed] [Google Scholar]
- 5.Filimonenko M, Stuffers S, Raiborg C, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179(3):485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miano B, Stoddard G, Davis S, Bromberg M. Inter-evaluator reliability of the ALS Functional Rating Scale. Amyotrophic Lateral Sclerosis & Other Motor Neuron Disorders. 2004;5(4):235–239. doi: 10.1080/14660820410021302. [DOI] [PubMed] [Google Scholar]
- 7.Cedarbaum J, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporated assessments of respiratory function. J Neurol Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 8.Kasarskis EJ, Dempsey-Hall L, Thompson MM, Lan Chi L, Mendiondo M, Kryscio R. Rating the severity of ALS by caregivers over the telephone using the ALSFRS-R. Amyotrophic Lateral Sclerosis & Other Motor Neuron Disorders. 2005;6(1):50–54. doi: 10.1080/14660820510027107. [DOI] [PubMed] [Google Scholar]
- 9.Simmons Z, Felgoise SH, Bremer BA, et al. The ALSSQOL: Balancing physical and nonphysical factors in assessing quality of life in ALS. Neurology. 2006;67(9):1659–1664. doi: 10.1212/01.wnl.0000242887.79115.19. [DOI] [PubMed] [Google Scholar]
- 10.Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item quick inventory of depressive symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biological Psychiatry. 2003;54(5):573–583. doi: 10.1016/s0006-3223(02)01866-8. [DOI] [PubMed] [Google Scholar]
- 11.Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73–82. doi: 10.1017/s0033291703001107. [DOI] [PubMed] [Google Scholar]
- 12.Brooks B. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis”. J Neurol Sci. 1994;124:96–107. doi: 10.1016/0022-510x(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 13.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50(3):163–70. [PubMed] [Google Scholar]
- 14.Jennison C, Turnbull BW. Group Sequential Methods with Applications to Clinical Trials. Chapmann & Hall/CRC; NY: 2000. pp. 145–169. [Google Scholar]
- 15.Schoenfeld DA. A simple algorithm for designing group sequential clinical trials. Biometrics. 2001;57(3):972–4. doi: 10.1111/j.0006-341x.2001.00972.x. [DOI] [PubMed] [Google Scholar]
- 16.Shin JH, Cho SI, Lim HR, et al. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol Pharmacol. 2007;71(4):965–75. doi: 10.1124/mol.106.030676. [DOI] [PubMed] [Google Scholar]
- 17.Feng HL, Leng Y, Ma CH, Zhang J, Ren M, Chuang DM. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience. 2008;155(3):567–72. doi: 10.1016/j.neuroscience.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dill J, Wang H, Zhou F, Li S. Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J Neurosci. 2008;28(36):8914–28. doi: 10.1523/JNEUROSCI.1178-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su H, Zhang W, Guo J, Guo A, Yuan Q, Wu W. Lithium enhances the neuronal differentiation of neural progenitor cells in vitro and after transplantation into the avulsed ventral horn of adult rats through the secretion of brain-derived neurotrophic factor. J Neurochem. 2009;108(6):1385–98. doi: 10.1111/j.1471-4159.2009.05902.x. [DOI] [PubMed] [Google Scholar]
- 20.Su H, Chu TH, Wu W. Lithium enhances proliferation and neuronal differentiation of neural progenitor cells in vitro and after transplantation into the adult rat spinal cord. Exp Neurol. 2007;206(2):296–307. doi: 10.1016/j.expneurol.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 21.Cudkowicz ME, Shefner JM, Schoenfeld DA, et al. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol. 2006;60(1):22–31. doi: 10.1002/ana.20903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1: Interim Analysis Flow Chart
Figure provides the details of the planned interim analysis for the trial.