

Abstract
Epoxyeicosatrienoic acids (EETs) are synthesized from arachidonic acid and EETs have a number of beneficial cardiovascular actions. This has led to the concept that EETs and its metabolic pathway can be therapeutically targeted for hypertension and other cardiovascular diseases. One approach has been to prevent the conversion of EETs to their inactive diols by inhibiting the soluble epoxide hydrolase (sEH) enzyme. Inhibition of sEH has been demonstrated to decrease blood pressure in certain experimental models of hypertension, decrease inflammation, and protect organs from damage associated with hypertension and other cardiovascular diseases. The development of sEH inhibitors has reached the point where they are being evaluated in humans. A second therapeutic approach has been to develop EET agonists. EET agonists have been essential for determining the structure function relationship for EETs and determining cell-signaling mechanisms by which EETs exert their cardiovascular actions. More recently, EET agonists have been administered chronically to experimental animal models of hypertension and metabolic syndrome and have been demonstrated to decrease blood pressure, improve insulin signaling, and improve vascular function. These experimental findings provide evidence for sEH inhibitors and EET agonists as a therapeutic approach for cardiovascular diseases, hypertension, and the associated end organ damage.
Introduction – Why target epoxyeicosatrienoic acids and soluble epoxide hydrolase?
Arachidonic acid metabolites, eicosanoids, are formed through three primary enzymatic pathways. Two of these pathways, the cyclooxygenase (COX) and the lipoxygenase (LOX) pathways have been successfully targeted for therapeutic applications.1,2 The third pathway is the cytochrome P450 (CYP) pathway consisting of two major enzymatic pathways. CYP hydroxylase enzymes convert arachidonic acid to the major biologically active metabolite, 20-hydroxysatetraenoic acid (20-HETE).3 Arachidonic acid is also metabolized by CYP expoygenase enzymes to biologically active epoxyeicsatrienoic acids (EETs) that are degraded to less active diols by soluble epoxide hydrolase (sEH).4,5 (Figure 1) In the past decade there has been extensive investigation as to the therapeutic potential for manipulating CYP hydroxylase or epoxygenase enzymatic pathways.
Figure 1.
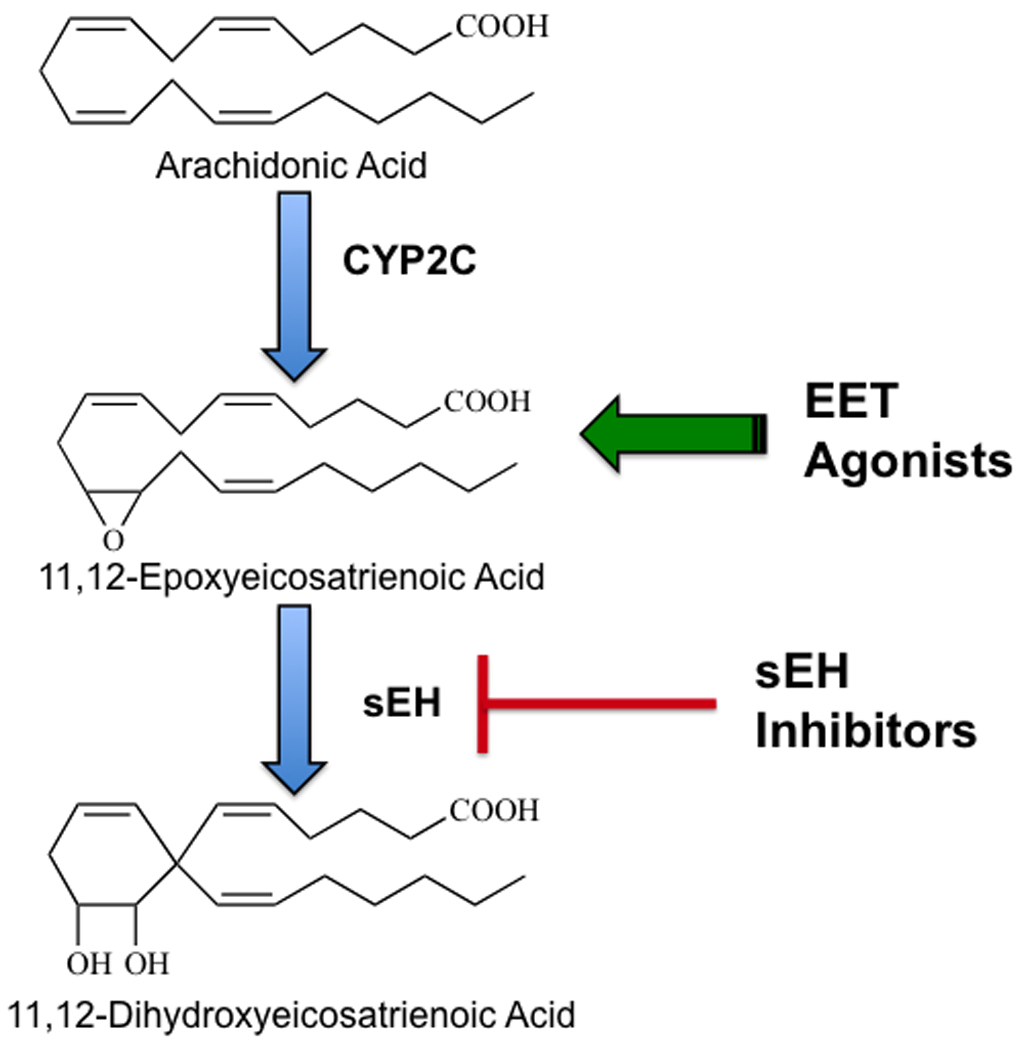
Therapeutic targeting for the epoxygenase pathway: Epoxyeicosatrienoic acids (EETs) are generated from arachidonic acid by cytochrome P450 (CYP2C) enzymes. EETs are converted to dihydroxyeicosatrienoic acids (DHETs) by the soluble epoxide hydrolase (sEH) enzyme. EET agonists and sEH inhibitors are two therapeutic targets for hypertension and cardiovascular diseases.
CYP epoxygenase metabolites have biological actions that implicate them as important contributors to cardiovascular function and blood pressure control. The first biological activity described for EETs was inhibition of renal tubular sodium reabsorption.6,7 Subsequently, EETs were determined to dilate blood vessels and were identified as endothelium-derived hyperpolarizing factors (EDHF).8,9,10 These biological actions are consistent with the idea that EETs would be eicosanoids that contribute to lowering of blood pressure and prevent salt-sensitive hypertension. This concept was further supported by a number of experimental studies in rodents demonstrating salt-sensitive hypertension in conditions where kidney CYP epoxygenase enzyme and EET levels were decreased.11,12,13 There is less evidence to support the concept that epoxygenase metabolites contribute to hypertension humans. A single nucleotide polymorphism in the CYP2J2 gene has been demonstrated to be associated with hypertension in Caucasion males and Caucasians without a family history of hypertension.14 These experimental findings in rodents and humans have generated interest in targeting the CYP epoxygenase pathway for the treatment of hypertension.
Even though EETs have actions on renal tubular transport and vascular function that are essential for blood pressure regulation it became apparent that additional biological actions ascribed to EETs made them an excellent therapeutic target for other cardiovascular diseases.4,15 These additional activities demonstrated for EETs included inhibition of platelet aggregation and anti-inflammation.16,17,18 EETs also have been found to have effects on vascular migration and proliferation including promoting angiogenesis.19–23 Thus EETs have become a therapeutic target for end organ damage associated with cardiovascular diseases, cardiac ischemic injury, atherosclerosis, and stroke.
One way to increase EET levels is to inhibit their degradation to the less active diols, dihydroxyeicosatrienoic acids (DHETs), by inhibiting sEH. (Figure 1) This approach to inhibit sEH has been used successfully in a number of rodent models of hypertension and other cardiovascular diseases.4,15 Pharmacological induction of CYP epoxygenase enzymes has also been used to elevate EET levels.11,12 Another approach to target EETs has been to regulate the CYP epoxygenase and sEH enzymes via genetic manipulation in mice.11,24 The latest approach that has been tested in vivo has been the development of agonistic analogs for the EETs.25,26 (Figure 1) This review article will focus on the therapeutic potential for targeting the CYP epoxygenase pathway in hypertension and other cardiovascular diseases.
EET Vascular Actions
EETs were first investigated for effects on vascular tone in mesenteric resistance arteries.27 Vasodilation has been the predominant EET action on blood vessels isolated from a number of different organs.8,9,10,25,28 In contrast, the DHETs are less active or inactive on vascular tone when direct comparisons to EETs have been conducted.28–30 EETs are generated by endothelial cells and relax the vascular smooth muscle cells through the activation of calcium-activated K+ (KCa) channels.8,9,10,30,31 These findings led to the identification of EETs as EDHFs and led to evaluation of epoxides for vascular actions beyond vascular tone.
It is now recognized that EETs have numerous vascular actions related to the maintenance of vascular homeostasis.12,20,26 These vascular actions include anti-inflammatory actions, vascular cell migration and proliferation, and anti-platelet aggregation actions.12,20,26 EETs prevent leukocyte adhesion to the vascular wall and decrease TNF-α activation of endothelial VCAM-1 expression.18,32 Several studies have provided evidence that the anti-inflammatory action of EETs is via inhibiting NFkB activation.18,32,33 Endothelial cell proliferation is another vascular consequence of increased EETs.22,34,35 Migration of vascular smooth muscle cells is inhibited by EETs or epoxygenase overexpression.23 5,6-EET, 11,12-EET, and 14,15-EET inhibit rat aortic smooth muscle cell migration in response to platelet-derived growth factor.23 The integrated vascular growth response is that EETs are pro-angiogenic and contribute to vascular endothelial growth factor-induced angiogensis.21,36 EETs have also been demonstrated to have anti-aggregatory properties in platelets.16,17,37 Arachidonic acid stimulation of platelet aggregation is inhibited by EETs and EETs inhibit platelet adhesion to endothelial cells.37 As a whole, these studies suggest that EETs can have a variety of vascular actions and that modulation of EETs has therapeutic potential for end organ damage associated with hypertension.
EET Cellular Mechanisms of Action
Vascular actions described for EETs have greatly expanded over the years and this has been associated with experimental studies determining cellular signal transduction pathways by which EETs act. EETs act as EDHFs through endothelial-dependent and –independent cell signaling mechanisms.38 One of the initial findings was that EETs increased endothelial Ca2+ levels by activating intracellular Ca2+ pools.39,40,41 This increase in endothelial Ca2+ has been linked to cell membrane hyperpolarization.38,40 Subsequent studies have provide evidence that EETs can also increase endothelial Ca2+ via activation of vanilloid (TRP) channels and that the increase in calcium activates small and intermediate KCa channels to cause cell membrane hyperpolarization.42,43 This endothelial cell hyperpolarization could be transmitted to smooth muscle cells via the release of K+ ions into the subendothelial space or by gap junctions that are electrically coupled to vascular smooth muscle cells.38 EETs can also act as a transferrable factor that hyperpolarizes the vascular smooth muscle cell to elicit relaxation.8,10,38 Release of EETs from endothelial cells hyperpolarizes vascular smooth muscle cells through activation of large-conductance KCa channels.38 Experimental studies have implicated several cell signaling mechanisms in EET activation of KCa channel activation. Although the identity for EET receptors remains elusive, EETs appear to activate vascular smooth muscle KCa channels via G protein (Gs) dependent mechanism.8,29,31,38 Other investigations provide evidence for increased cAMP and protein kinase A as a key signaling molecules required for KCa activation and smooth muscle cell hyperpolarization.31,44,45 There are differences in potency and activity of the EET regioisomers and cell signaling mechanisms utilized among the various vascular tissues. Many of these apparent differences may be resolved with the identification of endothelial or vascular smooth muscle cell EET receptors.
There are some commonalities with regard to the cell signaling pathways by which EETs exert vascular actions other than EDHF-mediated dilation. Cell signaling pathways implicated in endothelial cell proliferation include the phosphotidyl inositol-3-kinase (PI3K)/Akt pathway.22,34,35 Mitogen-activated protein kinase (MAPK) and cAMP / PKA pathways also contribute to endothelial cell proliferation.22,34,35 EET anti-migratory actions in vascular smooth muscle cells are also dependent on cAMP / PKA cell signaling.22,34 EETs also contribute to vascular endothelial growth factor-mediated angiogenesis by activation Akt and MAPK in endothelial cells.21,36 Like endothelial and vascular smooth muscle cells, EETs hyperpolarize platelet cell membranes.17 EET hyperpolarization of human platelets is through activation of KCa channels.17 Overall, these cell-signaling mechanisms by which EETs exert vascular actions suggests that EET agonists have the potential to identify one or more EET binding sites or receptors.
Given the vascular actions attributed to EETs and the cell signaling mechanisms utilized it has been postulated that modulation of EETs in cardiovascular diseases has potential therapeutic value to prevent end organ damage. As mentioned previously, there have been two primary approaches for targeting epoxides therapeutically; sEH inhibition or EET agonists. Limitations for these approaches need to be taken into consideration. Inhibiting sEH will result in an overall increase in EET regioisomers and could alter arachidonic acid metabolism through other enzymatic pathways. The development of EET agonists is limited by the fact that there are no known receptors and thus it is difficult to determine the selectivity of EET agonist. Even with these limitations, significant progress has been made with these two approaches and the promise for protecting from end organ damage will be discussed.
Soluble Epoxide Hydrolase Inhibitors
The development of sEH inhibitors (sEHIs) advanced rapidly over the past decade to the point where they have been tested in human clinical trials. Although there were sEHIs that were available for many years, these sEHIs had limited use in vitro and were not extensively evaluated for their effects on vascular function.4,5 The development carbamate urea sEHIs and the finding that injection of a single dose of N,N’-dicyclohexylurea (DCU) lowered blood pressure in spontaneously hypertensive rats (SHR) peaked interest in sEHIs and blood pressure control.46 Subsequent formulations improved solubility and bioavailability to the extent that sEHIs could be administered orally via drinking water over long periods of time.4,13,47 This allowed for preclinical testing in animal models of hypertension and other cardiovascular diseases. This led to the eventual development of the sEHI, AR9281 that went through to completion of a Phase IIa clinical trial in patients with mild to moderate hypertension and impaired glucose tolerance (http://clinicaltrials.gov/ct2/show/NCT00847899). Although this clinical trial has ended, the final outcomes have not been made publically available.
Interest was generated by numerous experimental hypertension studies that demonstrated blood pressure lowering when sEHIs were chronically administered. (Figure 2) Blood pressure lowering was observed in SHR, angiotensin hypertension and deoxycorticosterone acetate (DOCA) salt hypertension.13,46,48,49 In general, these studies demonstrated a lowering of blood pressure that did not return values down to normal. As additional experimental studies were reported it became apparent that there was an inability of sEHIs to lower blood pressure in some animal models of hypertension. Blood pressure was not decreased by sEHI treatment in various strains of the SHR including the stroke-prone SHR or in Goto-Kakizaki diabetic rats with angiotensin infused to induce hypertension.50–52 Mice with a genetic deficiency in sEH, Ephx2 −/− mice also had variable results when blood pressure was assessed.53–55 More recently, sEHI treatment to Ren-2 transgenic rats was shown to delay the progression of hypertension but did not decrease the maximum increase in blood pressure.56 An interesting aspect of the hypertension studies is that despite variable effects on blood pressure, sEHIs or Ephx2 gene deletion was anti-inflammatory and provided end organ protection.13,48,51,52,54
Figure 2.
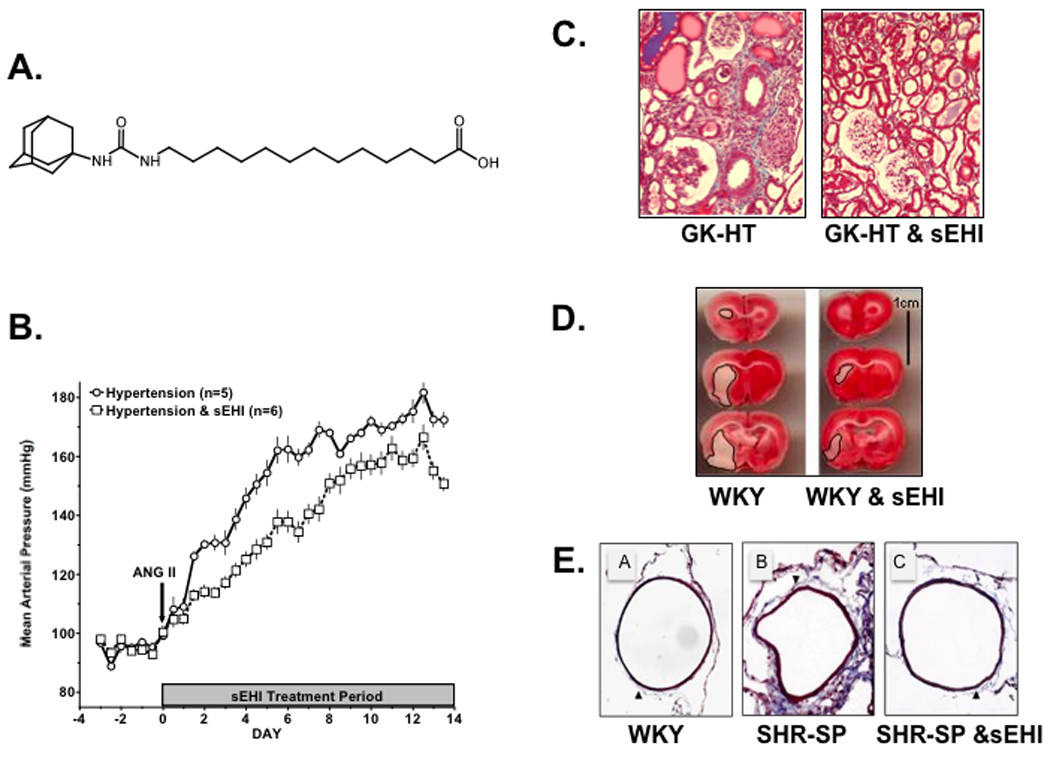
Blood pressure and cardiovascular protective actions for soluble epoxide hydrolase inhibition (sEHI): Panel A.) chemical structure for 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA); Panel B.) sEHI decreases blood pressure in angiotensin salt-sensitive hypertension; Panel C.) sEHI decreases renal injury in hypertensive and diabetic Goto-Kakizaki rats (GK-HT); Panel D.) sEHI decreases cerebral ischemic injury in Wistar-Kyoto rats (WKY); Panel E.) sEHI decreases the cerebral artery wall to lumen ratio and collagen deposition in stroke-prone spontaneously hypertensive rats (SHR-SP). Figure adapted from references 13, 51, 52.
Administration of sEHIs has consistently been demonstrated to decrease kidney, heart, and brain damage associated with hypertension and other cardiovascular diseases.4,15 (Figure 2) Renal injury associated with hypertension is greatly attenuated by sEHI treatment. Glomerular barrier function and renal vascular function were improved in angiotensin and DOCA-salt hypertension following sEHI administration.13,54 Decreased renal injury was observed when sEHI administration occurred during the development of hypertension or after hypertension was established.13,57 Experimental studies in the diabetic Goto-Kakizaki rats with angiotensin hypertension clearly established that sEHI treatment could provide renal end organ protection without lowering blood pressure.51 These studies also demonstrated that sEHIs decreased renal macrophage infiltration associated with hypertension.13,51,57 Reduced cardiac hypertrophy was also observed following sEHI treatment in DOCA-salt hypertension.48 Other studies have demonstrated that sEHIs decreased cardiac hypertrophy and decreased cardiac ischemia reperfusion injury.58–63 Protection from ischemic reperfusion injury by sEHI administration or in Ephx2 −/− mice is dependent on EET mediated STAT3 signaling in cardiac myocytes.60 Like the kidney and heart, sEHI administration protects the brain from ischemic damage.52,64 Cerebral ischemic injury in stroke-prone SHR was greatly attenuated by administration of the sEHI, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA).52 The ability of sEHIs to protect the brain from injury in cardiovascular disease is due to its dual action to improve vascular function and remodeling and by protecting the neurons from cell death.52,64,65 Taken together these studies demonstrate that sEHI has the ability to protect organs from injury in hypertension and relates to actions not only on blood vessels but other cell types.
One area that is becoming increasingly controversial is how to determine the selectivity of sEHIs. Determining selective inhibition for sEHIs is difficult to discern since the proposed active metabolites, EETs are difficult to measure in plasma and tissue and there are questions as to which EET regioisomer or other fatty acid epoxide is mediating the cardiovascular action. Administration of sEHIs such as AUDA results in plasma and tissue levels that are sufficient to inhibit the enzyme.13,52 Experimental studies have also demonstrated that the plasma and urinary arachidonic acid epoxide to diol ratio increases in rodents treated with sEHIs.13,33,57 These studies have also demonstrated increases in the plasma linoleic epoxide to diol ratio.13,33,57 Overall, experimental studies have demonstrated a trend for 14,15-EET and 14,15-DHET to be the primary arachidonate epoxide to diol ratio that increases in response to sEHI administration.4,15 However many studies do not report the individual regioisomeric profile since there is large variability from animal to animal, between different rodent strains, and differences based on diet. Preclinical studies in rodents have been hampered by the lack of in vivo inhibitors for EETs. This makes it very difficult to demonstrate that a cardiovascular phenotype in response to sEHI administration is related to EET biological activity. Measurements in human urine or plasma have also been very difficult. Although mass spectrometry analysis is becoming more common, the ability to measure epoxides in human samples is still very difficult and relatively expensive. Additional issues such as epoxides binding to plasma proteins, incorporation into cell membranes, and changes in fatty acid profiles depending on dietary intake provide further difficulties when trying to ensure sufficient dosage following sEHI therapy. Currently an ex vivo blood assay is used to ensure a proper dosage of an sEHI has been administered.66,67 Lastly, there is also the likelihood that urinary and tissue levels may not accurately reflex changes in blood vessel and tissue epoxide to diol ratios.
EET Agonists
EET agonists have been used extensively in vitro since 1996; however, their successful use in vivo has been very recent.25,26,68,69 The development of EET analogs that mimic the actions of endogenous EETs has been useful since the synthesis and storage of EETs is less than ideal for experimental studies. A first generation of EETs was developed to resist β-oxidation, resist esterification, and improve solubility.25,26 A subsequent generation of EET agonistic analogs was designed by modifying the epoxide to resist metabolism by sEH.26 Other EET analog modifications include elongation and shortening of carbon chain length and removal of olefin bonds.25,26 Agonistic analogs have been generated for all four EET regioisomers and these have provided valuable information on structure activity relationships for biological actions.
EET agonistic analogs of 14,15-EET have been demonstrated to dilate coronary arteries.70,71 Coronary artery dilation in response to 14,15-EET agonists is dependent on vascular smooth muscle cell K+ channel activation and the same cell signaling pathways that are utilized by endogenous 14,15-EET.25,71 Development of 14,15-EET agonists led to the discovery of EET antagonists. 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE and 14,15-EEZE methylsulfonamide (14,15-EEZE-SI) were determined to antagonize EET-induced biological actions.72,73 14,15-EEZE inhibits the relaxation to all regioisomeric EETs whereas 14,15-EEZE-SI inhibits the actions of 14,15-EET and 5,6-EET.72 The use of 14,15-EET agonists and EET antagonists has been limited to in vitro experimental settings. On the other hand, agonistic analogs of 11,12-EET have been developed further and have been successfully employed for in vivo experiments with chronic administration.68,69 (Figure 3) 11,12-EET analogs have been demonstrated to have vasodilatory properties and inhibit TNF-α induced vascular VCAM-1 expression.32,44,74 Like 14,15-EET agonists, 11,12-EET agonistic analogs have biological actions through the same cell signaling mechanisms as 11,12-EET.44 More recently, the actions of 11,12-EET agonists in hypertension and cardiometabolic syndrome have been explored.
Figure 3.
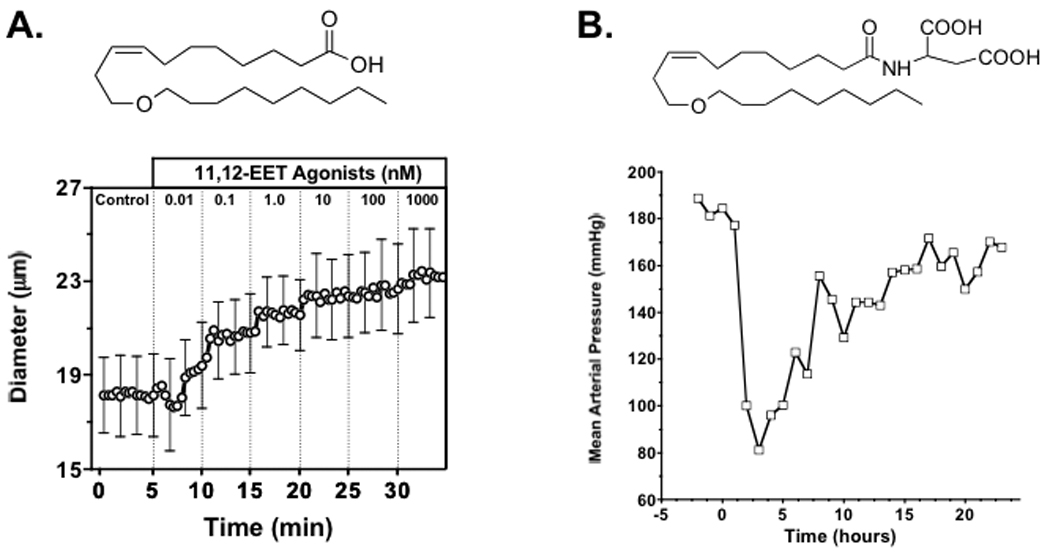
Vascular and blood pressure responses to EET agonists: Panel A.) chemical structure for 11-nonyloxy-undec-8(Z)-enoic acid and afferent arteriolar dilation; Panel B.) chemical structure for 11-nonyloxy-undec-8(Z)-enoic acid structure with aspartic acid amidation (NUDSA) and time course for decrease in blood pressure to an NUDSA i.p. injection (1 mg, n=1) to an angiotensin hypertension rat. Figure adapted from references 44, 69.
The first demonstration that an 11,12-EET agonists could improve vascular function in hypertension was the demonstration that 11,12-EET-SI reversed the enhanced vascular reactivity to angiotensin in hypertension.74 11,12-EET-SI does not resist metabolism by sEH and would not be useful for chronic administration. Several 11,12-EET agonistic analogs were generated with better solubility that resisted β-oxidation and sEH metabolism.26 These agonists were variations on the 11-nonyloxy-undec-8(Z)-enoic acid structure. Although many of these 11,12-EET agonistic analogs failed to alter the progression of hypertension in SHR or angiotensin dependent hypertension, there were a couple of 11,12-EET agonists that did ameliorate the progression of hypertension.68,69 Amidation of the carboxylic group on 11-nonyloxy-undec-8(Z)-enoic acid structure with aspartic acid provided the most promising 11,12-EET agonist (NUDSA). (Figure 3) This 11,12-EET agonist lowers blood pressure in SHR and angiotensin dependent hypertension.69 NUDSA has also been demonstrated to reverse the metabolic syndrome phenotype observed in heme-oxygenase 2 (HO-2) gene deficient mice. HO-2 −/− mice have increased body weight, hypertension, and elevated blood glucose levels.68 These metabolic syndrome phenotypes in HO-2 mice are associated with decreased EET levels and impaired vascular function. HO-2 −/− mice administered NUDSA for two weeks decreased body weight, decreased blood pressure, decreased blood glucose, and improved endothelial dependent dilation.68 One interesting finding in this study was that NUDSA increased renal Cyp2c44 epoxygenase expression.68 This raises the possibility that regulation of epoxygenase enzymes could contribute to the biological actions of EET agonists during chronic administration. Overall, the findings with EET agonists such as NUDSA suggest that EET agonists have potential therapeutic benefit for the treatment of hypertension and other cardiovascular diseases.
Selectivity will be a significant hurdle to overcome for EET agonists since EET antagonists have not been developed to the point where they can be administered chronically. EET antagonists if developed could be used to demonstrate that the EET agonists are acting in a manner similar to endogenous EETs. EET agonists and antagonists have been used to determine structure activity relationships and provided clues to the identity of EET receptors.32,44,75 Identification of EET of EET receptors would allow for screening of compounds that could act on the different receptors. There is experimental evidence that the discovery of EET receptors is on the horizon but until that occurs it will be very difficult to demonstrate selectivity for EET agonistic analogs. On the other hand, biochemical analysis methods have been developed to measure EET agonists in biological fluids.76 These methods have determined that intraperitoneal injection of NUDSA results in sufficient plasma NUDSA to exert a biological action.76 Future experimental studies will have to evaluate EET agonist plasma levels as well as evaluate the many cardiovascular actions attributed to EETs as a means to demonstrate proper selectivity for this therapeutic.
Conclusions
Mounting evidence gathered in experimental animal models of hypertension and cardiovascular diseases suggests that sEHIs or EET agonists have potential therapeutic benefits. There are therapeutic areas still to be explored for sEHIs and EET agonists. The fact that sEHIs can synergize with COX-2 inhibitors could potentially be used for anti-hyperalgesic and anti-inflammatory actions without the cardiotoxicity associated with COX-2 inhibitors.77 EET agonists and sEHIs have the potential to be used in combination with anti-diabetics, anti-hypertensives, and hypolipidemic therapies. Additionally, the combination of a 20-HETE inhibitor and sEHI or EET agonist is a possibility in hypertension. A recently published study demonstrated that combined inhibition of 20-HETE formation and sEHI decreased blood pressure and protected the heart and kidney from hypertension-induced injury in Ren-2 transgenic rats.56 Future exploration is required to determine the therapeutic potential for sEHIs and EET agonists alone or in combination in cardiovascular diseases and hypertension.
It does appear as if some of the EET agonists like NUDSA may also inhibit sEH and increase CYP2C epoxygenase expression.68 This type of combinational activity described for NUDSA could provide added beneficial effects. On the other hand, there is also potential for unwanted side effects with chronic sEHI or EET agonist administration. Angiogenesis is a potential consequence of chronic sEHI or EET agonist administration through activation of vascular endothelial growth factor.21 Promotion of angiogenesis could promote vascularization and tumor growth. In contrast, there are cardiovascular diseases where angiogenesis would be beneficial.
The therapeutic potential for EET agonists and sEHIs could extend beyond hypertension and cardiovascular diseases. Neural protection from ischemic injury has been attributed to sEHI actions on blood vessels and neurons.52,64 There is growing evidence that sEHIs provide protection from ischemic damage in the brain and heart through effects on apoptotic signaling cascades.4,52,64 EET agonists and sEHIs have also been demonstrated to modulate pain in various experimental animal models.78,79 Other possible therapeutic applications for EET agonists and sEHIs are sure to be discovered when these agents are tested in other disease models.
Acknowledgments
Funding
This work was supported by NIH grants HL59699 and DK38226 and Advancing a Healthier Wisconsin.
References
- 1.Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro JD, Toro AA, Baracat EC. Antileukotrienes in the treatment of asthma and allergic rhinitis. J Pediatr (Rio J) 2006;82:S213–S221. doi: 10.2223/JPED.1553. [DOI] [PubMed] [Google Scholar]
- 3.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 4.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson HR, Corona S, Capdevila JH, Chacos N, Manna S, Womack A, Falck JR. Effects of epoxyeicosatrienoic acids on ion transport in the rabbit cortical collecting tubule. In: Braquet P, Garay RP, Frohlich JC, Nicosia S, editors. Prostaglandins, and Membrane Ion Transport. New York: Raven Press; 1985. pp. 311–318. [Google Scholar]
- 7.Sakairi Y, Jacobson HR, Noland TD, Capdevila JH, Falck JR, Breyer MD. 5,6-EET inhibits ion transport in collecting duct by stimulating endogenous prostaglandin synthesis. Am J Physiol. 1995;268:F931–F939. doi: 10.1152/ajprenal.1995.268.5.F931. [DOI] [PubMed] [Google Scholar]
- 8.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 9.Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation. 2003;107:769–776. doi: 10.1161/01.cir.0000047278.28407.c2. [DOI] [PubMed] [Google Scholar]
- 10.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 11.Capdevila JH, Falck JR, Imig JD. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007;72:683–689. doi: 10.1038/sj.ki.5002394. [DOI] [PubMed] [Google Scholar]
- 12.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496–F503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 13.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46:975–981. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imig JD. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc Drug Rev. 2006;24:169–188. doi: 10.1111/j.1527-3466.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 15.Heizer ML, McKinney JS, Ellis EF. 14,15-Epoxyeicosatrienoic acid inhibits platelet aggregation in mouse cerebral arterioles. Stroke. 1991;22:1389–1393. doi: 10.1161/01.str.22.11.1389. [DOI] [PubMed] [Google Scholar]
- 16.Krotz F, Riexinger T, Buerkle MA, Nithipatikom K, Gloe T, Sohn HY, Campbell WB, Pohl U. Membrane-potential-dependent inhibition of platelet adhesion to endothelial cells by epoxyeicosatrienoic acids. Arterioscler Thromb Vasc Biol. 2004;24:595–600. doi: 10.1161/01.ATV.0000116219.09040.8c. [DOI] [PubMed] [Google Scholar]
- 17.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA. 2002;99:2222–2227. doi: 10.1073/pnas.261710799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleming I. Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediat. 2007;82:60–67. doi: 10.1016/j.prostaglandins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Yang S, Wei S, Pozzi A, Capdevila JH. The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch. Biochem Biophys. 2009;489:82–91. doi: 10.1016/j.abb.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potente M, Fisslthaler B, Busse R, Fleming I. 11,12-Epoxyeicosatrienoic acid-induced inhibition of FOXO factors promotes endothelial proliferation by down-regulating p27Kip1. J Biol Chem. 2003;278:29619–29625. doi: 10.1074/jbc.M305385200. [DOI] [PubMed] [Google Scholar]
- 22.Sun J, Sui X, Bradbury JA, Zeldin DC, Conte MS, Liao JK. Inhibition of vascular smooth muscle cell migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ Res. 2002;90:1020–1027. doi: 10.1161/01.res.0000017727.35930.33. [DOI] [PubMed] [Google Scholar]
- 23.Athirakul K, Bradbury JA, Graves JP, DeGraff LM, Ma J, Zhao Y, Couse JF, Quigley R, Harder DR, Zhao X, Imig JD, Pedersen TL, Newman JW, Hammock BD, Conley AJ, Korach KS, Coffman TM, Zeldin DC. Increased blood pressure in mice lacking cytochrome P450 2J5. Faseb J. 2008;22:4096–4108. doi: 10.1096/fj.08-114413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gauthier KM, Falck JR, Reddy LM, Campbell WB. 14,15-EET analogs: characterization of structural requirements for agonist and antagonist activity in bovine coronary arteries. Pharmacol Res. 2004;49:515–524. doi: 10.1016/j.phrs.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Sudhahar V, Shaw S, Imig JD. Epoxyeicosatrienoic Acid Analogs and Vascular Function. Curr Med Chem. 2010;17:1181–1190. doi: 10.2174/092986710790827843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proctor KG, Falck JR, Capdevila J. Intestinal vasodilation by epoxyeicosatrienoic acids: arachidonic acid metabolites produced by a cytochrome P450 monooxygenase. Circ Res. 1987;60:50–59. doi: 10.1161/01.res.60.1.50. [DOI] [PubMed] [Google Scholar]
- 27.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol. 1996;7:2364–2370. doi: 10.1681/ASN.V7112364. [DOI] [PubMed] [Google Scholar]
- 28.Larsen BT, Miura H, Hatoum OA, Campbell WB, Hammock BD, Zeldin DC, Falck JR, Gutterman DD. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(Ca) channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol Heart Circ Physiol. 2006;290:H491–H499. doi: 10.1152/ajpheart.00927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol. 1992;263:H519–H525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 30.Dimitropoulou C, West L, Field MB, White RE, Reddy LM, Falck JR, Imig JD. Protein phosphatase 2A and Ca2+-activated K+ channels contribute to 11,12-epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat. 2007;83:50–61. doi: 10.1016/j.prostaglandins.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Falck JR, Reddy LM, Reddy YK, Bondlela M, Krishna UM, Ji Y, Sun J, Liao JK. 11,12-epoxyeicosatrienoic acid (11,12-EET): structural determinants for inhibition of TNF-alpha-induced VCAM-1 expression. Bioorg Med Chem Lett. 2003;13:4011–4014. doi: 10.1016/j.bmcl.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 32.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem. 2002;277:15671–15676. doi: 10.1074/jbc.M110806200. [DOI] [PubMed] [Google Scholar]
- 34.Yan G, Chen S, You B, Sun J. Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovasc Res. 2008;78:308–314. doi: 10.1093/cvr/cvn006. [DOI] [PubMed] [Google Scholar]
- 35.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R, Falck JR, Capdevila JH. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–27146. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
- 36.Fitzpatrick FA, Ennis MD, Baze ME, Wynalda MA, McGee JE, Liggett WF. Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids. Influence of stereochemistry. J Biol Chem. 1986;261:15334–15338. [PubMed] [Google Scholar]
- 37.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch - Eur J Physiol. 2010;459:881–895. doi: 10.1007/s00424-010-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graier WF, Simecek S, Sturek M. Cytochrome P450 mono-oxygenase-regulated signaling of Ca2+ entry in human and bovine endothelial cells. J Physiol. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoebel BG, Kostner GM, Graier WF. Activation of microsomal cytochrome P450 mono-oxygenase by Ca2+ store depletion and its contribution to Ca2+ entry in porcine aortic endothelial cells. Br J Pharmacol. 1997;121:1579–1588. doi: 10.1038/sj.bjp.0701304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mombouli JV, Holzmann S, Kostner GM, Graier WF. Potentiation of Ca2+ signaling in endothelial cells by 11,12-epoxyeicosatrienoic acid. J Cardiovasc Pharmacol. 1999;33:779–784. doi: 10.1097/00005344-199905000-00015. [DOI] [PubMed] [Google Scholar]
- 41.Weston AH, Félétou M, Vanhoutte PM, Falck JR, Campbell WB, Edwards G. Bradykinin-induced, endothelium-dependent responses in porcine coronary arteries: involvement of potassium channel activation and epoxyeicosatrienoic acids. Br J Pharmacol. 2005;145:775–784. doi: 10.1038/sj.bjp.0706256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R, Nilius B. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908–915. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- 43.Imig JD, Dimitropoulou C, Reddy DS, White RE, Falck JR. Afferent arteriolar dilation to 11, 12-EET analogs involves PP2A activity and Ca2+-activated K+ Channels. Microcirculation. 2008;15:137–150. doi: 10.1080/10739680701456960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imig JD, Inscho EW, Deichmann PC, Reddy KM, Falck JR. Afferent arteriolar vasodilation to the sulfonimide analog of 11, 12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension. 1999;33:408–413. doi: 10.1161/01.hyp.33.1.408. [DOI] [PubMed] [Google Scholar]
- 45.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 46.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loch D, Hoey A, Morisseau C, Hammock BO, Brown L. Prevention of hypertension in DOCA-salt rats by an inhibitor of soluble epoxide hydrolase. Cell Biochem Biophys. 2007;47:87–98. doi: 10.1385/cbb:47:1:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 49.Fornage M, Hinojos CA, Nurowska BW, Boerwinkle E, Hammock BD, Morisseau CH, Doris PA. Polymorphism in soluble epoxide hydrolase and blood pressure in spontaneously hypertensive rats. Hypertension. 2002;40:485–490. doi: 10.1161/01.hyp.0000032278.75806.68. [DOI] [PubMed] [Google Scholar]
- 50.Olearczyk JJ, Quigley JE, Mitchell BC, Yamamoto T, Kim IH, Newman JW, Luria A, Hammock BD, Imig JD. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond) 2009;116:61–70. doi: 10.1042/CS20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, Hammock BD, Imig JD. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol. 2009;174:2086–2095. doi: 10.2353/ajpath.2009.080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang H, Gill R, Morisseau C, Newman JW, Hammock BD. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem. 2007;282:2891–2898. doi: 10.1074/jbc.M608057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297:F740–F448. doi: 10.1152/ajprenal.00098.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–40510. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 55.Certikova Chabova V, Walkowska A, Kompanowska-Jezierska E, Sadowski J, Kujal P, Vernerova Z, Vanourkova Z, Kopkan L, Kramer HJ, Falck JR, Imig JD, Hammock BD, Vaneckova I, Cervenka L. Combined inhibition of 20-hydroxyeicosatetraenoic acid formation and of epoxyeicosatrienoic acids degradation attenuates hypertension and hypertension-induced end-organ damage in Ren-2 transgenic rats. Clin Sci (Lond) 2010;118:617–632. doi: 10.1042/CS20090459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 57.Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, Zhang Y, Chiamvimonvat N, Shyy JY, Hammock BD, Zhu Y. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:564–569. doi: 10.1073/pnas.0811022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gross GJ, Gauthier KM, Moore J, Falck JR, Hammock BD, Campbell WB, Nithipatikom K. Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am J Physiol Heart Circ Physiol. 2008;294:H2838–H2844. doi: 10.1152/ajpheart.00186.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merkel MJ, Liu L, Cao Z, Packwood W, Young J, Alkayed NJ, Van Winkle DM. Inhibition of soluble epoxide hydrolase preserves cardiomyocytes: role of STAT3 signaling. Am J Physiol Heart Circ Physiol. 2010;298:H679–H687. doi: 10.1152/ajpheart.00533.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Motoki A, Merkel MJ, Packwood WH, Cao Z, Liu L, Iliff J, Alkayed NJ, Van Winkle DM. Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H2128–H2134. doi: 10.1152/ajpheart.00428.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seubert JM, Sinal CJ, Graves J, DeGraff LM, Bradbury JA, Lee CR, Goralski K, Carey MA, Luria A, Newman JW, Hammock BD, Falck JR, Roberts H, Rockman HA, Murphy E, Zeldin DC. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–450. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA. 2006;103:18733–18738. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, DeBarber AE, Koop DR, Alkayed NJ. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–2078. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simpkins AN, Rudic RD, Roy S, Tsai HJ, Hammock BD, Imig JD. Soluble epoxide hydrolase inhibition modulates vascular remodeling. Am J Physiol Heart Circ Physiol. 2010;298:H795–H806. doi: 10.1152/ajpheart.00543.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen HC, Ding FX, Wang S, Deng Q, Zhang X, Chen Y, Zhou G, Xu S, Chen HS, Tong X, Tong V, Mitra K, Kumar S, Tsai C, Stevenson AS, Pai LY, Alonso-Galicia M, Chen X, Soisson SM, Roy S, Zhang B, Tata JR, Berger JP, Colletti SL. Discovery of a Highly Potent, Selective, and Bioavailable Soluble Epoxide Hydrolase Inhibitor with Excellent Ex Vivo Target Engagement. J Med Chem. 2009;52:5009–5012. doi: 10.1021/jm900725r. [DOI] [PubMed] [Google Scholar]
- 66.Marino JP., Jr Soluble epoxide hydrolase, a target with multiple opportunities for cardiovascular drug discovery. Curr Top Med Chem. 2009;9:452–463. doi: 10.2174/156802609788340805. [DOI] [PubMed] [Google Scholar]
- 67.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther. 2009;331:906–916. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Imig JD, Falck JR. Compositions and methods for the treatment of renal and cardiovascular disease. #7,550,617. US Patent. 2009
- 69.Falck JR, Kodela R, Manne R, Atcha KR, Puli N, Dubasi N, Manthati VL, Capdevila JH, Yi XY, Goldman DH, Morisseau C, Hammock BD, Campbell WB. 14,15-Epoxyeicosa-5,8,11-trienoic Acid (14,15-EET) Surrogates Containing Epoxide Bioisosteres: Influence upon Vascular Relaxation and Soluble Epoxide Hydrolase Inhibition. J Med Chem. 2009;52:5069–5075. doi: 10.1021/jm900634w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Falck JR, Krishna UM, Reddy YK, Kumar PS, Reddy KM, Hittner SB, Deeter C, Sharma KK, Gauthier KM, Campbell WB. Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am J Physiol Heart Circ Physiol. 2003;284:H337–H349. doi: 10.1152/ajpheart.00831.2001. [DOI] [PubMed] [Google Scholar]
- 71.Gauthier KM, Jagadeesh SG, Falck JR, Campbell WB. 14,15-epoxyeicosa-5(Z)-enoic-mSI: a 14,15- and 5,6-EET antagonist in bovine coronary arteries. Hypertension. 2003;42:555–561. doi: 10.1161/01.HYP.0000091265.94045.C7. [DOI] [PubMed] [Google Scholar]
- 72.Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, Campbell WB. 14,15-Epoxyeicosa-5(Z)-enoic acid: a selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circ Res. 2002;90:1028–1036. doi: 10.1161/01.res.0000018162.87285.f8. [DOI] [PubMed] [Google Scholar]
- 73.Imig JD, Zhao X, Falck JR, Wei S, Capdevila JH. Enhanced renal microvascular reactivity to angiotensin II in hypertension is ameliorated by the sulfonimide analog of 11,12-epoxyeicosatrienoic acid. J Hypertens. 2001;19:983–992. doi: 10.1097/00004872-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 74.Yang W, Holmes BB, Gopal VR, Kishore RV, Sangras B, Yi XY, Falck JR, Campbell WB. Characterization of 14,15-epoxyeicosatrienoyl-sulfonamides as 14,15-epoxyeicosatrienoic acid agonists: use for studies of metabolism and ligand binding. J Pharmacol Exp Ther. 2007;321:1023–1031. doi: 10.1124/jpet.107.119651. [DOI] [PubMed] [Google Scholar]
- 75.Shaw S, Shrestha B, Nithripatikom K, Imig J. LC-MS measurement of the epoxyeicosatrienoic acid mimetic EET-8-ZE-D in rat serum. FASEB J. 2009;23:1019.13. http://www.fasebj.org/cgi/content/meeting_abstract/23/1_MeetingAbstracts/1019.13. [Google Scholar]
- 76.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Terashvili M, Tseng LF, Wu HE, Narayanan J, Hart LM, Falck JR, Pratt PF, Harder DR. Antinociception produced by 14,15-epoxyeicosatrienoic acid is mediated by the activation of beta-endorphin and met-enkephalin in the rat ventrolateral periaqueductal gray. J Pharmacol Exp Ther. 2008;326:614–622. doi: 10.1124/jpet.108.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79:2311–2319. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]