

Abstract
Nucleobase radicals are the major reactive intermediates produced when hydroxyl radical reacts with nucleic acids. 5,6-Dihydrouridin-6-yl radical (1) was independently generated from a ketone precursor via Norrish Type I photocleavage in a dinucleotide, single stranded, and double stranded RNA. This radical is a model of the major hydroxyl radical adduct of uridine. Tandem lesions resulting from addition of the peroxyl radical derived from 1 to the 5′-adjacent nucleotide are observed by ESI- MS. Radical 1 produces direct strand breaks at the 5′-adjacent nucleotide and at the initial site of generation. The preference for cleavage at these two positions depends upon the secondary structure of the RNA and whether O2 is present or not. Varying the identity of the 5′-adjacent nucleotide has little effect on strand scission. In general, strand scission is significantly more efficient under anaerobic conditions than when O2 is present. Strand scission is more than twice as efficient in double stranded RNA than in a single stranded oligonucleotide under anaerobic conditions. Internucleotidyl strand scission occurs via β-fragmentation following C2′-hydrogen atom abstraction by 1. The subsequently formed olefin cation radical ultimately yields products containing 3′-phosphate or 3′-deoxy-2′-ketouridine termini. These end groups are proposed to result from competing deprotonation pathways. The dependence of strand scission efficiency from 1 on secondary structure under anaerobic conditions suggests that this reactivity may be useful for extracting additional RNA structural information from hydroxyl radical reactions.
Keywords: DNA damage, reaction mechanism, free radicals
Nucleobase radicals constitute the major family of reactive species produced in nucleic acids that are exposed to hydroxyl radical (OH•), which is produced by γ-radiolysis and metal complexes (e.g. Fe•EDTA). Consequently, these radicals’ fates are significant when determining the effects of these damaging agents on DNA and RNA. For many years RNA was used as a substrate for studying the chemistry of γ-radiolysis and significant advances in our understanding of nucleic acid radical reactivity were achieved by combining a variety of analytical techniques with random generation of reactive species by ionizing radiation within the biopolymer.1,2 Although DNA damage chemistry was also studied in this way, elucidation of the reactivity of radicals formed in this biopolymer has benefited from an approach in which reactive intermediates and metastable lesions are independently generated from modified nucleotides at specific sites within synthetic biopolymers.3–14 Synthetically controlled formation of reactive intermediates and DNA lesions has helped resolve mechanistic controversies,7,10 and uncover novel reaction pathways.11,12 Independent RNA radical generation from modified nucleotides to produce reactive intermediates has been employed less frequently.6,15 However, the increasing use of OH• cleavage to probe RNA structure and folding kinetics,16–19 and the importance of RNA damage in diseases have provided greater impetus to further unravel the underlying chemistry of its oxidative damage.20,21
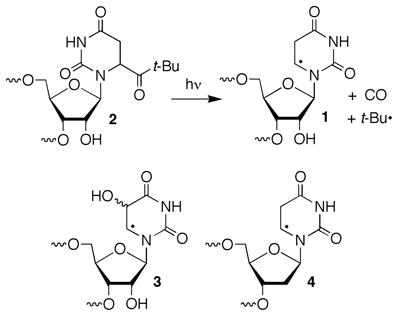 |
(1) |
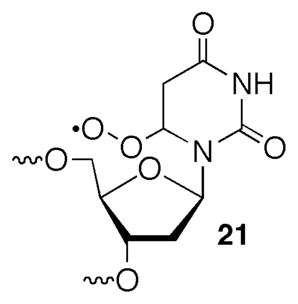
Recently, we reported on the formation of direct strand breaks from 5,6-dihydrouridin-6-yl radical (1) following its photochemical generation from 2 (eqn. 1).22 Norrish type I cleavage has been used to generate several nucleic acid radicals.5,13,14,23 The ketone (2) produces 5,6-dihydrouridin-6-yl radical (1) along with an equivalent of t-butyl radical and carbon monoxide via type I cleavage. Decarbonylation of the intermediate, acyl radical(s) (≥105 s−1) is rapid and the diffusible t-butyl radical is produced in subnanomolar concentration.24 5,6-Dihydrouridin-6-yl radical (1) was chosen as a synthetically expedient analogue of the major OH• adduct of uridine (3).25 Addition to the C5-position of uridine accounts for ~70% of the reactions between OH• and the pyrimidine ring in poly(U).2 Direct strand breaks from 1 were produced at the 5′-adjacent nucleotide (internucleotidyl) and at the nucleotide where the radical was originally produced (intranucleotidyl). Strand scission was most efficient in double-stranded RNA under anaerobic conditions and was reduced significantly in the presence of O2. Under anaerobic conditions direct strand scission was more than 2-fold greater under degassed conditions when 1 was flanked by uridines in a duplex compared to a single-stranded oligonucleotide. This RNA nucleobase radical reactivity was distinct from a similar DNA radical (4), which does not yield significant amounts of direct strand scission under either of the above conditions regardless of secondary structure.12,26,27 Isotopic labeling and product studies on a dinucleotide in which 1 was produced indicated internucleotidyl strand scission followed C2′-hydrogen atom abstraction (5, Scheme 1). Subsequent cleavage by β-elimination from 5 yields radical cation 6, consistent with observations in DNA.5,28 Internucleotidyl strand scission via C2′-hydrogen atom abstraction by 1 and not 4 was ascribed to the significant reduction in the bond dissociation energy of the corresponding carbon-hydrogen bond in RNA compared to DNA.29
Scheme 1.

Observations reported in the above study indicate that oxidative RNA damage may have physiological consequences and possible utility for studying biopolymer structure and folding. RNA structure determination by OH• cleavage is typically carried out under aerobic conditions. If this chemistry occurs in a variety of sequences, the preliminary results summarized above suggest that it may be possible to extract more structural information from such experiments by comparing cleavage patterns under aerobic and anaerobic conditions. Enhanced cleavage at a particular site in the absence of O2 relative to that observed under aerobic conditions will indicate duplex structure. In addition, if C6-pyrimidine nucleobase radicals account for ~70% of the reactions between OH• and uridine, the preliminary data suggest that RNA may be more susceptible to cleavage in O2 deficient cells. This would be an exception to the generally accepted oxygen enhancement effect.1 Such consequences prompted us to more completely examine the reactivity of 1 in RNA.
Results and Discussion
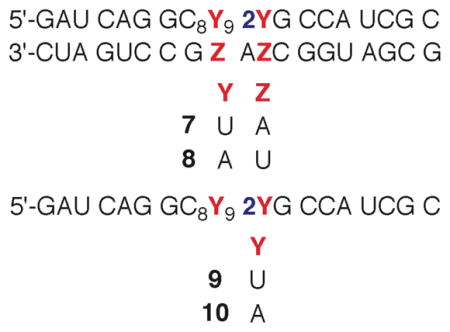
Product analysis from 5,6-dihydrouridin-6-yl (1) flanked by uridine in duplex RNA under anaerobic conditions
Initial studies showed that direct strand breaks were produced most efficiently when 5,6-dihydrouridin-6-yl (1) was generated in duplex RNA under anaerobic conditions.22 Denaturing polyacrylamide gel electrophoresis (PAGE) analysis coupled with enzymic end group analysis of photolyzed 3′-32P-7 (Please note that the radical-containing strand is radiolabeled in all experiments.) revealed two products each containing 5′-phosphate groups. Comparison with independently synthesized oligonucleotide markers showed that the major and minor products resulted from oxidation at the 5′-adjacent (internucleotidyl) and nucleobase radical containing (intranucleotidyl) nucleotides respectively. Similar analysis of 5′-32P-7 identified the corresponding fragments containing 3′-phosphate termini, as well as a product containing a ribose fragment that resulted from internucleotidyl oxidation.22 MALDI-TOF MS analysis of the crude photolysate of 7 confirmed the formation of the 4 phosphorylated fragmentation products (12–15, Figure 1). Oligonucleotide 12 corresponds to the major product observed by gel electrophoresis when 5′-32P-7 is photolyzed under anaerobic conditions. It results from internucleotidyl hydrogen atom abstraction by 1 as discussed below (Scheme 2). Similarly, 14 corresponds to the 3′-phosphate product resulting from intranucleotidyl hydrogen atom abstraction.
Figure 1.

MALDI-TOF MS analysis of crude photolysate of 7.
Scheme 2.

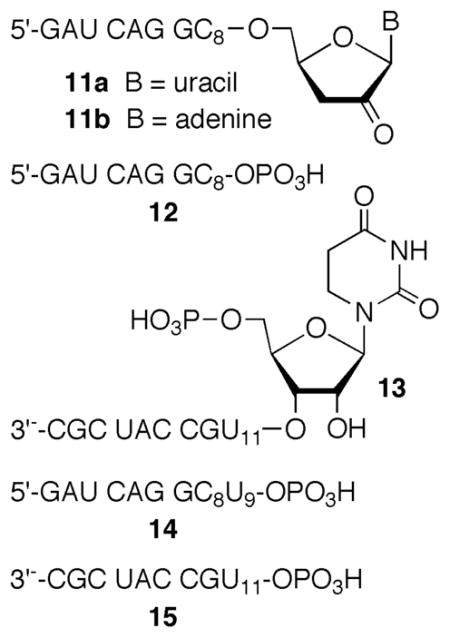
Similarly, 13 and 15 correspond to the 3′-fragments containing 5′-phosphates that are observed by gel electrophoresis when 3′-32P-7 is photolyzed. An additional ion (m/z = 2835.4) corresponding to 11a (or another species containing the same m/z) was also observed. Treatment of crude 5′-32P-7 photolysate with NaBH4 resulted in a mobility shift in 11a, consistent with its assignment as a ketone.30 Although other products with this molecular weight would react similarly with NaBH4, mechanistic considerations (vide infra) lead us to propose that 11a is the internucleotidyl cleavage product containing a ribose fragment.
Direct strand scission under anaerobic conditions from 5,6-dihydrouridin-6-yl (1) flanked by adenosines
Nucleobase radical reactivity in single (10) and double stranded (8) substrates when flanked by adenosine was examined under anaerobic conditions in order to probe the generality of the chemistry observed when 1 is flanked by uridine (Table 1). Adenosine was chosen over cytidine in order to provide a larger structural distinction from uridine. Guanosine was not employed due to possible complications arising from interactions between the nucleobase and the photoexcited ketone.31 The qualitative product distribution was very similar when 1 was produced in duplex RNA in which it was flanked by A (8) as previously observed for U (7). Direct strand scission resulting from oxidation of the 5′-adjacent nucleotide and the position at which the original radical was generated yielded 3′-fragments containing phosphate groups at the 5′-termini. 5′-Fragments containing 3′-phosphates formed at both nucleotide positions. In addition, a slower moving product consistent with a sugar fragment formed in conjunction with oxidation of the 5′-adjacent nucleotide was observed. Although this product was not detected by mass spectrometry, denaturing gel electrophoresis revealed that it reacted with NaBH4 which is consistent with its assignment as 11b.30
Table 1.
Direct strand scission under anaerobic conditions from 5,6-dihydrouridin-6-yl (1) flanked by adenosine.
| Absolute yield (%): | Inter. | Intra. | Total | % Inter. | ||
|---|---|---|---|---|---|---|
| Substratea | Phosphate | Ribose frag. | Phosphate | Ribose frag. | ||
| ds (8) | 27.7 ± 4.5 | 4.9 ± 1.0 | 4.9 ± 0.9 | - | 38.3 ± 3.3 | 85.8 ± 4.4 |
| ss (10) | 5.1 ± 0.6 | 3.2 ± 0.6 | 4.1 ± 0.1 | 3.3 ± 0.4 | 15.7 ± 0.4 | 52.4 ± 2.2 |
The substrate is designated by its secondary structure (ss, ds).
Quantitative analysis of strand scission reinforces the similarities between duplexes in which A (Table 1) or U flanks the 5,6-dihydrouridin-6-yl radical (1).22 The percentage of strand scission that results from oxidation of the 5′-adjacent nucleotide is within experimental error when 1 is flanked by A or U (86.8 ± 3.7 %) within a duplex. In addition, the ratio of internucleotidyl cleavage products containing 3′-phosphate termini relative to those containing the 3′-ribose fragment (11a,b) are also within experimental error of one another (U1U = 5.8 ± 1.2; A1A = 5.9 ± 2.4). The relative amount of internucleotidyl and intranucleotidyl strand scission products in single stranded substrates was the most significant difference in products formed from 1 flanked by A (10) or U (9). The original report on 5,6-dihydrouridin-6-yl radical (1) reactivity showed that intranucleotidyl strand scission accounted for more than 80% of direct strand breaks when 1 is flanked by uridines in a single stranded substrate.22 In contrast, internucleotidyl oxidation accounts for ~50% of the direct strand breaks in single stranded RNA when 1 is flanked by adenosines (Table 1). Despite this change in the proportion of cleavage products in single stranded RNA (ssRNA), the overall preference for direct strand scission when 1 is produced within a duplex (dsRNA) compared to ssRNA was very similar in the two flanking sequences (dsRNA/ssRNA: U1U = 2.2, A1A = 2.4).
 |
(2) |
5,6-Dihydrouridin-6-yl radical (1) reactivity flanked by uridine under aerobic conditions
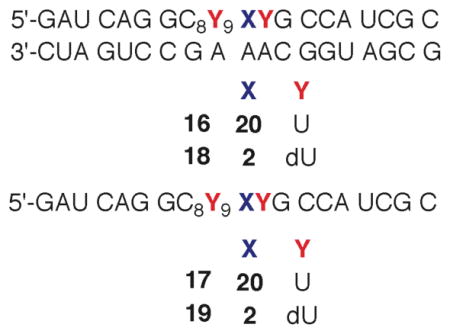
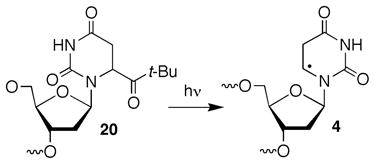
Direct strand scission is significantly reduced under aerobic conditions.22 O2 is expected to react with 1 and any subsequent alkyl radical formed much faster (k ~ 2 × 109 M−1s−1, [O2] ~ 0.2 mM) than these species abstract hydrogen atoms from the ribose backbone and/or undergo β-elimination (< 2 × 103 s−1).32 Hence, any direct strand breaks were expected to result from peroxyl radicals. In contrast to cleavage under anaerobic conditions, strand scission from 5,6-dihydrouridin-6-yl radical (1) was lower in double stranded RNA (7) than the single stranded substrate (9, Table 2). With the exception of intranucleotidyl cleavage in single stranded RNA (9), the observed strand scission was very low when 2 was photolyzed under aerobic conditions. Consequently, we utilized additional oligonucleotide probes in order to substantiate that cleavage was not an artifact due to sample handling. Substituting 5,6-dihydro-2′-deoxyuridin-6-yl radical (4) generated from the previously reported ketone (20, eqn. 2) for the ribose analogue virtually eliminated intranucleotidyl strand scission in single (17) and double stranded (16) RNA.26,33 This is consistent with previous observations regarding the reactivity of 4 in single and double stranded oligodeoxynucleotides.12,26 Intranucleotidyl strand scission was restored in single stranded substrate (19) upon generation of 1 flanked by 2′-deoxyuridine. These observations suggest that intranucleotidyl hydrogen atom abstraction by the peroxyl radical of 1 (21) in single stranded substrates occurs and result in strand breaks. Blocking intranucleotidyl strand scission in single stranded RNA does not result in an increase in cleavage at the 5′-adjacent nucleotide. However, it is possible that deactivating the intranucleotidyl pathway may increase hydrogen atom abstraction from the 5′-adjacent nucleotide but via a position (e.g. C1′) that does not lead to direct strand scission.12,34,35
Table 2.
Direct strand scission under aerobic conditions from 5,6-dihydrouridin-6-yl (1) flanked by uridine.
| Substratea | Absolute yield (%) |
% Inter. | Rel. to Anaerobic | ||
|---|---|---|---|---|---|
| Inter. | Intra. | Total | |||
| ds (7) | 2.2 ± 1.0 | 3.0 ± 1.1 | 5.2 ± 0.9 | 43.2 ± 20.0 | 0.1 |
| ss (9) | 1.3 ± 0.3 | 6.2 ± 1.0 | 7.5 ± 1.2 | 17.3 ± 1.5 | 0.4 |
| ds (16) | 5.2 ± 2.2 | 0.7 ± 0.5 | 5.9 ± 1.7 | 85.3 ± 15.4 | 0.2 |
| ss (17) | 1.4 ± 0.6 | 0.9 ± 1.1 | 2.3 ± 1.6 | 69.6 ± 21.6 | 0.2 |
| ds (18) | 1.1 ± 0.3 | 2.0 ± 0.7 | 3.1 ± 0.5 | 36.2 ± 11.6 | 0.2 |
| ss (19) | 1.1 ± 0.8 | 9.2 ± 1.1 | 10.3 ± 1.7 | 10.1 ± 6.1 | 1.4 |
The substrate is designated by its secondary structure (ss, ds).
The low strand scission yields when 1 was produced under aerobic conditions placed additional limits on product analysis. For instance, we were only able to detect the phosphate end group products resulting from intranucleotidyl cleavage by mass spectrometry.30 Enzymic end group analysis confirmed that the 5′-termini from single and double stranded substrates are exclusively phosphate groups.30 Kinase analyses revealed that the 3′-termini of the 5′-fragments are comprised mostly of phosphates, although there is a small amount of a product containing an unidentified ribose fragment.30
Mechanism of 3′-end group product formation
The previous report on the reactivity of 1 provided substantial evidence that direct strand scission at the 5′-adjacent uridine resulted from C2′-hydrogen atom abstraction.22 β-Fragmentation of phosphate from the resulting C2′-radical to produce a strand break was consistent with a large body of work initially by von Sonntag and subsequently corroborated by other studies in oligonucleotides and model compounds.2,5,28,36–38 Observation of a 3′-fragment containing a 5′-phosphate was expected for this mechanism. However, additional chemistry was required to produce the 3′-termini (3′-phosphate (12) and 3′-deoxy-2′-ketouridine (11a)) observed on the 5′-fragment. We considered the possibility that the 3′-phosphate terminus resulted from C4′-deprotonation of 6, followed by β-fragmentation of the 5′-phosphate (22, Scheme 2). Deprotonation of olefin cation radicals to produce allylic radicals can be very rapid.39 A previously reported model of chorismate synthase in which an allylic radical undergoes phosphate fragmentation provides additional precedent for producing RNA fragments containing 3′-phosphate termini.40 The requisite subsequent fragmentation step is similar to the reaction that led directly to strand scission and cleavage observed from C4′-radicals in DNA.36
Formation of 11a within 7 (and related molecules in different sequences) competes with 3′-phosphate formation. One electron reduction of the olefin cation radical (Scheme 3) would directly yield the enol tautomer (23) of this product. Based upon known oxidation potentials of enol ethers and dG, electron transfer from the purine should be thermodynamically downhill, with one caveat.41–43 Hydrogen bonding to 6 could stabilize it and reduce the driving force for electron transfer from guanosine.44 There is ample precedence for hole migration in DNA and this process was recently characterized in duplex RNA as well.7,45–48 We investigated this possibility by measuring the ratio of 3′-end groups in a series of duplexes containing varying amounts of guanosine in the vicinity of the nucleotide where the radical cation forms (Table 3). More facile electron transfer should result in a smaller ratio of 3′-phosphate to ketone (11) end products compared to the original duplex (7). The ease of electron transfer was expected to correlate with the density of guanosines proximal to the cation radical that would form at U9 following fragmentation (24 < 25 < 7 < 26). The overall strand scission yield in the duplexes were within experimental error of one another, suggesting that the photochemical generation of 5,6-dihydrouridin-6-yl (1) and subsequent strand scission are unaffected by the changes in sequence. Although the ratio of the products varied as a function of sequence, there was no clear trend with respect to the anticipated driving force for electron transfer.
Table 3.
Effect of proximal guanosines on the ratio of 3′-termini products resulting from strand cleavage by 5,6-dihydrouridin-6-yl (1)
| Strand scission (%) | Phosphate: Ketone (11) | |
|---|---|---|
| 5′-32P-(GAU CAU AA8U9 2UG UAA UCG C) 3′-(CUA GUA U U A AAC AUU AGC G) 24 |
39.6 ± 2.1 | 6.7 ± 0.1 |
| 5′-32P-(GAU CAU AC8U9 2UG UAA UCG C) 3′-(CUA GUA U G A AAC AUU AGC G) 25 |
45.7 ± 4.5 | 3.2 ± 0.8 |
| 5′-32P-(GAU CAG GC8U9 2UG CCA UCG C) 3′-(CUA GUC C G A AAC GGU AGC G) 7 |
35.2 ± 8.4 | 5.8 ± 1.2 |
| 5′-32P-(GAU CAG GG8U9 2UG UAA UCG C) 3′-(CUA GUC C C A AAC AUU AGC G) 26 |
37.1 ± 3.6 | 4.9 ± 0.7 |
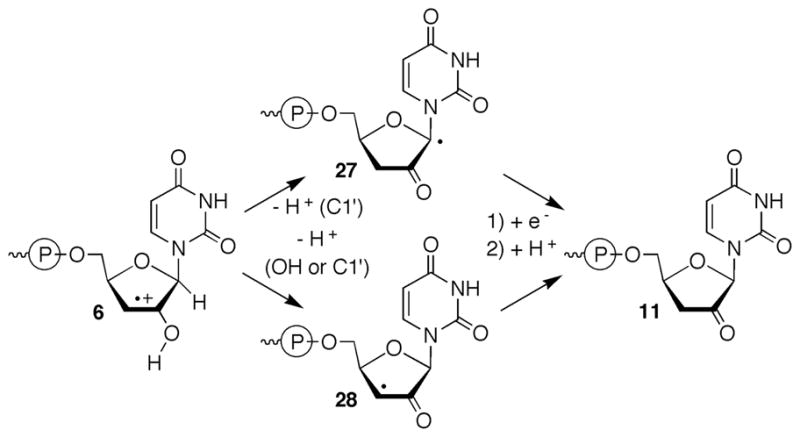
Consequently, we turned our attention to an alternative mechanism to explain formation of 11 (Scheme 4). We considered the possibility that deprotonation of the C1′-carbon or the hydroxyl group to yield α-keto radicals (27, 28, Scheme 4) from 6 compete with proton loss from C4′ to yield the product containing a 3′-phosphate terminus (e.g. 12, see Scheme 2). Subsequent reduction and protonation of the α-keto radicals would yield 11. Although we are unsure what the reducing agent would be in this system, relative deprotonation rate constants from C1′, C4′, and the 2′-hydroxyl group could be affected by pH. Changes in the relative rate constants of these product-determining steps would be reflected in the ratio of 3′-end group products as the pH is varied. The combined yield of the strand scission products resulting from internucleotidyl hydrogen atom abstraction decreased less than 7% over the pH range between 7.2 and 3.8 (Figure 2a). However, the ratio of 3′-phosphate containing product to 11a varied significantly, with the largest change occurring between pH 6.2 and 5.2 (Figure 2b). When the pH is reduced to 3.8 the relative ordering of products is reversed, and formation of the 3′-ketone containing cleavage product (11a) becomes favored over 3′-phosphate (12). Reducing the pH further to 1.9 resulted in a drastic reduction in the overall yield of strand scission products.
Scheme 3.

Figure 2.

Effect of pH on (A) strand scission yield and (B) 3′-end group ratio from 5,6-dihydrouridin-6-yl (1) generated from 5′-32P-7.
These data are consistent with competitive deprotonation and based upon the response of the product ratio to pH suggest that C4′-deprotonation in 6 is faster than those of the C1′- and hydroxyl group protons at higher pH. However, there is a reversal in the relative ordering of rate constants at lower pH. It is difficult to determine whether this is what one should expect because pKa values of structurally similar cation radicals in H2O are not available. Moreover, relevant kinetic constants are also scarce. Although it is well known that cation radicals are far more acidic than their neutral parent molecules, the limited data available is mostly for aromatic systems and pKa’s are often measured in CH3CN. For instance, the pKa’s of a series of cation radicals derived from stabilized enols range from 1.3 – 6.2 in CH3CN.49,50 In contrast, the allylic proton in a lipid cation radical was calculated to have a pKa ~0 in the same solvent.51 Although these data do not address the deprotonation kinetics, they are consistent with competing deprotonations from 6 to form the final 3′-termini products.
Hydrogen atom abstraction is the rate determining step in direct strand scission
The rate constant for strand cleavage (kCleave) from 5,6-dihydrouridin-6-yl (1) was estimated in single stranded (9) and double stranded (7) RNA by measuring the effect of BME on the cleavage yield. The rate constant was extracted from linear plots of the ratio of trapped product to cleavage product versus BME concentration.30 The trapping product was defined as the difference between the amount of intact RNA in the presence of BME and the absence of thiol. The slopes of these plots were the ratio of the rate constant for BME trapping (kTrap = 2.6 ± 0.5 × 106 M−1s−1) determined in a previous study.25 Cleavage from 1 in double stranded RNA (7) was faster (kCleave = 31 ± 10 s−1) than in single stranded (9, kCleave = 13 ± 4 s−1) substrate.22,30 Assuming that strand scission from 1 involves sequential hydrogen atom abstraction and cleavage (Scheme 1), evaluation of the data in the context of previously reported fragmentation rate constants provides insight into the rate determining step for this process. Radical fragmentation to yield a strand break is faster in single stranded than in double stranded RNA.32 Oxidized abasic site cleavage is also faster in single stranded than in double stranded DNA.52,53 Faster cleavage in single stranded substrates may be a result of the complementary strand in a duplex acting as a template for the reverse reaction, resulting in a slower observed rate for strand scission. The opposite trend is observed in our system. We interpret slower strand scission in single stranded RNA (9) as an indication that hydrogen atom abstraction by 1 is the rate determining step. Hydrogen atom abstraction by 1 could be faster in the duplex (7) where the reactive partner is held in a more rigid conformation.
Formation of products from 5,6-dihydrouridin-6-yl (1) that do not involve strand scission
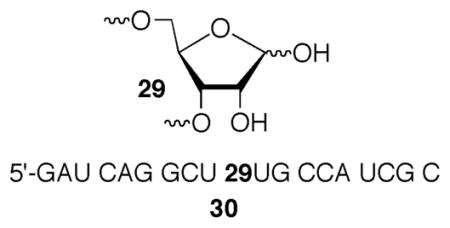
Nucleobase damage and abasic sites that do not result in direct strand scission are detected in DNA and discerned from one another via alkaline treatments of varying degrees of harshness.26 The inherent lability of RNA to alkaline conditions is incompatible with this approach, thus hindering its product analysis. However, RNA abasic sites (29) can be detected via the formation of strand breaks upon treatment with aniline.34 Various 5-32P-labeled RNA substrates containing 2 were photolyzed under aerobic or anaerobic conditions. The remaining uncleaved oligonucleotide was separated by denaturing polyacrylamide gel electrophoresis, recovered, desalted, and treated with aniline. We analyzed single stranded and double stranded RNA molecules in which 2 was flanked by U or A. Abasic sites were not detected in any of these substrates following generation of 1.30 The method was validated by subjecting 5′-32P-30, which was prepared using a variation of Leumann’s method, to aniline cleavage treatment.30,34
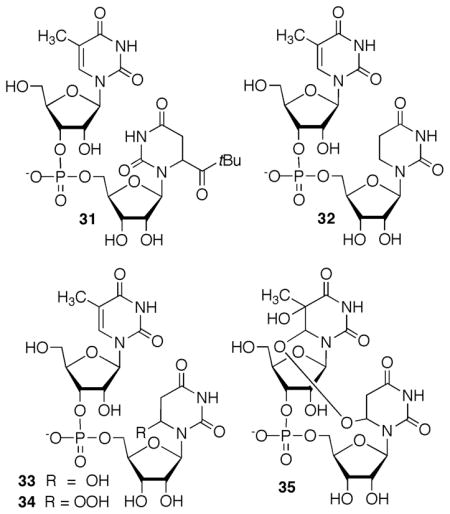
Products ascribable to 1 containing modified nucleobases were searched for via ESI-MS following photolysis of dinucleotide 31.30 Methyl uridine was used as the 5′-adjacent nucleotide in order to potentially remove any ambiguity in product assignment. The expected product (32) that contained 5,6-dihyrouridine was observed following photolysis of 31 (75 μM) under anaerobic conditions in the presence of β-mercaptoethanol (BME, 1 mM). Similarly, a mixture of the hydroxylated product (33) and the respective hydroperoxide (34) were cleanly produced when the dinucleotide was photolyzed under aerobic conditions in the presence of 480 μM BME. These latter products exhibited the expected shifts in m/z when 18O2 was used, indicating that both products result from dioxygen trapping of 1. Tandem lesion 35 was observed when 31 was photolyzed under aerobic conditions in the absence of BME. MS/MS analysis alternately in the presence of O2 or 18O2 revealed that 35 resulted from peroxyl radical (21) addition to the 5′-adjacent methyl uracil ring. This is consistent with what was observed from the reactivity of the analogous DNA radical (4) under aerobic conditions.12 Finally, the experiments were unaffected by the stereochemistry of 2 in the dinucleotide, consistent with the formation of products from a common intermediate, 5,6-dihydrouridin-6-yl (1).
Conclusions
As the major family of reactive species formed when OH• reacts with pyrimidines in RNA, a significant level of effort has been spent studying nucleobase radical reactivity.1,54–58 A number of pathways involving transfer of spin from a nucleobase radical to a proximal (or distal) ribose ring were proposed. By independently generating a nucleobase radical at defined positions in synthetic RNA, we unambiguously established that 5,6-dihydrouridin-6-yl radical (1) produces direct strand breaks in an intranucleotidyl manner and at the 5′-adjacent phosphate. Direct strand scission is more efficient under anaerobic conditions because O2 trapping of the radical(s) is much faster than hydrogen atom abstraction and subsequent fragmentation. A potentially important outcome of this research is the preference for strand scission in double stranded RNA under anaerobic conditions. This preference over single stranded RNA is attributable to faster hydrogen atom abstraction by 1 in the duplex and is consistent with competitive kinetic studies. Because nucleobase radicals such as 1 are the major species produced by OH•, preferential cleavage by 5,6-dihydrouridin-6-yl radical (1) in double stranded RNA by may be useful for distinguishing between single and double stranded RNA in hydroxyl radical cleavage experiments. If the preference for direct strand scission under anaerobic conditions in duplex RNA by 1 is general for nucleobase radicals, single and double stranded RNA will be distinguished from one another by comparing relative cleavage intensities in hydroxyl radical reactions (e.g. Fe•EDTA) under aerobic and anaerobic conditions. Double stranded regions will be discernible by relative increases in strand scission under anaerobic conditions. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) is currently the most general chemical method available for determining secondary RNA structure.59,60
Enhanced cleavage under anaerobic conditions by 1 is also contrary to the generally accepted oxygen enhancement by ionizing radiation, which is often associated with the ability of O2 to “fix” (prevent repair by thiols) DNA radicals.1 Overall strand scission in poly(U) induced by ionizing radiation increases under aerobic conditions. This suggests that nucleobase radicals, despite their relatively high yield are not major contributors to direct strand scission under aerobic conditions.
Supplementary Material
Scheme 4.
Acknowledgments
We are grateful for support of this research by the National Institute of General Medical Sciences (GM-054996).
Footnotes
Supporting Information Available. Experimental procedures, mass spectral data, and sample autoradiograms. These materials are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.von Sonntag C. The Chemical Basis of Radiation Biology. Taylor & Francis; London: 1987. [Google Scholar]
- 2.von Sonntag C. Free-Radical-Induced DNA Damage and Its Repair. Springer-Verlag; Berlin: 2006. [Google Scholar]
- 3.Pitié M, Pratviel G. Chem Rev. 2010;110:1018–1059. doi: 10.1021/cr900247m. [DOI] [PubMed] [Google Scholar]
- 4.Gates KS. Chem Res Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giese B, Beyrich-Graf X, Erdmann P, Giraud L, Imwindelried P, Müller SN, Schwitter U. J Am Chem Soc. 1995;117:6146–6147. [Google Scholar]
- 6.Strittmatter H, Dussy A, Schwitter U, Giese B. Angew Chem Int Ed. 1999;38:135–137. [Google Scholar]
- 7.Giese B. Acc Chem Res. 2000;33:631–636. doi: 10.1021/ar990040b. [DOI] [PubMed] [Google Scholar]
- 8.Meggers E, Michel-Beyerle ME, Giese B. J Am Chem Soc. 1998;120:12950–12955. [Google Scholar]
- 9.Greenberg MM, Barvian MR, Cook GP, Goodman BK, Matray TJ, Tronche C, Venkatesan H. J Am Chem Soc. 1997;119:1828–1839. [Google Scholar]
- 10.Bales BC, Pitié M, Meunier B, Greenberg MM. J Am Chem Soc. 2002;124:9062–9063. doi: 10.1021/ja026970z. [DOI] [PubMed] [Google Scholar]
- 11.Hong IS, Greenberg MM. J Am Chem Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 12.Hong IS, Carter KN, Sato K, Greenberg MM. J Am Chem Soc. 2007;129:4089–4098. doi: 10.1021/ja0692276. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg MM. Org Biomol Chem. 2007;5:18–30. doi: 10.1039/b612729k. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg MM. In: Wiley Ser React Intermed Chem Biol. Greenberg MM, editor. Vol. 2. 2009. pp. 135–162. [Google Scholar]
- 15.Crich D, Mo X-S. J Am Chem Soc. 1997;119:249–250. [Google Scholar]
- 16.Adilakshmi T, Bellur DL, Woodson SA. Nature. 2008;455:1268–1272. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodson SA. Curr Opin Chem Biol. 2008;12:667–673. doi: 10.1016/j.cbpa.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlatterer JC, Brenowitz M. Methods. 2009;49:142–147. doi: 10.1016/j.ymeth.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tullius TD, Greenbaum JA. Curr Opin Chem Biol. 2005;9:127–134. doi: 10.1016/j.cbpa.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Moreira PI, Nunomura A, Nakamura M, Takeda A, Shenk JC, Aliev G, Smith MA, Perry G. Free Radical Biol Med. 2008;44:1493–1505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Dedon PC, DeMott MS, Elmquist CE, Prestwich EG, McFaline JL, Pang B. Biomarkers in Medicine. 2007;1:293–312. doi: 10.2217/17520363.1.2.293. [DOI] [PubMed] [Google Scholar]
- 22.Jacobs AC, Resendiz MJE, Greenberg MM. J Am Chem Soc. 2010;132:3668–3669. doi: 10.1021/ja100281x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tronche C, Goodman BK, Greenberg MM. Chem Biol. 1998;5:263–271. [PubMed] [Google Scholar]
- 24.Fischer H, Paul H. Acc Chem Res. 1987;20:200–206. [Google Scholar]
- 25.Newman CA, Resendiz MJE, Sczepanski JT, Greenberg MM. J Org Chem. 2009;74:7007–7012. doi: 10.1021/jo9012805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carter KN, Greenberg MM. J Am Chem Soc. 2003;125:13376–13378. doi: 10.1021/ja036629u. [DOI] [PubMed] [Google Scholar]
- 27.Hong IS, Carter KN, Greenberg MM. J Org Chem. 2004;69:6974–6978. doi: 10.1021/jo0492158. [DOI] [PubMed] [Google Scholar]
- 28.Dizdaroglu M, Von Sonntag C, Schulte-Frohlinde D. J Am Chem Soc. 1975;97:2277–2278. doi: 10.1021/ja00841a051. [DOI] [PubMed] [Google Scholar]
- 29.Li M-J, Liu L, Wei K, Fu Y, Guo Q-X. J Phys Chem B. 2006;110:13582–13589. doi: 10.1021/jp060331j. [DOI] [PubMed] [Google Scholar]
- 30.See Supporting Information.
- 31.Adam W, Arnold MA, Nau WM, Pischel U, Saha-Moeller CR. J Am Chem Soc. 2002;124:3893–3904. doi: 10.1021/ja017600y. [DOI] [PubMed] [Google Scholar]
- 32.Giese B, Dussy A, Meggers E, Petretta M, Schwitter U. J Am Chem Soc. 1997;119:11130–11131. [Google Scholar]
- 33.Carter KN, Greenberg MM. J Org Chem. 2003;68:4275–4280. doi: 10.1021/jo034038g. [DOI] [PubMed] [Google Scholar]
- 34.Kupfer PA, Leumann CJ. Nucleic Acids Res. 2007;35:58–68. doi: 10.1093/nar/gkl948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trzupek JD, Sheppard TL. Org Lett. 2005;7:1493–1496. doi: 10.1021/ol050120h. [DOI] [PubMed] [Google Scholar]
- 36.Giese B, Beyrich-Graf X, Erdmann P, Petretta M, Schwitter U. Chem & Biol. 1995;2:367–375. doi: 10.1016/1074-5521(95)90217-1. [DOI] [PubMed] [Google Scholar]
- 37.Newcomb M, Miranda N, Huang X, Crich D. J Am Chem Soc. 2000;122:6128–6129. doi: 10.1021/ja015602c. [DOI] [PubMed] [Google Scholar]
- 38.Horner JH, Taxil E, Newcomb M. J Am Chem Soc. 2002;124:5402–5410. doi: 10.1021/ja0177399. [DOI] [PubMed] [Google Scholar]
- 39.Taxil E, Bagnol L, Horner JH, Newcomb M. Org Lett. 2003;5:827–830. doi: 10.1021/ol034061o. [DOI] [PubMed] [Google Scholar]
- 40.Giese B, Almstead NG. Tetrahedron Lett. 1994;35:1677–80. [Google Scholar]
- 41.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 42.Rathore R, Kochi JK. Tetrahedron Lett. 1994;35:8577–80. [Google Scholar]
- 43.Schmittel M, Lal M, Lal R, Roeck M, Langels A, Rappoport Z, Basheer A, Schlirf J, Deiseroth H-J, Floerke U, Gescheidt G. Tetrahedron. 2009;65:10842–10855. [Google Scholar]
- 44.Lal M, Langels A, Deiseroth H-J, Schlirf J, Schmittel M. J Phys Org Chem. 2003;16:373–379. [Google Scholar]
- 45.Schuster GB. Acc Chem Res. 2000;33:253–260. doi: 10.1021/ar980059z. [DOI] [PubMed] [Google Scholar]
- 46.Lewis FD, Letsinger RL, Wasielewski MR. Acc Chem Res. 2001;34:159–170. doi: 10.1021/ar0000197. [DOI] [PubMed] [Google Scholar]
- 47.Delaney S, Barton JK. J Org Chem. 2003;68:6475–6483. doi: 10.1021/jo030095y. [DOI] [PubMed] [Google Scholar]
- 48.Maie K, Miyagi K, Takada T, Nakamura M, Yamana K. J Am Chem Soc. 2009;131:13188–13189. doi: 10.1021/ja902647j. [DOI] [PubMed] [Google Scholar]
- 49.Mattay J, Schmittel M. Electron Transfer I. Vol. 169. Springer; Berlin/Heidelberg: 1994. pp. 183–230. [Google Scholar]
- 50.Schepp NP. J Org Chem. 2004;69:4931–4935. doi: 10.1021/jo049603+. [DOI] [PubMed] [Google Scholar]
- 51.Huvaere K, Cardoso DR, Homem-de-Mello P, Westermann S, Skibsted LH. J Phys Chem B. 2010;114:5583–5593. doi: 10.1021/jp9121744. [DOI] [PubMed] [Google Scholar]
- 52.Kim J, Gil JM, Greenberg MM. Angew Chem Int Ed. 2003;42:5882–5885. doi: 10.1002/anie.200352102. [DOI] [PubMed] [Google Scholar]
- 53.Chen J, Stubbe J. Biochemistry. 2004;43:5278–5286. doi: 10.1021/bi0495376. [DOI] [PubMed] [Google Scholar]
- 54.Isildar M, Schuchmann MN, Schulte-Frohlinde D, Von Sonntag C. Int J Radiat Biol. 1981;40:347–354. doi: 10.1080/09553008114551301. [DOI] [PubMed] [Google Scholar]
- 55.Deeble DJ, Schulz D, Von Sonntag C. Int J Radiat Biol. 1986;49:915–926. doi: 10.1080/09553008514553151. [DOI] [PubMed] [Google Scholar]
- 56.Schulte-Frohlinde D, Behrens G, Önal A. Int J Radiat Biol. 1986;50:103–110. doi: 10.1080/09553008614550481. [DOI] [PubMed] [Google Scholar]
- 57.Bothe E, Behrens G, Böhm E, Sethuram B, Schulte-Frohlinde D. Int J Radiat Biol. 1986;49:57–66. doi: 10.1080/09553008514552231. [DOI] [PubMed] [Google Scholar]
- 58.Hildenbrand K, Schulte-Frohlinde D. Int J Radiat Biol. 1989;55:725–738. doi: 10.1080/09553008914550781. [DOI] [PubMed] [Google Scholar]
- 59.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 60.Wilkinson KA, Merino EJ, Weeks KM. J Am Chem Soc. 2005;127:4659–4667. doi: 10.1021/ja0436749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.