

Abstract
A better understanding of the neurobiology of mood disorders, informed by preclinical research and bidirectionally translated to clinical research is critical for the future development of new and effective treatments. Recently, diverse new targets/compounds have been specifically tested in preclinical models and in proof-of-concept studies, with potential relevance as treatments for mood disorders. Most of the evidence comes from case reports, case series, or controlled proof-of-concept studies, some with small sample sizes. These include (1) the opioid neuropeptide system, (2) the purinergic system, (3) the glutamatergic system, (4) the tachykinin neuropeptide system, (5) the cholinergic system (muscarinic system), and (6) intracellular signaling pathways. These targets may be of substantial interest in defining future directions in drug development, as well as in developing the next generation of therapeutic agents for the treatment of mood disorders. Overall, further study of these and similar drugs may lead to a better understanding of relevant and clinically useful drug targets in the treatment of these devastating illnesses.
Keywords: cholinergic, depression, glutamate, mood disorders, novel treatments, pathophysiology
Introduction
Worldwide, bipolar disorder (BPD) and major depressive disorder (MDD) are among the most disabling of all non-communicable illnesses. These heterogeneous, chronic, and severe disorders are associated with persistent subsyndromal symptoms and frequent episode relapses and recurrences [1–3]. However, treatment of both disorders remains inadequate for many patients. For instance, the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study noted that only one-third of patients with MDD achieved remission after an adequate trial with a traditional antidepressant agent [4]. Similarly, a large-scale study funded by the National Institute of Mental Health (NIMH) failed to find significant benefit to adding antidepressants to mood stabilizers in patients with BPD-I or II depression over the course of 26 weeks [4; 5]. Thus, the development of new, effective, and better-tolerated therapeutic approaches with a more rapid onset of action is critical.
To achieve this objective, a variety of compounds targeting diverse new systems have been proposed and have been or are being tested; many of these may ultimately result in new and improved treatments for mood disorders (see Figure 1). Here, we describe studies evaluating some of these potentially promising targets. Specifically, we review: (1) the opioid neuropeptide system, (2) the purinergic system, (3) the glutamatergic system, (4) the tachykinin neuropeptide system, (5) the cholinergic system (muscarinic system), and (6) intracellular signaling pathways. Other putative targets not covered in this review include the melatonergic system, the glucocorticoid system and bioenergetics and oxidative stress. This paper reviews data on new compounds that have shown antimanic or antidepressant effects in subjects with mood disorders or similar effects in preclinical animal models. It is important to mention at the outset that the extrapolation of animal studies to humans requires cautious interpretation. However, such studies may provide a better understanding of the neurobiology of mood disorders. This bi-directional research approach—whereby preclinical work can be translated to clinical research is—critical for the future development of new and effective treatments.
Figure 1. Putative therapeutic targets for mood disorders.
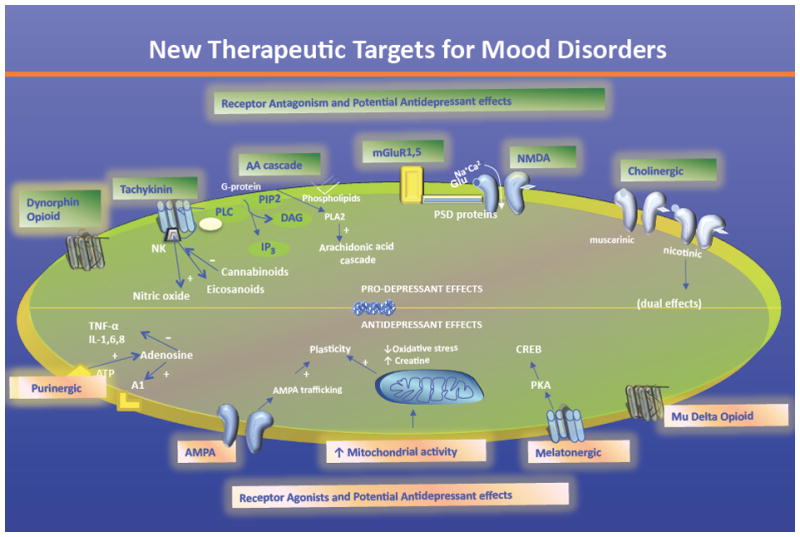
The dynorphin opioid system, the purinergic system, the glutamatergic system (AMPA receptors, NMDA receptors, and mGluRs), the tachykinin neuropeptide system, and the cholinergic system are all promising targets for the development of novel therapeutics for the treatment of mood disorders. Other promising targets not reviewed in this chapter include the AA cascade, the melatonergic system, the glucocorticoid system, oxidative stress, bioenergetics, and mitochondrial activity.
Abbreviations: AA: arachidonic acid; AMPA: α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate; CREB: cyclic AMP response element-binding; DAG: diacylglycerol; IP3: inositol triphosphate; mGluR: metabotropic glutamate receptor; NK: neurokinin; NMDA: N-methyl-D-aspartate; PIP-2: Phosphatidylinositol 4,5-bisphosphate; PKA: protein kinase A; PLA-2: phospolipase A2; PLC: phospholipase C; PSD: post-synaptic density; TNF: tumor necrosis factor.
1. The Opioid Neuropeptide System
The dynorphin opioid neuropeptide family modulates diverse behavioral mechanisms such as mood, endocrine, motor, and cognitive function, and opioid peptides and their receptors are potential candidates for the development of novel treatments for mood disorders. Three types of opioid receptors have been described in the pathophysiology of mood disorders: delta (δ), mu (μ), and kappa (κ). These receptors are coupled to different intracellular effector systems and are widespread in the ventral tegmental area (VTA), nucleus accumbens (NAc), and prefrontal cortex (PFC). Notably, opioid receptors are coexpressed in brain areas implicated in the pathophysiology of mood disorders [6]. For instance, patients with depression and anxiety were found to have lower serum β-endorphin levels [7]. A significant decrease in pro-dynorphin mRNA expression levels was also noted in subjects with BPD [8]. In addition, antidepressants have been shown to reverse stress-related changes in dynorphin levels in diverse limbic brain areas [9; 10].
Kappa opiate receptor activiation induces depressive symptoms in humans and animals [11], as well as psychotomimetic effects [12; 13]. In preclinical models, the kappa opioid receptor agonist Salvinorin-A (Salvia divinorum) induced depressive-like behaviors [14], while the receptor antagonist MCL-144B caused antidepressant-like effects [15; 16]. These agents have also been associated with preclinical anticonvulsant and antiepileptic effects (reviewed in [6]).
Kappa opiate receptor stimulation also decreases dopaminergic function in different brain areas [17]. In animal models of depression, the opioid antagonist naloxone blocked the antidepressant-like effects of tryciclic antidepressants and the serotonin norepinephrine reuptake inhibitor (SNRI) venlafaxine [18; 19]. In clinical studies, the partial kappa agonist pentazocine (Talwin) adjunctively showed rapid antimanic effects in subjects with BPD without arousing depressive symptoms [20]. No selective kappa agonists have been clinically evaluated as monotherapy in subjects with BPD.
Few studies have described the efficacy of opioid agonists such as oxymorphone and buprenorphine in treatment-resistant MDD [21–23]. Buprenorphine—which appears to exert antidepressant effects in certain individuals [21]—is often described as a mixed mu/kappa antagonist, although compelling evidence suggests it is actually a partial kappa agonist. Mu receptors are densely present in diverse brain regions involved in stress response and emotional regulation. Animal studies suggest that mu opioid receptor agonists have antidepressant-like effects [24–26]. For instance, the synthetic opioid tramadol, an atypical agent that binds weakly to mu receptors, showed antidepressant-like effects in diverse animal models [27–29]. Small clinical studies of individuals with treatment-resistant MDD further noted that it appeared to have antidepressant/antisuicidal effects [30; 31] and similar antidepressant effects to venlafaxine [32]. In terms of circuitry of depression and opioid receptors, a PET study by Kennedy and colleagues [33] reported dysregulated endogenous opioid emotion regulation circuitry in women with MDD. They found that sustained sadness was associated with significant decreases in mu-opioid receptor binding potential in the left inferior temporal cortex; these findings correlated with negative affect ratings. In addition, it was observed that those patients who did not respond to a 10-week course of the antidepressant fluoxetine had increased mu-opioid system activation, while responders displayed responses more closely related to those of controls (mean deactivations of opioid neurotransmission) [33].
Delta receptors have a high affinity for the endogenous opioid peptide proenkephalin. In animal models, proenkephalin-knockout mice displayed increased anxiety- and depressive-like behaviors, and diverse delta receptor agonists appear to have antidepressant-like properties [34], including SNC80 [35]. Interestingly, acute treatment of rodents with a delta opioid agonist simultaneously increased expression of brain derived neurotrophic factor (BDNF) and produced antidepressant-like effects [36]. Selective delta-opioid receptor agonists have antidepressant-like effects in models such as the forced swim test [36–39]. Receptor localization studies have shown that delta-opioid receptors reside in areas of the brain implicated in mood regulation [40–43]. For example, localization of the delta-opioid receptor in the amygdala is consistent with the modulation of anxiety states, whereas localization in cortex and hippocampus, regions associated with depression, is consistent with potential antidepressant action. Chronic but not acute treatment with imipramine significantly decreased the density and affinity of delta-opioid receptor agonist-opioid receptors in striatum of the rat brain. The decrease was confirmed by delta-opioid receptor immunostaining [44]. RB101, an inhibitor of enkephalin-degrading enzymes, produces antinociceptive, antidepressant, and anxiolytic effects in rodents, without typical opioid-related negative side effects [45]. The antidepressant-like and anxiolytic effects of RB101 appear to be mediated only through the delta-opioid receptor.
2. The Purinergic System
Purinergic neurotransmission is regulated by the neurotransmitter adenosine triphosphate (ATP) and the neuromodulator adenosine. The system modulates dopamine, gamma aminobutyric acid (GABA), and serotonin [46], and purines control sleep, motor activity, appetite, cognition, memory, and social interaction [47]. Adenosine acts mostly through adenosine 1 and 2A receptors. In animal models, it induces antidepressant-like and anticonvulsant effects [46; 48]. In animal studies, adenosine agonists exert sedative, anticonvulsant, anti-aggressive, and antipsychotic properties [49]. In contrast, adenosine antagonists such as caffeine increase irritability, anxiety, and insomnia in individuals with BPD [50].
Purinergic system dysfunction has long been implicated in the pathophysiology of BPD [47]. Early clinical studies showed that enhanced excretion of uric acid was associated with remission of manic symptoms [51]. Plasma uric acid levels have also been found to be higher during the manic phase of BPD, but not during the depressive or euthymic phases [52], suggesting that increased uric acid may represent a state rather than a trait marker of mania in BPD. Recently, increased uric acid levels were described in drug-naive subjects with BPD who were experiencing their first manic episode [53]. In addition, a recent nationwide population-based study involving more than 24,000 subjects with BPD found an increased risk of gout among patients with BPD [54]. Relatedly, the P2RX7 (purinergic receptor P2X, ligand-gated ion channel, 7) gene has been described as a candidate gene for both MDD and BPD, because of repeated associations with the rs2230912 (Gln460Arg) polymorphism [55; 56].
The purinergic modulator allopurinol, widely used to treat gout, was recently shown to induce antimanic effects in three different clinical studies [57–59]. One study assessed the antimanic effects of adjunctive allopurinol for eight weeks [57], and noted a difference between allopurinol and placebo at endpoint. Another recent, double-blind, placebo-controlled study evaluated the efficacy of allopurinol, dipyridamole, and placebo added to lithium in 180 subjects with BPD during a manic episode [59]; antipsychotics were not allowed during the trial. Allopurinol significantly decreased manic symptoms after four weeks compared to dipyridamole and placebo, and the antimanic effects of allopurinol were directly correlated with decreased plasma uric acid levels [59].
3. The Glutamatergic System
Glutamate is the most abundant excitatory neurotransmitter in the brain, and is present in three different cell compartments: the pre- and post-synaptic neurons and glial cells; together these comprise the “tripartite glutamatergic synapse” [60] (see Figure 2). Glutamate modulates several functions, including memory, learning, and synaptic plasticity [61–64].
Figure 2. Pathophysiological basis and potential therapeutic targets for mood disorders involving glutamatergic neurotransmission.
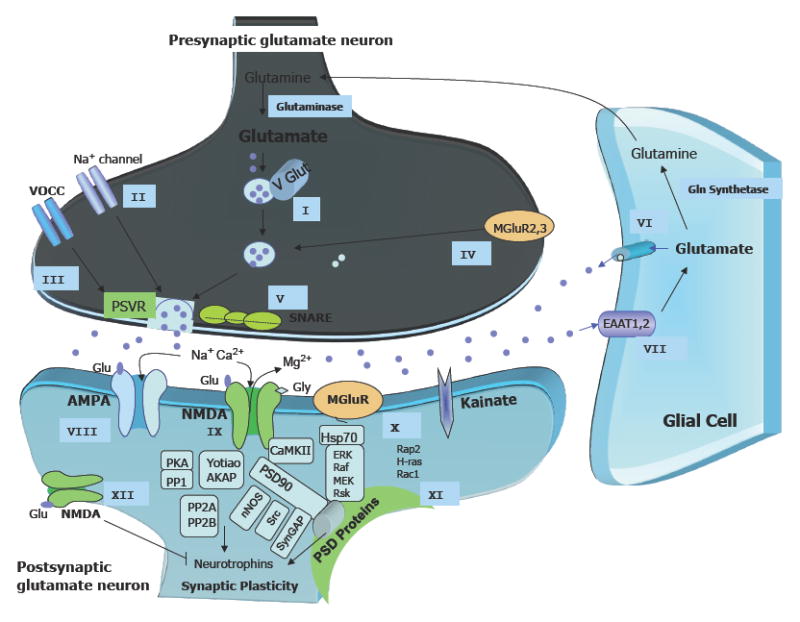
Ketamine preferentially targets postsynaptic AMPA/NMDA receptors, while riluzole’s antidepressant effects occur through direct regulation, mostly at presynaptic voltage-operated channels and glia. All potential targets for new, improved glutamatergic agents are described below.
Presynaptic targets: Before release, Glu is packaged into vesicles by VGLUTs, which control glutamate concentration produced in the synaptic vesicles (Target I). Activation of voltage-gated sodium channels (Target II) and VOCCs (Target III) depolarizes the plasma membrane, allowing for the influx of sodium (through depolarization of axon terminals) and calcium (through interaction with the SNARE proteins) (Target IV), leading to the fusion of synaptic vesicles and the consequent release of Glu into the synaptic cleft. The type II mGluRs (mGlu2/3) are also present presynaptically (Target V), and may directly limit the synaptic release of Glu. These mGluRs also mediate feedback inhibition of Glu release, thus decreasing the activity of Glu synaptic terminals when the activation of presynaptic receptors reaches a certain level.
Glia: Glu can also be directly released from glial cells, regulating synaptic activity pre-and post-synaptically (Target VI). After its release, large amounts of Glu are rapidly distributed across the synaptic cleft, where it can bind to glutamate receptors in the postsynaptic regions. The remaining unbound Glu is rapidly removed from the synaptic cleft to the presynaptic neuron mostly by glia and EAATs. The glial transporters EAAT1 (also known in rodents as GLAST) and EAAT2 (or GLT-1) are essential for blocking pathological increases in Glu levels (Target VII). Glial Glu transporter expression is upregulated by neuronal activity. Notably, the expression of Glu transporters by glial cells and their anatomical arrangement within the synaptic cleft can be dynamically and reversibly modified, and can directly regulate their ability to scavenge Glu.
Postsynaptic neuron: AMPARs (GluR1–4) mediate fast Glu neurotransmission and play a major role in learning and memory through critical regulation of calcium metabolism, plasticity, and oxidative stress. When activated, these receptors open the transmembrane pore, thus allowing the influx of sodium and the consequent depolarization of the neuronal membrane (Target VIII).
The NMDAR channel includes the subunits NR1, NR2 (NR2A–NR2D), and NR3 (NR3A and NR3B). Glu’s binding sites have been described mostly in the NR2 subunit, whereas the NR1 subunit is the site for its co-agonist, glycine. NR2A and NR2B subunit receptors are both highly expressed in brain areas implicated in mood regulation, but NMDA receptors containing NR2A mediate faster neurotransmission than NR2B receptors (Target IX).
The mGluRs include eight receptor subtypes (mGluR1 to mGluR8) classified in three groups (I-III) based on their sequence homology and effectors. The mGluRs in Group I, including mGluR1 and mGluR5, stimulate the breakdown of phosphoinositide phospholipids in the cell plasma membrane. mGluRs in Groups II (including mGLuR2/3) and III (including mGluRs 4, 6, 7, and 8) limit the generation of cAMP by activating inhibitory G- proteins. While Group I receptors are coupled to the phospholipase C signal transduction pathway, Group II and III mGluRs are both coupled in an inhibitory manner to the adenylyl cyclase pathway, which is involved in regulating the release of Glu or other neurotransmitters such as GABA (Target X).
KARs are involved in excitatory neurotransmission by activating postsynaptic receptors, and in inhibitory neurotransmission by modulating GABA release. There are five types of KAR subunits: GluR5 (GRIK1), GluR6 (GRIK2), GluR7 (GRIK3), KA1 (GRIK4), and KA2 (GRIK5). GluR5–7 can form heteromers; KA1 and KA2 can only form functional receptors by binding with one of the GluR5–7 subunits. KARs have a more limited distribution in the brain than AMPARs and NMDARs, and their function is not well defined; they are believed to affect synaptic signaling and plasticity less than AMPARs (Target XI).
Cytoplasmic PSD-enriched intracellular molecules (e.g. PSD93) interact with Glu receptors at the synaptic membrane to modulate receptor activity and signal transduction. PSD95 and similar scaffolding molecules link the NMDARs with intracellular enzymes that regulate signaling, and also provide a physical connection between diverse neurotransmitter systems to synchronize information from different effectors (Target XII).
Finally, recent preclinical work suggests that the mammalian target of rapamycin (mTOR)-dependent synapse formation underlies ketamine’s rapid antidepressant properties [121] (not shown).
Abbreviations: AMPA: -amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; cAMP: cyclic adenosine monophosphate); EAAT: excitatory amino-acid transporters; GABA: gamma-aminobutyric acid; Glu: Glutamate; Gly: Glycine; KA: kainate; NMDA: N-methyl-D-aspartate; PSD: postsynaptic density; PSVR: presynaptic voltage-operated release; mGluRs: metabotropic Glu receptors; SNARE: soluble N-ethylmaleimide-sensitive factor attachment receptor; VGLUTs: vesicular Glu transporters.
Glutamate receptor subtypes include a group of pharmacologically distinct ligand-gated ion channels (N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors) and the eight G-protein coupled metabotropic receptors. Increased synaptic levels of glutamate can lead to excitotoxic levels after an insult, leading to increased extrasynaptic glutamate [65]. Several glutamate ionotropic receptors and their respective subunits have been identified: NMDA (NR1, NR2, NR2B, NR2C, NR2D, NR3A, and NR3B subunits), AMPA (GluR1, GluR2, GluR3, and GluR4) and kainate (GluR5, GluR6, GluR7, KA1, and KA2).
The Effects of Mood Stabilizers on the Glutmatergic System
Notably, several mood stabilizers—typically used to treat patients with BPD—indirectly or directly affect the glutamatergic system. For instance, the classic mood stabilizer lithium alters glutamate uptake [66], glutamate receptor expression and function [67–71], vesicular glutamate transporter 1 (VGLUT1) expression, and presynaptic glutamate release [72–74]. Similarly, the mood stabilizer valproate (VPA) alters VGLUT1 expression and presynaptic glutamate release [75; 76], increases excitatory amino-acid transporter (EAAT) expression [77; 78], and alters glutamate receptor expression and function [67; 79–85].
In addition, prolonged treatment—both in vitro and in vivo—with therapeutically relevant concentrations of either lithium and VPA downregulates AMPA GluR1 synaptic expression in hippocampus [86]. Additional support for the therapeutic relevance of these data is provided by recent studies indicating that AMPA receptor antagonists attenuate several “manic-like” behaviors produced by amphetamine administration [87].
Of all the medications currently used to treat mood disorders, lamotrigine—an anticonvulsant that also has mood stabilizing properties—probably has the most direct effect on the glutamatergic system. Lamotrigine inhibits the release of glutamate in the hippocampus of rats [88], and increases AMPA subunit receptor expression [79]. Lamotrigine may also inhibit the excessive release of glutamate via its limiting effects at P-type and N-type calcium channels, potassium channels, and sodium channels [89]. However, it is important to note that while lamotrigine is associated with modest efficacy in the treatment of bipolar depression [90], its clinical utility has been questioned [91].
Ionotropic Glutamate Receptors: NMDA
Preclinical evidence suggests that NMDA receptor antagonists display antidepressant-like effects in different models (reviewed in [92; 93]). For instance, the NMDA anatagonists dizocilpine (MK-801) and CGP 37849 have consistently shown antidepressant-like effects as monotherapy or when combined with traditional antidepressants [94–98]. Also, NR2A knockout mice showed highly robust anxiolytic- and antidepressant-like phenotypes [99], and GluR1 knockout mice displayed increased depressive-like behaviors (learned helplessness) [100].
Ketamine
The high affinity NMDA receptor antagonist ketamine increases presynaptic release of glutamate by increasing glutamatergic neuron firing after disinhibiting gamma aminobutyric acid (GABA)ergic inputs [101]. In animal models, ketamine displayed anxiolytic and antidepressant effects [102–106].
Notably, ketamine’s antidepressant effects have been tested in several clinical trials for both MDD and bipolar depression. In the first small, preliminary investigation [107], significant antidepressant effects were noted within 72 hours after ketamine infusion in treatment-resistant patients with MDD. In a double-blind, crossover, placebo-controlled study evaluating patients with treatment-resistant MDD, a rapid (within two hours) and relatively sustained (one to two weeks) antidepressant effect was noted after a single infusion of ketamine [108]. More than 70% of patients responded to ketamine (50% improvement) at 24 hours after infusion, and 35% maintained a sustained response after one week of treatment. It is important to note that the response rates (71%) 24 hours after infusion with ketamine were similar to those observed after six to eight weeks of therapy with standard monoaminergic-based antidepressants (65%) [109]. Ketamine was superior to placebo on the 21-item HAM-D from 110 minutes through seven days after the single infusion. This finding has since been replicated in several other, albeit uncontrolled, studies [110–112]; the magnitude and time-frame of response to ketamine in these studies was similar to the previous controlled studies. Several variables have been found to predict initial antidepressant response to ketamine, including family history of alcohol dependence [111], and increased pretreatment rostral ACC activity [113]. More recently, anterior cingulate desynchronization and functional connectivity with the amygdala during a working memory task was found to predict antidepressant response to ketamine [114]. In contrast, plasma BDNF levels—which other studies have noted is associated with antidepressant response to chronic treatment with standard antidepressants [115]—were not associated with initial antidepressant response to a single dose of ketamine [116].
More recently, ketamine has been studied in bipolar depression [117]. In that study, subjects receiving ketamine had significant improvement in depressive symptoms compared to patients receiving placebo within 40 minutes after infusion; these effects remained significant through Day 3, and up to Day 7 in those who completed both phases of the study. Of the 17 subjects treated with ketamine, 56% met response criteria at 40 minutes post-infusion, and 44% met response criteria and 31% met remission criteria the day following ketamine infusion. Seventy-one percent of all subjects responded to ketamine at some point during the study.
Ketamine has also been found to have significant antisuicidal effects [118; 119]. In the study by Price and colleagues, 26 patients with treatment-resistant MDD where found to have significant reductions on the suicidality item of the MADRS 24 hours after a single infusion of ketamine. In the study by Diazgranados and colleagues, 33 subjects with treatment-resistant MDD received a single open-label infusion of ketamine and were rated at baseline and at 40, 80, 120, and 230 minutes post-infusion. Suicidal ideation scores significantly decreased on the Scale for Suicide Ideation (SSI) and suicidality items of depression rating scales within 40 minutes after infusion, and this effect remained significant through the first four hours post-infusion. Measures of depression, anxiety, and hopelessness were significantly improved at all time points. In addition to confirming the findings of the first study, the second study demonstrated that suicidal ideation was improved as soon as 40 minutes post-infusion, a finding with enormous public health implications. Controlled studies are necessary to confirm ketamine’s rapid antisuicidal properties.
Because of the inherent propensity of ketamine to produce cognitive deficits and psychotomimetic effects, its use at this time remains limited to the research setting. Studies with more selective subtype NMDA antagonists are underway, in order to determine whether these have antidepressant effects that can occur safely without causing ketamine’s undesirable side effects. A recent, double-blind, randomized, placebo-controlled clinical trial evaluating the NMDAR-2B subunit-selective antagonist CP-101,606 found that this agent induced significant and relatively rapid antidepressant effects (by Day 5) in patients with treatment-resistant MDD, but with evidence of psychomimetic properties [120]. As a result, the CP-101,606 research program was discontinued. However, additional clinical studies with subunit NMDR-2A and −2B antagonists are underway or completed and include AZD6765 (AstraZeneca Pharmaceuticals, completed) and EVT 101 (Evotec Neurosciences; ongoing). In addition to determining the efficacy of these agents in treatment-resistant MDD, these studies should also shed some light on the issue of rapidity of onset of antidepressant action.
It is also interesting to note that ketamine’s rapid antidepressant effects may occur by increasing AMPA throughput, as suggested by behavioral studies showing that blockade of AMPA activation limit the antidepressant effects of ketamine [104]. It is thus possible that targeting AMPA receptors more directly, or AMPA’s downstream pathways, would yield similar antidepressant effects to ketamine but without its psychomimetic effects. This preclinical work has been further extended to suggest that mammalian target of rapamycin (mTOR)-dependent synapse formation underlies ketamine’s rapid antidepressant properties [121]. In an elegant series of studies, investigators from the Duman laboratory demonstrated that ketamine rapidly activates the mTOR pathway, leading to increased synaptic signaling proteins and increased number and function of new spine synapses in the prefrontal cortex of rats.
Ionotropic Glutamate Receptors: AMPA
AMPA receptors are a subfamily of ionotropic glutamatge receptors that mediate the fast excitatory component of neurotransmission. AMPA receptor potentiators have been tested in preclinical models of mood disorders. This class of agents limits receptor desensitization and/or deactivation rates [122]. It includes AMPAkines, which have displayed antidepressant-like effects in different models [79; 123; 124]. AMPA receptor trafficking is believed to modulate the antidepressant-like effects of AMPA potentiators by exerting a key role in regulating synaptic strength and behavioral plasticity [125].
Interestingly, in animal studies the AMPAkine Ampalex showed antidepressant-like effects [126]. Clinically, AMPA receptor antagonists such as GYKI 53773 and LY 300164 have been studied as potential anticonvulsants in Phase III trials. The competitive AMPA receptor antagonist NS1209 was found to induce more rapid and consistent anticonvulsant effects than diazepam in animal models [127], and showed good central nervous system (CNS) bioavailability and tolerability in Phase I/II clinical trials. It has been also evaluated in refractory status epilepticus [128]. Clinical testing of NS-1209 in the treatment of refractory status epilepticus has been initiated, but no results are yet available [129].
Riluzole
Riluzole is blood-brain-penetrant glutamatergic agent with well-defined neuroprotective properties and is FDA-approved for the treatment of amyotrophic lateral sclerosis (ALS). Riluzole increases membrane insertion and AMPA trafficking (mostly GluR1 and GluR2), thus limiting glutamate release. Riluzole also increases glutamate reuptake and enhances neurotrophin synthesis [130; 131].
In preclinical studies, pretreatment with 10 mg/kg riluzole (but not 3 mg/kg) moderately decreased amphetamine-induced hyperlocomotion [132], suggesting a potential antimanic effect. Enhanced 13C-glucose metabolism in the hippocampus and prefrontal cortex was also described after 21 days of treatment with riluzole [133].
In open-label clinical studies, riluzole showed significant antidepressant efficacy and good tolerability in patients with treatment-resistant MDD as well as those with bipolar depression. In MDD, 13 patients (68%) completed the trial, and all displayed significant improvement in both depressive and anxiety symptoms at Week 6 [134]. Another study found that riluzole induced antidepressant effects after one week of treatment, decreasing HAM-D scores 36% among completers [135]. Riluzole was also tested in 14 patients with bipolar depression as add-on therapy to lithium for eight weeks. Patients in that study had a significant decrease in MADRS scores [136]. Another recent, small, open-label, add-on study of 14 subjects with bipolar depression found that riluzole significantly reduced HAM-D scores [137].
Metabotropic Glutamate Receptors (mGluRs)
There is considerable interest in the potential psychiatric applications of mGluR agonists and antagonists (reviewed in [138]). The mGluR family includes eight receptor subtypes (mGluR1 to GluR8) separated into three groups based on their agonist selectivity, second messenger systems coupled-receptor, and sequence homology. Group I mGluRs (mGluR1 and mGluR5) are coupled to the phospholipase C signal transduction pathway, while Group II (mGluR2 and mGluR3) and III (mGluR4 and mGluR6 to mGluR8) receptor activity inhibits the adenylyl cyclase signal transduction pathway.
Group I mGLuRs have functionally important interactions with NMDA receptors to modulate posynaptic excitability. The mGluRs are involved in the early phase of memory formation and the mechanism of long-term depression [139–141]. The Group I mGluR5 antagonists MPEP (2-methyl-6-[phenylethynyl]-pyridine) and MTEP ([(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine) have shown antidepressant-like activity in the modified forced swim test in rats [142], tail suspension test in mice [142], and olfactory bulbectomized rats [143]. Furthermore, MPEP treatment was recently found to increase hippocampal mRNA levels [144]. The mGluR1 antagonist EMQMCM ([3-ethyl-2-methyl-quinolin-6-yl]-(4-methyoxy-cyclohexyl)-methanone methanesulfonate) was active in the modified forced swim test in rats and in the tail suspension test in mice [145].
The mode of antidepressant-like activity of mGluR1 or −5 antagonists is uncertain; some have suggested that mGluR5 inhibitors might produce a final effect that is similar to that evoked by NMDA antagonists, which are known to display antidepressant-like effects. It is also possible that mGluR5 antagonists could exert antidepressant properties by acting on Group 1 mGluRs on glial cells and reducing the hypothesized overactivation of extrasynaptic NMDA receptors [146]. However, studies of the non-benzodiazepine anxiolytic fenobam, a potent and selective mGluR5 antagonist, were discontinued because of psychostimulant effects that occurred with its use [147]. The mGluR5 positive allosteric modulator 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB) was found to be brain penetrant and to reverse amphetamine-induced locomotor activity and amphetamine-induced deficits in prepulse inhibition in rats, two models thought to be sensitive to antipsychotic treatment [148]. Should this compound result in an antipsychotic drug for clinical use, it is likely that it will be used clinically during manic episodes. Also currently underway is a trial for treatment-resistant MDD with an mGluR5 allosteric antagonist (RO4917523, Roche); this trial is also examining the rapidity of onset of antidepressant action.
Group II mGluR2, mGluR2/3 are negatively linked to the adenylyl cyclase signal transduction pathway and decrease glutamate release, especially under conditions of glutamate excess in the synapse; moreover, they regulate glutamate transmission by post-synaptic mechanisms. Group II mGluRs agonists (e.g., LY341495) dose-dependently decreased the immobility time of mice in the tail suspension test and reduced immobility time and increased swimming behavior without affecting climbing behavior in rats [149]. In addition, MGS-0039 has been reported to be effective in the learned helplessness model of depression [150] and to increase cell proliferation in the adult mouse hippocampus [151]. AMPA receptor activation is believed to be responsible at least in part for the antidepressant-like activity of Group II mGluR antagonists; the AMPA antagonist NBQX blocked the antidepressant-like activity of MGS-0039 in the tail suspension test in mice [152]. Consistent with this notion, a recent study 24-week, randomized, double-blind, placebo-controlled trial of adjunctive NAC, a drug that indirectly increases mGluR2 stimulation by raising extrasynaptic glutamate levels [153], found that this agent had antidepressant effects in patients with BPD [154].
Pilc and colleagues demonstrated that a selective Group III mGluR agonist (ACPT-I, [1S,3R,4S]-1-aminocyclo-pentane-1,3,4-tricarboxylic acid) and an mGluR8 agonist (RS-PPG, [RS]-4-phosphonophenylglycine) for this receptor had antidepressant-like effects in the forced swim test in rats [155]. Additional proof for the involvement of Group II mGluRs comes from studies showing that mGluR7 knockout in mice produced antidepressant-like effects in the forced swim test and in the tail suspension test [156], and that Group III mGluR agonists had antidepressant effects in the behavioral despair test [157]. While the mGluR5 antagonists and Group II mGluR antagonists are promising compounds with considerable potential antidepressant-like activity, no Group III mGluR agonists have been clinically tested or studied in animal models of mania,
4. The Tachykinin Neuropeptide System
Several preclinical and clinical studies have investigated the potential therapeutic role of the tachykinin neuropeptide system in mood disorders. These neuropeptides include substance P (SP), neurokinin A, neurokinin B, and their respective receptors (NK1, NK2, NK3). The activity of these neuropeptides involves G-protein coupled receptor activity, increasing intracellular calcium via phospholipase C, and the diacylglycerol (DAG) signaling cascade [158]. These act as neuromodulators and neurotransmitters, and interact directly with the monoaminergic system in diverse brain areas implicated in mood regulation and emotion-processing [159]. Alterations in this system have been implicated in the pathophysiology of BPD. For instance, increased SP has been described in the CSF and serum of subjects experiencing a major depressive episode [159].
In preclinical models, the use of tachykinin antagonists—especially those associated with NK2 receptor regulation—induced antidepressant-like effects in several paradigms [160–163]. Interestingly, some of these agents are believed to potentiate the therapeutic effects of selective serotonin reuptake inhibitors (SSRIs). In addition, the TAC1 gene (tachykinin 1, SP) located at 7q21.3 encodes SP and neurokinin A, and TAC1 knockout mice showed decreased depression- and anxiety-related behaviors in diverse paradigms [164].
Several neurokinin receptor antagonists have been tested in Phase II and III trials, with conflicting results. Three different NK1 receptor antagonists—MK869, L759274, and CP122721—reduced depressive symptoms compared to placebo in randomized, double-blind, Phase II clinical trials; both MK869 and CP122721 were associated with fewer adverse effects than the active comparator (paroxetine or fluoxetine) [165; 166]. However, the efficacy of MK869 was not replicated in another multi-site, placebo-controlled, Phase III trial [167]. Three other studies that investigated NK1 and NK3 receptor antagonists found that these agents had no antidepressant efficacy compared to active agents or placebo [166; 168]. The NK1 receptor antagonist orvepitant (GW823296; GlaxoSmithKline) study in MDD was terminated to allow assessment of isolated events of seizure during the program. Similarly, a six-week Phase II trial with the NK3 receptor antagonist SR142801 (osanetant) found that it was no more effective than placebo or paroxetine [169]. However, in recent Phase III trials, the NK2 receptor antagonist SR48968 (saredudant) showed significant antidepressant effects during depressive episodes in a group of adult and elderly patients with MDD [159].
5. The Cholinergic System
Several decades ago, Janowsky proposed the “cholinergic-adrenergic imbalance hypothesis” of BPD, which suggested that cholinergic dysfunction was key to the pathophysiology of mood disorders [170]. On a genetic level, there is still little evidence that cholinergic system dysregulation is involved in mood disorders. One study found an association between 19 cholinergic genes and BPD [171]. In addition, decreased muscarinic type 2 receptor binding was noted in the anterior cingulate cortex of individuals with BPD in a PET study [172]. Animal studies found that that rats bred for increased muscarinic receptor sensitivity showed a behavioral phenotype similar to patients with depression (e.g., lethargy, lack of pleasure, and behavioral despair) [173]. In humans, cholinergic hyperactivity results in a worsening of depressive symptoms in patients with MDD [170]. Experiments add further support to this theory, as neuroendocrine and pupillary responses to cholinergic activity were found to be enhanced in depressed subjects [174] and attenuated in manic subjects [175]; improvement of manic symptoms with lithium and valproate led to normalization of pupillary responses.
Early clinical studies by Kasper and colleagues (1981) found that the anticholinergic drug biperiden had significant antidepressant properties [176]. A small controlled trial with the short-acting cholinesterase inhibitor physostigmine in mania showed that this agent had rapid, but not sustained, antimanic efficacy after single or multiple injections [177; 178]. Also, the long-acting cholinesterase inhibitor donepezil, added on to mood stabilizers, demonstrated rapid and significant antimanic effects in more than half of subjects with treatment-resistant mania [179]. Two recent double-blind studies investigated the antimuscarinic drug scopolamine hydrobromide in MDD and bipolar depression [180; 181]. In both trials, rapid antidepressant effects (within three to five days) were noted. Finally, it is of note that scopolamine appears to produce larger antidepressant and antianxiety effects in women than in men [182]. When reviewing the results of trials with agents like these that produce noticeable effects, it is important to note that future studies would be expected to include active comparators, given that these types of agents demonstrate effects that could potentially unmask the blinding.
6. Intracellular Signaling Pathways
Glycogen Synthase Kinase-3 (GSK-3)
GSK-3 is a highly active and multifunctional serine/threonine kinase that regulates diverse signaling pathways (eg, the phosphoinositide 3-kinase pathway, the Wnt pathway, protein kinase A (PKA), and protein kinase C (PKC)). In general, elevated activity of GSK-3 is proapoptotic, whereas GSK-3 inhibition attenuates or prevents apoptosis. Lithium and other mood stabilizers have been shown to directly target GSK-3 activity [183; 184]. Lithium also induces neurotrophic and neuroprotective effects in rodents, in part due to GSK-3 inhibition (reviewed in [185]). Mice overexpressing a constitutively active form of GSK-3β in the brain displayed enhanced locomotor activity as well as decreased habituation in the open field test. In contrast, a recent study focusing specifically on GSK-3α found that GSK-3α knockout mice showed decreased exploratory activity, decreased immobility time in the forced swim test, and reduced aggressive behavior, among other phenotypes [186]. Another study found that the GSK-3 inhibitor AR-A014418 induced significant antidepressant-like effects in the forced swim test and attenuated D-amphetamine-induced hyperlocomotion [187; 188].
In monocytes from a small group of symptomatic patients with BPD, serine phosphorylation of both GSK-3α and GSK-3β was lower than in healthy controls [189]. Notably, reduced serine phosphorylation of GSK-3 correlated significantly with severity of manic and depressive symptoms, suggesting that GSK-3 activity was affected by mood states. Studies of several other cohorts measuring the expression of GSK-3 mRNA and proteins generated inconsistent results. One study found that total GSK-3 levels tended to be higher in BPD-I than healthy controls in a Chinese population (unpublished data), while another study found that GSK-3 mRNA levels were lower in the peripheral blood mononuclear cells (PBMCs) of teenage suicide victims, and that GSK-3 protein levels were lower in platelets of patients with BPD [190]. Li and colleagues recently reported that levels of GSK-3α and GSK-3β in a group of bipolar manic subjects were higher than healthy controls [191]. They also noted that the improvement of manic symptoms during eight weeks of treatment with lithium, valproate, and atypical antipsychotics was associated with a significant increase in the inhibitory serine phosphorylation of GSK-3, but not total GSK-3 levels. This finding is in line with preclinical data suggesting that GSK-3 inhibition via increased serine phosphorylation is a response of GSK-3 to the psychotropics used to treat BPD, and supports the notion that GSK-3 is a promising molecular target in the pharmacological treatment of BPD.
Presently, no blood-brain-penetrant GSK-selective inhibitor has been clinically tested in proof-of-principle studies in mood disorders. It is important to mention, however, that the use of GSK-3 inhibitors would be limited by GSK-3’s involvement with diverse pathways and multiple substrates that may induce side effects and/or toxicity [192].
PKC Signaling Cascade
PKC plays a key role in regulating neuronal excitability, neurotransmitter release, and plasticity. PKC isoforms differ in their structure, subcellular localization, tissue specificity, mode of activation, and substrate specificity. Diverse studies support the direct involvement of PKC and its substrates in the pathophysiology and therapeutics of mood disorders [193–198]. In preclinical models, lithium and valproate have been found to directly regulate PKC, and the PKC inhibitor tamoxifen was found to limit amphetamine-induced hyperactivity in a large open field [199].
A recent clinical trial provided further evidence for the involvement of this system in bipolar mania. Although well known for its anti-estrogenic properties, tamoxifen is also a potent PKC inhibitor at high concentrations. A single-blind study showed that tamoxifen had significant antimanic effects in five of seven BPD subjects [200]. Another four-week, three-arm, double-blind, placebo-controlled, add-on study involving 13 women compared the antimanic efficacy of tamoxifen with medroxyprogesterone acetate and placebo. Greater improvement in manic and positive symptoms of psychosis was noted in patients receiving tamoxifen than in those receiving placebo; all patients received concomitant lithium or valproate [201]. These preliminary results were subsequently confirmed in two three-week, double-blind, placebo-controlled, monotherapy studies [202; 203]. Both studies showed that tamoxifen had superior antimanic efficacy compared to placebo, and that these therapeutic effects were not associated with sedative effects or increased risk of depression. However, some of tamoxifen’s antimanic effects may be due to its anti-estrogen effects (see [204]). There is considerable enthusiasm for developing PKC inhibitors; one recently synthesized agent is endoxifen, which exhibits a fourfold higher PKC potency than tamoxifen [205]. However, it is not clear whether it also has anti-estrogen effects, a property that limits tamoxifen’s usefulness in the long-term treatment of BPD.
Conclusion
Recent preclinical studies have investigated diverse targets/compounds and neurotransmitter and neuromodulatory systems that are now at the proof-of-concept stage. Here, we reviewed several agents for which there are recent and promising data. Ultimately, these may result in putative novel treatments for mood disorders. These include (1) the opioid neuropeptide system, (2) the purinergic system, (3) the glutamatergic system, (4) the tachykinin neuropeptide system, (5) the cholinergic system (muscarinic system), and (6) intracellular signaling pathways. It is important to note that none of these treatments have yet been shown to be safe and effective in the treatment of mood disorders. Also, important methodological differences exist regarding how specific compounds are evaluated. For instance, most of the evidence presented here comes from case reports, case series, or proof-of-concept studies, some with very small sample sizes. Key to future studies are such variables as what dose/plasma levels and time frames are consistent with clinical therapeutic effect, localization to brain regions implicated in the neurobiology of the disorder under consideration, and association with human genetic findings. However, given the global impact of these disorders, and the dearth of rapid-acting and effective treatments, a new generation of agents for the treatment of mood disorders is clearly needed. The evidence reviewed here can guide important future research in drug development for mood disorders and, ultimately, have a tremendous positive impact on public health worldwide.
Acknowledgments
Funding for this work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health, Department of Health and Human Services (IRP-NIMH-NIH-DHHS). Ioline Henter provided invaluable editorial assistance.
Footnotes
Disclosure
Dr. Zarate is listed as a co-inventor on a patent for the use of ketamine in major depression. Dr. Zarate has assigned his patent rights on ketamine to the U.S. government. The author(s) declare that, except for income received from our primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. Dr Machado-Vieira would like to thank FAPESP and ABADHS, Sao Paulo, Brazil.
References
- 1.Fagiolini A, Kupfer DJ, Masalehdan A, et al. Functional impairment in the remission phase of bipolar disorder. Bipolar Disord. 2005;7(3):281–5. doi: 10.1111/j.1399-5618.2005.00207.x. [DOI] [PubMed] [Google Scholar]
- 2.Gitlin MJ, Mintz J, Sokolski K, et al. Subsyndromal depressive symptoms after symptomatic recovery from mania are associated with delayed functional recovery. J Clin Psychiatry. 2010 doi: 10.4088/JCP.09m05291gre. [DOI] [PubMed] [Google Scholar]
- 3.Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530–7. doi: 10.1001/archpsyc.59.6.530. [DOI] [PubMed] [Google Scholar]
- 4.Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 5.Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356(17):1711–22. doi: 10.1056/NEJMoa064135. [DOI] [PubMed] [Google Scholar]
- 6.Schwarzer C. 30 years of dynorphins--new insights on their functions in neuropsychiatric diseases. Pharmacol Ther. 2009;123(3):353–70. doi: 10.1016/j.pharmthera.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darko DF, Risch SC, Gillin JC, Golshan S. Association of beta-endorphin with specific clinical symptoms of depression. Am J Psychiatry. 1992;149(9):1162–7. doi: 10.1176/ajp.149.9.1162. [DOI] [PubMed] [Google Scholar]
- 8.Hurd YL. Subjects with major depression or bipolar disorder show reduction of prodynorphin mRNA expression in discrete nuclei of the amygdaloid complex. Mol Psychiatry. 2002;7(1):75–81. doi: 10.1038/sj.mp.4000930. [DOI] [PubMed] [Google Scholar]
- 9.Chartoff EH, Papadopoulou M, MacDonald ML, et al. Desipramine reduces stress-activated dynorphin expression and CREB phosphorylation in NAc tissue. Mol Pharmacol. 2009;75(3):704–12. doi: 10.1124/mol.108.051417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirayama Y, Ishida H, Iwata M, et al. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90(5):1258–68. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- 11.Barber A, Gottschlich R. Novel developments with selective, non-peptidic kappa-opioid receptor agonists. Expert Opin Investig Drugs. 1997;6(10):1351–68. doi: 10.1517/13543784.6.10.1351. [DOI] [PubMed] [Google Scholar]
- 12.Rimoy GH, Wright DM, Bhaskar NK, Rubin PC. The cardiovascular and central nervous system effects in the human of U-62066E. A selective opioid receptor agonist. Eur J Clin Pharmacol. 1994;46(3):203–7. doi: 10.1007/BF00192549. [DOI] [PubMed] [Google Scholar]
- 13.Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology (Berl) 2001;157(2):151–62. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- 14.Carlezon WA, Jr, Beguin C, DiNieri JA, et al. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316(1):440–7. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- 15.Mague SD, Pliakas AM, Todtenkopf MS, et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305(1):323–30. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 16.Reindl JD, Rowan K, Carey AN, et al. Antidepressant-like effects of the novel kappa opioid antagonist MCL-144B in the forced-swim test. Pharmacology. 2008;81(3):229–35. doi: 10.1159/000112867. [DOI] [PubMed] [Google Scholar]
- 17.Carlezon WA, Jr, Beguin C, Knoll AT, Cohen BM. Kappa-opioid ligands in the study and treatment of mood disorders. Pharmacol Ther. 2009;123(3):334–43. doi: 10.1016/j.pharmthera.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berrocoso E, Rojas-Corrales MO, Mico JA. Non-selective opioid receptor antagonism of the antidepressant-like effect of venlafaxine in the forced swimming test in mice. Neurosci Lett. 2004;363(1):25–8. doi: 10.1016/j.neulet.2004.03.041. [DOI] [PubMed] [Google Scholar]
- 19.Devoize JL, Rigal F, Eschalier A, et al. Influence of naloxone on antidepressant drug effects in the forced swimming test in mice. Psychopharmacology (Berl) 1984;84(1):71–5. doi: 10.1007/BF00432028. [DOI] [PubMed] [Google Scholar]
- 20.Cohen BM, Murphy B. The effects of pentazocine, a kappa agonist, in patients with mania. Int J Neuropsychopharmacol. 2008;11(2):243–7. doi: 10.1017/S1461145707008073. [DOI] [PubMed] [Google Scholar]
- 21.Bodkin JA, Zornberg GL, Lukas SE, Cole JO. Buprenorphine treatment of refractory depression. J Clin Psychopharmacol. 1995;15(1):49–57. doi: 10.1097/00004714-199502000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Nyhuis PW, Gastpar M, Scherbaum M. Opiate treatment in depression refractory to antidepressants and electroconvulsive therapy. J Clin Psychopharmacol. 2008;28:593–595. doi: 10.1097/JCP.0b013e31818638a4. [DOI] [PubMed] [Google Scholar]
- 23.Stoll AL, Rueter S. Treatment augmentation with opiates in severe and refractory major depression. Am J Psychiatry. 1999;156(12):2017. doi: 10.1176/ajp.156.12.2017. [DOI] [PubMed] [Google Scholar]
- 24.Besson A, Privat AM, Eschalier A, Fialip J. Effects of morphine, naloxone and their interaction in the learned-helplessness paradigm in rats. Psychopharmacology (Berl) 1996;123(1):71–8. doi: 10.1007/BF02246283. [DOI] [PubMed] [Google Scholar]
- 25.Rojas-Corrales MO, Berrocoso E, Gibert-Rahola J, Mico JA. Antidepressant-like effects of tramadol and other central analgesics with activity on monoamines reuptake, in helpless rats. Life Sci. 2002;72(2):143–52. doi: 10.1016/s0024-3205(02)02220-8. [DOI] [PubMed] [Google Scholar]
- 26.Tejedor-Real P, Mico JA, Maldonado R, et al. Implication of endogenous opioid system in the learned helplessness model of depression. Pharmacol Biochem Behav. 1995;52(1):145–52. doi: 10.1016/0091-3057(95)00067-7. [DOI] [PubMed] [Google Scholar]
- 27.Rojas-Corrales MO, Berrocoso E, Gibert-Rahola J, Mico JA. Antidepressant-like effect of tramadol and its enantiomers in reserpinized mice: comparative study with desipramine, fluvoxamine, venlafaxine and opiates. J Psychopharmacol. 2004;18(3):404–11. doi: 10.1177/026988110401800305. [DOI] [PubMed] [Google Scholar]
- 28.Rojas-Corrales MO, Gibert-Rahola J, Mico JA. Tramadol induces antidepressant-type effects in mice. Life Sci. 1998;63(12):PL175–80. doi: 10.1016/s0024-3205(98)00369-5. [DOI] [PubMed] [Google Scholar]
- 29.Yalcin I, Aksu F, Bodard S, et al. Antidepressant-like effect of tramadol in the unpredictable chronic mild stress procedure: possible involvement of the noradrenergic system. Behav Pharmacol. 2007;18(7):623–31. doi: 10.1097/FBP.0b013e3282eff109. [DOI] [PubMed] [Google Scholar]
- 30.Shapira NA, Verduin ML, DeGraw JD. Treatment of refractory major depression with tramadol monotherapy. J Clin Psychiatry. 2001;62(3):205–6. doi: 10.4088/jcp.v62n0312b. [DOI] [PubMed] [Google Scholar]
- 31.Spencer C. The efficacy of intramuscular tramadol as a rapid-onset antidepressant. Aust N Z J Psychiatry. 2000;34(6):1032–3. doi: 10.1177/000486740003400101. [DOI] [PubMed] [Google Scholar]
- 32.Reeves RR, Cox SK. Similar effects of tramadol and venlafaxine in major depressive disorder. South Med J. 2008;101:193–195. doi: 10.1097/SMJ.0b013e3181616e66. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy SE, Koeppe RA, Young EA, Zubieta JK. Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Arch Gen Psychiatry. 2006;63(11):1199–208. doi: 10.1001/archpsyc.63.11.1199. [DOI] [PubMed] [Google Scholar]
- 34.Jutkiewicz EM. The antidepressant -like effects of delta-opioid receptor agonists. Mol Interv. 2006;6(3):162–9. doi: 10.1124/mi.6.3.7. [DOI] [PubMed] [Google Scholar]
- 35.Saitoh A, Yamada M, Takahashi K, et al. Antidepressant-like effects of the delta-opioid receptor agonist SNC80 ([(+)-4-[(alphaR)-alpha-[(2S,5R)-2,5-dimethyl-4-(2-propenyl)-1-piperazinyl ]-(3-methoxyphenyl)methyl]-N,N-diethylbenzamide) in an olfactory bulbectomized rat model. Brain Res. 2008;1208:160–9. doi: 10.1016/j.brainres.2007.07.095. [DOI] [PubMed] [Google Scholar]
- 36.Torregrossa MM, Jutkiewicz EM, Mosberg HI, et al. Peptidic delta opioid receptor agonists produce antidepressant-like effects in the forced swim test and regulate BDNF mRNA expression in rats. Brain Res. 2006;1069(1):172–81. doi: 10.1016/j.brainres.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broom DC, Jutkiewicz EM, Folk JE, et al. Nonpeptidic delta-opioid receptor agonists reduce immobility in the forced swim assay in rats. Neuropsychopharmacology. 2002;26(6):744–55. doi: 10.1016/S0893-133X(01)00413-4. [DOI] [PubMed] [Google Scholar]
- 38.Jutkiewicz EM, Rice KC, Traynor JR, Woods JH. Separation of the convulsions and antidepressant-like effects produced by the delta-opioid agonist SNC80 in rats. Psychopharmacology (Berl) 2005;182(4):588–96. doi: 10.1007/s00213-005-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tejedor-Real P, Mico JA, Smadja C, et al. Involvement of delta-opioid receptors in the effects induced by endogenous enkephalins on learned helplessness model. Eur J Pharmacol. 1998;354(1):1–7. doi: 10.1016/s0014-2999(98)00423-3. [DOI] [PubMed] [Google Scholar]
- 40.Cahill CM, McClellan KA, Morinville A, et al. Immunohistochemical distribution of delta opioid receptors in the rat central nervous system: evidence for somatodendritic labeling and antigen-specific cellular compartmentalization. J Comp Neurol. 2001;440(1):65–84. doi: 10.1002/cne.1370. [DOI] [PubMed] [Google Scholar]
- 41.Goodman RR, Snyder SH, Kuhar MJ, Young WS., 3rd Differentiation of delta and mu opiate receptor localizations by light microscopic autoradiography. Proc Natl Acad Sci U S A. 1980;77(10):6239–43. doi: 10.1073/pnas.77.10.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mansour A, Thompson RC, Akil H, Watson SJ. Delta opioid receptor mRNA distribution in the brain: comparison to delta receptor binding and proenkephalin mRNA. J Chem Neuroanat. 1993;6(6):351–62. doi: 10.1016/0891-0618(93)90010-2. [DOI] [PubMed] [Google Scholar]
- 43.Quirion R, Zajac JM, Morgat JL, Roques BP. Autoradiographic distribution of mu and delta opiate receptors in rat brain using highly selective ligands. Life Sci. 1983;33 (Suppl 1):227–30. doi: 10.1016/0024-3205(83)90484-8. [DOI] [PubMed] [Google Scholar]
- 44.Varona A, Gil J, Saracibar G, et al. Effects of imipramine treatment on delta-opioid receptors of the rat brain cortex and striatum. Arzneimittelforschung. 2003;53(1):21–5. doi: 10.1055/s-0031-1297065. [DOI] [PubMed] [Google Scholar]
- 45.Jutkiewicz EM. RB101-mediated protection of endogenous opioids: potential therapeutic utility? CNS Drug Rev. 2007;13(2):192–205. doi: 10.1111/j.1527-3458.2007.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87(2):659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- 47.Machado-Vieira R, Lara DR, Souza DO, Kapczinski F. Purinergic dysfunction in mania: an integrative model. Med Hypotheses. 2002;58(4):297–304. doi: 10.1054/mehy.2001.1543. [DOI] [PubMed] [Google Scholar]
- 48.Kaster MP, Rosa AO, Rosso MM, et al. Adenosine administration produces an antidepressant-like effect in mice: evidence for the involvement of A1 and A2A receptors. Neurosci Lett. 2004;355(1–2):21–4. doi: 10.1016/j.neulet.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 49.Lara DR, Dall’Igna OP, Ghisolfi ES, Brunstein MG. Involvement of adenosine in the neurobiology of schizophrenia and its therapeutic implications. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(4):617–29. doi: 10.1016/j.pnpbp.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 50.Ogawa N, Ueki H. Secondary mania caused by caffeine. Gen Hosp Psychiatry. 2003;25(2):138–9. doi: 10.1016/s0163-8343(02)00273-6. [DOI] [PubMed] [Google Scholar]
- 51.Anumonye A, Reading HW, Knight F, Ashcroft GW. Uric-acid metabolism in manic-depressive illness and during lithium therapy. Lancet. 1968;1(7555):1290–3. doi: 10.1016/s0140-6736(68)92300-3. [DOI] [PubMed] [Google Scholar]
- 52.De Berardis D, Conti CM, Campanella D, et al. Evaluation of plasma antioxidant levels during different phases of illness in adult patients with bipolar disorder. J Biol Regul Homeost Agents. 2008;22(3):195–200. [PubMed] [Google Scholar]
- 53.Salvadore G, Viale CI, Luckenbaugh DA, et al. Increased uric acid levels in drug-naive subjects with bipolar disorder during a first manic episode. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(6):819–21. doi: 10.1016/j.pnpbp.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chung KH, Huang CC, Lin HC. Increased risk of gout among patients with bipolar disorder: A nationwide population-based study. Psychiatry Res. 2010 doi: 10.1016/j.psychres.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 55.Barden N, Harvey M, Gagne B, et al. Analysis of single nucleotide polymorphisms in genes in the chromosome 12Q24.31 region points to P2RX7 as a susceptibility gene to bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(4):374–82. doi: 10.1002/ajmg.b.30303. [DOI] [PubMed] [Google Scholar]
- 56.Lucae S, Salyakina D, Barden N, et al. P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum Mol Genet. 2006;15(16):2438–45. doi: 10.1093/hmg/ddl166. [DOI] [PubMed] [Google Scholar]
- 57.Akhondzadeh S, Milajerdi MR, Amini H, Tehrani-Doost M. Allopurinol as an adjunct to lithium and haloperidol for treatment of patients with acute mania: a double-blind, randomized, placebo-controlled trial. Bipolar Disord. 2006;8(5 Pt 1):485–9. doi: 10.1111/j.1399-5618.2006.00363.x. [DOI] [PubMed] [Google Scholar]
- 58.Machado-Vieira R, Lara DR, Souza DO, Kapczinski F. Therapeutic efficacy of allopurinol in mania associated with hyperuricemia. J Clin Psychopharmacol. 2001;21(6):621–2. doi: 10.1097/00004714-200112000-00017. [DOI] [PubMed] [Google Scholar]
- 59.Machado-Vieira R, Soares JC, Lara DR, et al. A double-blind, randomized, placebo-controlled 4-week study on the efficacy and safety of the purinergic agents allopurinol and dipyridamole adjunctive to lithium in acute bipolar mania. J Clin Psychiatry. 2008;69(8):1237–45. doi: 10.4088/jcp.v69n0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machado-Vieira R, Manji HK, Zarate CA. The role of the tripartite glutamatergic synapse in the pathophysiology and therapeutics of mood disorders. Neuroscientist. 2009;15(5):525–39. doi: 10.1177/1073858409336093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bannerman DM, Good MA, Butcher SP, et al. Distinct components of spatial learning revealed by prior training and NMDA receptor blockade. Nature. 1995;378(6553):182–6. doi: 10.1038/378182a0. [DOI] [PubMed] [Google Scholar]
- 62.Collingridge GL. Long-term potentiation. A question of reliability. Nature. 1994;371(6499):652–3. doi: 10.1038/371652a0. [DOI] [PubMed] [Google Scholar]
- 63.Collingridge GL, Bliss TV. Memories of NMDA receptors and LTP. Trends Neurosci. 1995;18(2):54–6. [PubMed] [Google Scholar]
- 64.Watkins J, Collingridge G. Phenylglycine derivatives as antagonists of metabotropic glutamate receptors. Trends Pharmacol Sci. 1994;15(9):333–42. doi: 10.1016/0165-6147(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 65.Soriano FX, Hardingham GE. Compartmentalized NMDA receptor signalling to survival and death. J Physiol. 2007;584(Pt 2):381–7. doi: 10.1113/jphysiol.2007.138875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dixon JF, Hokin LE. Lithium stimulates accumulation of second-messenger inositol 1,4,5-trisphosphate and other inositol phosphates in mouse pancreatic minilobules without inositol supplementation. Biochem J. 1994;304:251–258. doi: 10.1042/bj3040251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Du J, Gray NA, Falke CA, et al. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci. 2004;24(29):6578–89. doi: 10.1523/JNEUROSCI.1258-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hashimoto R, Hough C, Nakazawa T, et al. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 2002;80(4):589–97. doi: 10.1046/j.0022-3042.2001.00728.x. [DOI] [PubMed] [Google Scholar]
- 69.Karkanias NB, Papke RL. Lithium modulates desensitization of the glutamate receptor subtype gluR3 in Xenopus oocytes. Neurosci Lett. 1999;277(3):153–6. doi: 10.1016/s0304-3940(99)00878-2. [DOI] [PubMed] [Google Scholar]
- 70.Ma J, Zhang GY. Lithium reduced N-methyl-D-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci Lett. 2003;348(3):185–9. doi: 10.1016/s0304-3940(03)00784-5. [DOI] [PubMed] [Google Scholar]
- 71.Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9(9):2081–4. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- 72.Dixon JF, Los GV, Hokin LE. Lithium stimulates glutamate “release” and inositol 1,4,5-trisphosphate accumulation via activation of the N-methyl-D-aspartate receptor in monkey and mouse cerebral cortex slices. Proc Natl Acad Sci U S A. 1994;91:8358–8362. doi: 10.1073/pnas.91.18.8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hokin LE, Dixon JF, Los GV. A novel action of lithium: stimulation of glutamate release and inositol 1,4,5 trisphosphate accumulation via activation of the N-methyl D-aspartate receptor in monkey and mouse cerebral cortex slices. Adv Enzyme Regul. 1996;36:229–244. doi: 10.1016/0065-2571(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 74.Moutsimilli L, Farley S, Dumas S, et al. Selective cortical VGLUT1 increase as a marker for antidepressant activity. Neuropharmacology. 2005;49(6):890–900. doi: 10.1016/j.neuropharm.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 75.Cunningham MO, Woodhall GL, Jones RS. Valproate modifies spontaneous excitation and inhibition at cortical synapses in vitro. Neuropharmacology. 2003;45:907–917. doi: 10.1016/s0028-3908(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 76.Kang TC, Kim DS, Kwak SE, et al. Valproic acid reduces enhanced vesicular glutamate transporter immunoreactivities in the dentate gyrus of the seizure prone gerbil. Neuropharmacology. 2005;49(6):912–21. doi: 10.1016/j.neuropharm.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 77.Hassel B, Iversen EG, Gjerstad L, Tauboll E. Up-regulation of hippocampal glutamate transport during chronic treatment with sodium valproate. J Neurochem. 2001;77(5):1285–92. doi: 10.1046/j.1471-4159.2001.00349.x. [DOI] [PubMed] [Google Scholar]
- 78.Ueda Y, Willmore LJ. Molecular regulation of glutamate and GABA transporter proteins by valproic acid in rat hippocampus during epileptogenesis. Exp Brain Res. 2000;133(3):334–9. doi: 10.1007/s002210000443. [DOI] [PubMed] [Google Scholar]
- 79.Du J, Suzuki K, Wei Y, et al. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology. 2007;32(4):793–802. doi: 10.1038/sj.npp.1301178. [DOI] [PubMed] [Google Scholar]
- 80.Ko GY, Brown-Croyts LM, Teyler TJ. The effects of anticonvulsant drugs on NMDA-EPSP, AMPA-EPSP, and GABA-IPSP in the rat hippocampus. Brain Res Bull. 1997;42:297–302. doi: 10.1016/s0361-9230(96)00268-7. [DOI] [PubMed] [Google Scholar]
- 81.Kunig G, Niedermeyer B, Deckert J, et al. Inhibition of [3H]alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid [AMPA] binding by the anticonvulsant valproate in clinically relevant concentrations: an autoradiographic investigation in human hippocampus. Epilepsy Res. 1998;31(2):153–7. doi: 10.1016/s0920-1211(98)00022-9. [DOI] [PubMed] [Google Scholar]
- 82.Loscher W. Effects of the antiepileptic drug valproate on metabolism and function of inhibitory and excitatory amino acids in the brain. Neurochem Res. 1993;18:485–502. doi: 10.1007/BF00967253. [DOI] [PubMed] [Google Scholar]
- 83.Steppuhn KG, Turski L. Modulation of the seizure threshold for excitatory amino acids in mice by antiepileptic drugs and chemoconvulsants. J Pharmacol Exp Ther. 1993;265(3):1063–70. [PubMed] [Google Scholar]
- 84.Turski L. The N-methyl-D-aspartate receptor complex. Various sites of regulation and clinical consequences. Arzneimittelforschung. 1990;40(5):511–4. [PubMed] [Google Scholar]
- 85.Zeise ML, Kasparow S, Zieglgansberger W. Valproate suppresses N-methyl-D-aspartate-evoked, transient depolarizations in the rat neocortex in vitro. Brain Res. 1991;544:345–348. doi: 10.1016/0006-8993(91)90078-a. [DOI] [PubMed] [Google Scholar]
- 86.Du J, Gray NA, Falke C, et al. Structurally dissimilar antimanic agents modulate synaptic plasticity by regulating AMPA glutamate receptor subunit GluR1 synaptic expression. Ann N Y Acad Sci. 2003;1003:378–380. doi: 10.1196/annals.1300.031. [DOI] [PubMed] [Google Scholar]
- 87.Du J, Creson T, Gray NA, et al. The role of hippocampal GluR1 and GluR2 receptors in manic-like behaviors. J Neurosci. 2008;28:68–79. doi: 10.1523/JNEUROSCI.3080-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ahmad S, Fowler LJ, Whitton PS. Effects of combined lamotrigine and valproate on basal and stimulated extracellular amino acids and monoamines in the hippocampus of freely moving rats. Naunyn Schmiedebergs Arch Pharmacol. 2005;371(1):1–8. doi: 10.1007/s00210-004-1008-4. [DOI] [PubMed] [Google Scholar]
- 89.Kugaya A, Sanacora G. Beyond monoamines: glutamatergic function in mood disorders. CNS Spectr. 2005;10(10):808–19. doi: 10.1017/s1092852900010403. [DOI] [PubMed] [Google Scholar]
- 90.Geddes JR, Calabrese JR, Goodwin GM. Lamotrigine for treatment of bipolar depression: independent meta-analysis and meta-regression of individual patient data from five randomised trials. Br J Psychiatry. 2009;194(1):4–9. doi: 10.1192/bjp.bp.107.048504. [DOI] [PubMed] [Google Scholar]
- 91.Ghaemi SN. The failure to know what isn’t known: negative publication bias with lamotrigine and a glimpse inside peer review. Evid Based Ment Health. 2009;12:65–68. doi: 10.1136/ebmh.12.3.65. [DOI] [PubMed] [Google Scholar]
- 92.Machado-Vieira R, Salvadore G, Ibrahim LA, et al. Targeting glutamatergic signaling for the development of novel therapeutics for mood disorders. Curr Pharm Des. 2009;15(14):1595–611. doi: 10.2174/138161209788168010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zarate CA, Quiroz J, Payne J, Manji HK. Modulators of the glutamatergic system: implications for the development of improved therapeutics in mood disorders. Psychopharmacol Bull. 2002;36(4):35–83. [PubMed] [Google Scholar]
- 94.Meloni D, Gambarana C, De Montis MG, et al. Dizocilpine antagonizes the effect of chronic imipramine on learned helplessness in rats. Pharmacol Biochem Behav. 1993;46(2):423–6. doi: 10.1016/0091-3057(93)90374-3. [DOI] [PubMed] [Google Scholar]
- 95.Padovan CM, Guimaraes FS. Antidepressant-like effects of NMDA-receptor antagonist injected into the dorsal hippocampus of rats. Pharmacol Biochem Behav. 2004;77(1):15–9. doi: 10.1016/j.pbb.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 96.Papp M, Moryl E. New evidence for the antidepressant activity of MK-801, a non-competitive antagonist of NMDA receptors. Pol J Pharmacol. 1993;45(5–6):549–53. [PubMed] [Google Scholar]
- 97.Skolnick P, Miller R, Young A, et al. Chronic treatment with 1-aminocyclopropanecarboxylic acid desensitizes behavioral responses to compounds acting at the N-methyl-D-aspartate receptor complex. Psychopharmacology (Berl) 1992;107(4):489–96. doi: 10.1007/BF02245261. [DOI] [PubMed] [Google Scholar]
- 98.Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185(1):1–10. doi: 10.1016/0014-2999(90)90204-j. [DOI] [PubMed] [Google Scholar]
- 99.Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31(11):2405–14. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- 100.Chourbaji S, Vogt MA, Fumagalli F, et al. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 2008;22(9):3129–34. doi: 10.1096/fj.08-106450. [DOI] [PubMed] [Google Scholar]
- 101.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17(8):2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aguado L, San Antonio A, Perez L, et al. Effects of the NMDA receptor antagonist ketamine on flavor memory: conditioned aversion, latent inhibition, and habituation of neophobia. Behav Neural Biol. 1994;61(3):271–81. doi: 10.1016/s0163-1047(05)80010-x. [DOI] [PubMed] [Google Scholar]
- 103.Garcia LS, Comim CM, Valvassori SS, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):140–4. doi: 10.1016/j.pnpbp.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 104.Maeng S, Zarate CA, Jr, Du J, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–52. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 105.Mickley GA, Schaldach MA, Snyder KJ, et al. Ketamine blocks a conditioned taste aversion (CTA) in neonatal rats. Physiol Behav. 1998;64(3):381–90. doi: 10.1016/s0031-9384(98)00097-3. [DOI] [PubMed] [Google Scholar]
- 106.Silvestre JS, Nadal R, Pallares M, Ferre N. Acute effects of ketamine in the holeboard, the elevated-plus maze, and the social interaction test in Wistar rats. Depress Anxiety. 1997;5(1):29–33. [PubMed] [Google Scholar]
- 107.Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–4. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 108.Zarate CA, Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 109.Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry. 2001;62(11):869–77. doi: 10.4088/jcp.v62n1106. [DOI] [PubMed] [Google Scholar]
- 110.aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67:139–145. doi: 10.1016/j.biopsych.2009.08.038. [DOI] [PubMed] [Google Scholar]
- 111.Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65(2):181–4. doi: 10.1016/j.biopsych.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Phelps LE, Brutsche N, Moral JR, et al. Family History of Alcohol Dependence and Initial Antidepressant Response to an N-methyl-D-aspartate Antagonist. Biol Psychiatry. 2008 Nov 7; doi: 10.1016/j.biopsych.2008.09.029. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Salvadore G, Cornwell BR, Colon-Rosario V, et al. Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry. 2009;65(4):289–95. doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Salvadore G, Cornwell BR, Sambataro F, et al. Anterior cingulate desynchronization and functional connectivity with the amygdala during a working memory task predict rapid antidepressant response to ketamine. Neuropsychopharmacology. 2010;35:1415–1422. doi: 10.1038/npp.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry. 2008;64:527–532. doi: 10.1016/j.biopsych.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Machado-Vieira R, Yuan P, Brutsche N, et al. Brain-derived neurotrophic factor and initial antidepressant response to an N-methyl-D-aspartate antagonist. J Clin Psychiatry. 2009;70:1662–1666. doi: 10.4088/JCP.08m04659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Diazgranados N, Ibrahim L, Brutsche NE, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67(8):793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Diazgranados N, Ibrahim LA, Brutsche NE, et al. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J Clin Psychiatry. 2010 doi: 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Price RB, Nock MK, Charney D, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66:522–529. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Preskorn SH, Baker B, Kolluri S, et al. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631–7. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- 121.Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bleakman D, Lodge D. Neuropharmacology of AMPA and kainate receptors. Neuropharmacology. 1998;37(10–11):1187–204. doi: 10.1016/s0028-3908(98)00139-7. [DOI] [PubMed] [Google Scholar]
- 123.Black MD. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology (Berl) 2005;179(1):154–63. doi: 10.1007/s00213-004-2065-6. [DOI] [PubMed] [Google Scholar]
- 124.Miu P, Jarvie KR, Radhakrishnan V, et al. Novel AMPA receptor potentiators LY392098 and LY404187: effects on recombinant human AMPA receptors in vitro. Neuropharmacology. 2001;40(8):976–83. doi: 10.1016/s0028-3908(01)00027-2. [DOI] [PubMed] [Google Scholar]
- 125.Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7(5):426–37. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Knapp RJ, Goldenberg R, Shuck C, et al. Antidepressant activity of memory-enhancing drugs in the reduction of submissive behavior model. Eur J Pharmacol. 2002;440(1):27–35. doi: 10.1016/s0014-2999(02)01338-9. [DOI] [PubMed] [Google Scholar]
- 127.Pitkanen A, Mathiesen C, Ronn LC, et al. Effect of novel AMPA antagonist, NS1209, on status epilepticus. An experimental study in rat. Epilepsy Res. 2007;74(1):45–54. doi: 10.1016/j.eplepsyres.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 128.Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res. 2006;69(3):273–94. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Perucca E. What is the promise of new antiepileptic drugs in status epilepticus? Focus on brivaracetam, carisbamate, lacosamide, NS-1209, and topiramate. Epilepsia. 2009;50 (Suppl 12):49–50. doi: 10.1111/j.1528-1167.2009.02366.x. [DOI] [PubMed] [Google Scholar]
- 130.Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24(1):123–8. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mizuta I, Ohta M, Ohta K, et al. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett. 2001;310(2–3):117–20. doi: 10.1016/s0304-3940(01)02098-5. [DOI] [PubMed] [Google Scholar]
- 132.Lourenco Da Silva A, Hoffmann A, Dietrich MO, et al. Effect of riluzole on MK-801 and amphetamine-induced hyperlocomotion. Neuropsychobiology. 2003;48(1):27–30. doi: 10.1159/000071825. [DOI] [PubMed] [Google Scholar]
- 133.Chowdhury GM, Banasr M, de Graaf RA, et al. Chronic riluzole treatment increases glucose metabolism in rat prefrontal cortex and hippocampus. J Cereb Blood Flow Metab. 2008;28(12):1892–7. doi: 10.1038/jcbfm.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zarate CA, Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171–4. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- 135.Sanacora G, Kendell SF, Levin Y, et al. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry. 2007;61(6):822–5. doi: 10.1016/j.biopsych.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zarate CA, Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430–2. doi: 10.1016/j.biopsych.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 137.Brennan BP, Hudson JI, Jensen JE, et al. Rapid enhancement of glutamatergic neurotransmission in bipolar depression following treatment with riluzole. Neuropsychopharmacology. 2010;35(3):834–46. doi: 10.1038/npp.2009.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Krystal JH, Mathew SJ, D’Souza DC, et al. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs. 2010;24:669–693. doi: 10.2165/11533230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 139.Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behav Brain Res. 2003;140(1–2):1–47. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- 140.Salinska E, Stafiej A. Metabotropic glutamate receptors (mGluRs) are involved in early phase of memory formation: possible role of modulation of glutamate release. Neurochem Int. 2003;43(4–5):469–74. doi: 10.1016/s0197-0186(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 141.Tan Y, Hori N, Carpenter DO. The mechanism of presynaptic long-term depression mediated by group I metabotropic glutamate receptors. Cell Mol Neurobiol. 2003;23(2):187–203. doi: 10.1023/A:1022949922364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li X, Need AB, Baez M, Witkin JM. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther. 2006;319(1):254–9. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]
- 143.Wieronska JM, Szewczyk B, Branski P, et al. Antidepressant-like effect of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist in the olfactory bulbectomized rats. Amino Acids. 2002;23(1–3):213–6. doi: 10.1007/s00726-001-0131-5. [DOI] [PubMed] [Google Scholar]
- 144.Legutko B, Szewczyk B, Pomierny-Chamiolo L, et al. Effect of MPEP treatment on brain-derived neurotrophic factor gene expression. Pharmacol Rep. 2006;58(3):427–30. [PubMed] [Google Scholar]
- 145.Belozertseva IV, Kos T, Popik P, et al. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur Neuropsychopharmacol. 2007;17(3):172–9. doi: 10.1016/j.euroneuro.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 146.D’Ascenzo M, Fellin T, Terunuma M, et al. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci U S A. 2007;104:1995–2000. doi: 10.1073/pnas.0609408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Porter RH, Jaeschke G, Spooren W, et al. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315(2):711–21. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- 148.Kinney GG, O’Brien JA, Lemaire W, et al. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313(1):199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 149.Chaki S, Yoshikawa R, Hirota S, et al. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46(4):457–67. doi: 10.1016/j.neuropharm.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 150.Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl) 2006;186(4):587–93. doi: 10.1007/s00213-006-0390-7. [DOI] [PubMed] [Google Scholar]
- 151.Yoshimizu T, Chaki S. Increased cell proliferation in the adult mouse hippocampus following chronic administration of group II metabotropic glutamate receptor antagonist, MGS0039. Biochem Biophys Res Commun. 2004;315(2):493–6. doi: 10.1016/j.bbrc.2004.01.073. [DOI] [PubMed] [Google Scholar]
- 152.Karasawa J, Shimazaki T, Kawashima N, Chaki S. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res. 2005;1042(1):92–8. doi: 10.1016/j.brainres.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 153.Krystal JH. Capitalizing on extrasynaptic glutamate neurotransmission to treat antipsychotic-resistant symptoms in schizophrenia. Biol Psychiatry. 2008;64:358–360. doi: 10.1016/j.biopsych.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 154.Berk M, Copolov DL, Dean O, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder--a double-blind randomized placebo-controlled trial. Biol Psychiatry. 2008;64(6):468–75. doi: 10.1016/j.biopsych.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 155.Gasparini F, Bruno V, Battaglia G, et al. (R,S)-4-phosphonophenylglycine, a potent and selective group III metabotropic glutamate receptor agonist, is anticonvulsive and neuroprotective in vivo. J Pharmacol Exp Ther. 1999;289(3):1678–87. [PubMed] [Google Scholar]
- 156.Cryan JF, Kelly PH, Neijt HC, et al. Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur J Neurosci. 2003;17(11):2409–17. doi: 10.1046/j.1460-9568.2003.02667.x. [DOI] [PubMed] [Google Scholar]
- 157.Palucha A, Tatarczynska E, Branski P, et al. Group III mGlu receptor agonists produce anxiolytic- and antidepressant-like effects after central administration in rats. Neuropharmacology. 2004;46(2):151–9. doi: 10.1016/j.neuropharm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 158.Khawaja AM, Rogers DF. Tachykinins: receptor to effector. Int J Biochem Cell Biol. 1996;28(7):721–38. doi: 10.1016/1357-2725(96)00017-9. [DOI] [PubMed] [Google Scholar]
- 159.Ebner K, Sartori SB, Singewald N. Tachykinin receptors as therapeutic targets in stress-related disorders. Curr Pharm Des. 2009;15(14):1647–74. doi: 10.2174/138161209788168074. [DOI] [PubMed] [Google Scholar]
- 160.Dableh LJ, Yashpal K, Rochford J, Henry JL. Antidepressant-like effects of neurokinin receptor antagonists in the forced swim test in the rat. Eur J Pharmacol. 2005;507(1–3):99–105. doi: 10.1016/j.ejphar.2004.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Louis C, Stemmelin J, Boulay D, et al. Additional evidence for anxiolytic- and antidepressant-like activities of saredutant (SR48968), an antagonist at the neurokinin-2 receptor in various rodent-models. Pharmacol Biochem Behav. 2008;89(1):36–45. doi: 10.1016/j.pbb.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 162.Micale V, Tamburella A, Leggio GM, et al. Behavioral effects of saredutant, a tachykinin NK2 receptor antagonist, in experimental models of mood disorders under basal and stress-related conditions. Pharmacol Biochem Behav. 2008;90(3):463–9. doi: 10.1016/j.pbb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 163.Steinberg R, Alonso R, Griebel G, et al. Selective blockade of neurokinin-2 receptors produces antidepressant-like effects associated with reduced corticotropin-releasing factor function. J Pharmacol Exp Ther. 2001;299(2):449–58. [PubMed] [Google Scholar]
- 164.Bilkei-Gorzo A, Racz I, Michel K, Zimmer A. Diminished anxiety- and depression-related behaviors in mice with selective deletion of the Tac1 gene. J Neurosci. 2002;22(22):10046–52. doi: 10.1523/JNEUROSCI.22-22-10046.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kramer MS, Cutler N, Feighner J, et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science. 1998;281(5383):1640–5. doi: 10.1126/science.281.5383.1640. [DOI] [PubMed] [Google Scholar]
- 166.Kramer MS, Winokur A, Kelsey J, et al. Demonstration of the efficacy and safety of a novel substance P (NK1) receptor antagonist in major depression. Neuropsychopharmacology. 2004;29(2):385–92. doi: 10.1038/sj.npp.1300260. [DOI] [PubMed] [Google Scholar]
- 167.Keller M, Montgomery S, Ball W, et al. Lack of efficacy of the substance p (neurokinin1 receptor) antagonist aprepitant in the treatment of major depressive disorder. Biol Psychiatry. 2006;59(3):216–23. doi: 10.1016/j.biopsych.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 168.Rupniak NM, Kramer MS. Discovery of the antidepressant and anti-emetic efficacy of substance P receptor (NK1) antagonists. Trends Pharmacol Sci. 1999;20(12):485–90. doi: 10.1016/s0165-6147(99)01396-6. [DOI] [PubMed] [Google Scholar]
- 169.Holmes A, Heilig M, Rupniak NM, et al. Neuropeptide systems as novel therapeutic targets for depression and anxiety disorders. Trends Pharmacol Sci. 2003;24(11):580–8. doi: 10.1016/j.tips.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 170.Janowsky DS, el-Yousef MK, Davis JM, Sekerke HJ. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2(7778):632–5. doi: 10.1016/s0140-6736(72)93021-8. [DOI] [PubMed] [Google Scholar]
- 171.Shi J, Hattori E, Zou H, et al. No evidence for association between 19 cholinergic genes and bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(6):715–23. doi: 10.1002/ajmg.b.30417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cannon DM, Carson RE, Nugent AC, et al. Reduced muscarinic type 2 receptor binding in subjects with bipolar disorder. Arch Gen Psychiatry. 2006;63(7):741–7. doi: 10.1001/archpsyc.63.7.741. [DOI] [PubMed] [Google Scholar]
- 173.Overstreet DH. The Flinders sensitive line rats: a genetic animal model of depression. Neurosci Biobehav Rev. 1993;17(1):51–68. doi: 10.1016/s0149-7634(05)80230-1. [DOI] [PubMed] [Google Scholar]
- 174.Dilsaver SC. Pharmacologic induction of cholinergic system up-regulation and supersensitivity in affective disorders research. J Clin Psychopharmacol. 1986;6(2):65–74. [PubMed] [Google Scholar]
- 175.Sokolski KN, DeMet EM. Cholinergic sensitivity predicts severity of mania. Psychiatry Res. 2000;95(3):195–200. doi: 10.1016/s0165-1781(00)00182-7. [DOI] [PubMed] [Google Scholar]
- 176.Kasper S, Moises HW, Beckmann H. The anticholinergic biperiden in depressive disorders. Pharmacopsychiatria. 1981;14(6):195–8. doi: 10.1055/s-2007-1019597. [DOI] [PubMed] [Google Scholar]
- 177.Davis KL, Berger PA, Hollister LE, Defraites E. Physostigmine in mania. Arch Gen Psychiatry. 1978;35(1):119–22. doi: 10.1001/archpsyc.1978.01770250121012. [DOI] [PubMed] [Google Scholar]
- 178.Khouzam HR, Kissmeyer PM. Physostigmine temporarily and dramatically reversing acute mania. Gen Hosp Psychiatry. 1996;18(3):203–4. doi: 10.1016/0163-8343(96)00004-7. [DOI] [PubMed] [Google Scholar]
- 179.Burt T, Sachs GS, Demopulos C. Donepezil in treatment-resistant bipolar disorder. Biol Psychiatry. 1999;45(8):959–64. doi: 10.1016/s0006-3223(98)00320-5. [DOI] [PubMed] [Google Scholar]
- 180.Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010 Jan 13; doi: 10.1016/j.biopsych.2009.11.021. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63(10):1121–9. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Furey ML, Khanna A, Hoffman EM, Drevets WC. Scopolamine produces larger antidepressant and antianxiety effects in women than in men. Neuropsychopharmacology. 2010;35:2479–2488. doi: 10.1038/npp.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–47. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- 184.Li X, Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology. 2010;35:2143–2154. doi: 10.1038/npp.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gould TD, Manji HK. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 2005;30(7):1223–37. doi: 10.1038/sj.npp.1300731. [DOI] [PubMed] [Google Scholar]
- 186.Kaidanovich-Beilin O, Lipina TV, Takao K, et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain. 2009;2(1):35. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol. 2004;7(4):387–90. doi: 10.1017/S1461145704004535. [DOI] [PubMed] [Google Scholar]
- 188.Gould TD, Picchini AM, Einat H, Manji HK. Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr Drug Targets. 2006;7(11):1399–409. doi: 10.2174/1389450110607011399. [DOI] [PubMed] [Google Scholar]
- 189.Polter A, Beurel E, Yang S, et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35(8):1761–74. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pandey GN, Ren X, Rizavi HS, Dwivedi Y. Glycogen synthase kinase-3beta in the platelets of patients with mood disorders: effect of treatment. J Psychiatr Res. 2010;44(3):143–8. doi: 10.1016/j.jpsychires.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 191.Li X, Liu M, Cai Z, et al. Regulation of glycogen synthase kinase-3 during bipolar mania treatment. Bipolar Disord. 2010;12:741–752. doi: 10.1111/j.1399-5618.2010.00866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rayasam GV, Tulasi VK, Sodhi R, et al. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol. 2009;156(6):885–98. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chen G, Manji HK, Hawver DB, et al. Chronic sodium valproate selectively decreases protein kinase C alpha and epsilon in vitro. J Neurochem. 1994;63(6):2361–4. doi: 10.1046/j.1471-4159.1994.63062361.x. [DOI] [PubMed] [Google Scholar]
- 194.Friedman E, Hoau Yan W, Levinson D, et al. Altered platelet protein kinase C activity in bipolar affective disorder, manic episode. Biol Psychiatry. 1993;33(7):520–5. doi: 10.1016/0006-3223(93)90006-y. [DOI] [PubMed] [Google Scholar]
- 195.Hahn CG, Friedman E. Abnormalities in protein kinase C signaling and the pathophysiology of bipolar disorder. Bipolar Disord. 1999;1(2):81–6. doi: 10.1034/j.1399-5618.1999.010204.x. [DOI] [PubMed] [Google Scholar]
- 196.Manji HK, Etcheberrigaray R, Chen G, Olds JL. Lithium decreases membrane-associated protein kinase C in hippocampus: selectivity for the alpha isozyme. J Neurochem. 1993;61(6):2303–10. doi: 10.1111/j.1471-4159.1993.tb07474.x. [DOI] [PubMed] [Google Scholar]
- 197.Manji HK, Lenox RH. Ziskind-Somerfeld Research Award. Protein kinase C signaling in the brain: molecular transduction of mood stabilization in the treatment of manic-depressive illness. Biol Psychiatry. 1999;46(10):1328–51. doi: 10.1016/s0006-3223(99)00235-8. [DOI] [PubMed] [Google Scholar]
- 198.Young LT, Wang JF, Woods CM, Robb JC. Platelet protein kinase C alpha levels in drug-free and lithium-treated subjects with bipolar disorder. Neuropsychobiology. 1999;40(2):63–6. doi: 10.1159/000026598. [DOI] [PubMed] [Google Scholar]
- 199.Einat H, Yuan P, Szabo ST, et al. Protein kinase C inhibition by tamoxifen antagonizes manic-like behavior in rats: implications for the development of novel therapeutics for bipolar disorder. Neuropsychobiology. 2007;55(3–4):123–31. doi: 10.1159/000106054. [DOI] [PubMed] [Google Scholar]
- 200.Bebchuk JM, Arfken CL, Dolan-Manji S, et al. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch Gen Psychiatry. 2000;57(1):95–7. doi: 10.1001/archpsyc.57.1.95. [DOI] [PubMed] [Google Scholar]
- 201.Kulkarni J, Garland KA, Scaffidi A, et al. A pilot study of hormone modulation as a new treatment for mania in women with bipolar affective disorder. Psychoneuroendocrinology. 2006;31(4):543–7. doi: 10.1016/j.psyneuen.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 202.Yildiz A, Guleryuz S, Ankerst DP, et al. Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry. 2008;65(3):255–63. doi: 10.1001/archgenpsychiatry.2007.43. [DOI] [PubMed] [Google Scholar]
- 203.Zarate CA, Jr, Singh JB, Carlson PJ, et al. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9(6):561–70. doi: 10.1111/j.1399-5618.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- 204.Goldstein JA. Danazol and the rapid-cycling patient. J Clin Psychiatry. 1986;47(3):153–4. [PubMed] [Google Scholar]
- 205.Ali SM, Ahmad A, Shahabuddin S, et al. Endoxifen is a new potent inhibitor of PKC: A potential therapeutic agent for bipolar disorder. Bioorg Med Chem Lett. 2010;20:2665–2667. doi: 10.1016/j.bmcl.2010.02.024. [DOI] [PubMed] [Google Scholar]