

Abstract
Niemann–Pick type C disease is a fatal lysosomal storage disorder caused by loss of NPC1 function. The disorder severely affects multiple body systems, particularly the nervous system. To test whether rescue of NPC1 activity in neurons, astrocytes, or other cell types can correct the neurological defects, a Tet-inducible Npc1-YFP transgene was introduced into Npc1−/− mice for the cell type-specific rescue of NPC1 loss. NPC1-YFP produced in neurons prevented neuron degeneration, slowed reactive glial activity, and ameliorated the disease. NPC1-YFP produced in astrocytes or in cells of visceral tissue did not. These results suggest that loss of NPC1 activity from neurons is the primary cause of the neuropathology and that rescue of NPC1 function in neurons is sufficient to mitigate the disease. The ability of neurons to survive and function in a cell-autonomous fashion allowed the use of this newly engineered rescue system to further define the brain regions or neuron populations required to ameliorate a neurological symptom. NPC1-YFP produced specifically in cerebellar Purkinje neurons reduced ataxia, increased weight, and prolonged life, but it did not prevent the eventual decline and premature death of Npc1−/− mice. Significant increase in lifespan correlated with sustained reduction of inflammation in the thalamus. Neuron rescue of other forebrain areas provided little benefit. Future work targeting increasingly discrete neuronal networks should reveal which CNS areas are critical for survival. This work may have broad implications for understanding the anatomical and cellular basis of neurological signs and symptoms of other neurodegenerative and lysosomal disorders.
Introduction
Niemann–Pick disease, type C (NPC), is an inherited cholesterol storage disorder. Clinical presentations mostly relate to the progressive widespread neurological deterioration (Imrie et al., 2007). Symptoms include, but are not limited to, dementia, ataxia, dysarthria, dystonia, and epilepsy (Fink et al., 1989). Patients suffer symptoms for many years before death, and the increasing severity of neurological problems adds to the health care burden.
The majority (95%) of NPC cases result from loss-of-function mutations in the NPC1 gene (Park et al., 2003). NPC1 encodes a sterol-binding 13-pass membrane protein required to prevent the intracellular accumulation of lipids (Infante et al., 2008). Although the genetic cause of the disease has been identified, an understanding of the cellular basis of the neurological symptoms remains limited.
Previous attempts to identify the cell types responsible for the disease pathology have used a mouse model with a mutant Npc1 allele, npcnih (Npc1−) (Loftus et al., 1997). Considerable disease correction was observed when functional NPC1 protein was produced in Npc1−/− mice using a prion (Loftus et al., 2002) or a glial fibrillary acidic protein (GFAP) promoter-driven transgene (Zhang et al., 2008; Donohue et al., 2009; Kapur et al., 2009). The prion promoter directed high expression of Npc1 in the brain, both in neurons and glia (Loftus et al., 2002). Although GFAP can be found in neurons (Hol et al., 2003), the GFAP promoter has been extensively used to drive transgene expression in astrocytes (Lee et al., 2008). Prior mammalian studies have revealed NPC1 predominantly localized in glia (Patel et al., 1999), and both astrocytes and microglia have been suggested to mediate inflammation and neurodegeneration in NPC mice (Baudry et al., 2003; Chen et al., 2007). Based on these data, it is reasonable to infer that glial loss of NPC1 function is the primary cause of the neuropathology.
Although glial dysfunction has been implicated in the disease process, two independent studies using a mouse chimera or a conditional knock-out of Npc1 have demonstrated the cell-autonomous death of NPC1-deficient neurons (Ko et al., 2005; Elrick et al., 2010). However, neuron-specific NPC1 rescue in an otherwise Npc1−/− animal had not been tested. We hypothesized that neuron survival could also be cell-autonomous, in which case, NPC1 function in neurons would be sufficient to correct neurological disorder. To test our hypothesis, we created a novel NPC rescue model that takes advantage of the powerful and versatile Tet system for inducible and cell type-specific gene expression in mice (Zhu et al., 2002).
To identify the cell type responsible for disease pathogenesis, we produced traceable and functional NPC1 protein in CNS neurons, astrocytes, or visceral tissue of Npc1−/− mice. We found that neuronal NPC1 function is sufficient for neuron survival and that neuron-specific rescue corrected neurological dysfunction in anatomically defined brain regions. Differences in the signs and symptoms of the disease and its rate of progression depended on which neuron populations were rescued.
Materials and Methods
Engineered tetO-Npc1-YFP reporter mouse line.
To build the tetO-Npc1-YFP transgene construct, the Npc1-FP (Ko et al., 2001) coding region in a pEYFP-N3 vector (Clontech) was inserted into a pcDNA3.1+ vector (Invitrogen) between the hCMV promoter and the BGH poly(A) site. The hCMV promoter was then replaced with a Tet-O CMVmin sequence from pUHD 10–3 (Gossen and Bujard, 1992). The FVB-Tg(tetO-Npc1-YFP) strain was produced in-house (Stanford Transgenic Research Center) by DNA microinjection into FVB/N embryos. Nine founders were identified, and only the one selected for this work exhibited good induction of NPC1-YFP specifically in the Purkinje neuron (PN) layer of the mouse cerebellum, when crossed with a Pcp2-tTA driver line.
Acquired mouse lines and backcross onto the FVB strain.
FVB/N, BALB/c-Npc1+/−, FVB-Tg(Pcp2-tTA), B6;CBA-Tg(Camk2a-tTA), B6.Cg-Tg(Eno2-tTA), B6.Cg-Tg(GFAP-tTA), and B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2) were obtained from The Jackson Laboratory. The BALB/c-Npc1+/− strain was backcrossed for >10 generations to develop a congenic FVB-Npc1+/−. The B6 transgenic lines were backcrossed five generations to develop incipient congenic FVB lines. For all congenic lines, strain coat color consistently remained albino as expected for a FVB stock, and PCR tests verified homozygosity for the retinal degeneration allele Pde6brd1/rd1 (Giménez and Montoliu, 2001), which is unique to FVB.
Mouse genotyping.
Ear clips (1–2 mm) were dissolved in 75 μl of 50 mm NaOH at 95°C for 30 min. Two microliters were used to run standard PCR: 35 cycles of 95°C for 30 s denaturing, 56°C for 30 s annealing, and 72°C for 30 s extension. For identifying homozygotes, 0.05 μg of genomic DNA was used in a 21-cycle PCR. Novel primer combinations used for genotyping are listed 5′ to 3′. Primers ACTTACAGATCGCCATTGAAAGCATC, CGATGCACATCTGGTTCCATCTAC, and TGTGTCTTTCAAGGTTGTTCCAGA produced a 325 bp amplicon for Npc1+ and 199 bp for Npc1−. GACGTAAACGGCCACAAGTTC and CTTCAGCTCGATGCGGTTCAC generated a 300 bp amplicon for YFP, and CAACCCGTAAACTCGCCCAG and GGCTCTGCACCTTGGTGATC generated a 450 bp amplicon for tTA. For ROSA-rtTA, see Jackson Laboratory genotyping protocols for strain name B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/J.
Mouse care, handling, and behavioral data analysis.
Under U.S. federal law concerning animal welfare, all procedures are in compliance with regulations set by Stanford's Institutional Animal Care and Use Committee. The behavioral assays undertaken have been chosen for their minimum mouse handling. Prism5 GraphPad software was used to generate graphs and perform statistics. Only the data from mice with verified NPC1-YFP expression at endpoint were incorporated into the analysis. To induce reporter expression with rtTA, Dox was administered via drinking water at 1 mg/ml in 5% sucrose every 3 d.
Parallel rod walkway test for ataxia.
The parallel rod floor test for ataxia (Kamens and Crabbe, 2007) was modified. A transparent narrow tunnel was suspended 30 cm above the floor. The floor of the tunnel was composed of evenly spaced parallel rods similar to the original floor test. The mouse, while inside the tunnel, traveled back and forth from one end to the other traversing the rods. Foot slips and distance traveled were recorded using a Sony HDR-HC9 camcorder. The data are reported as errors/cm × 100 as previously described (Kamens et al., 2005).
Weight and survival measurements.
Mouse weight was recorded every 2–7 d. Lifespan was calculated as the average age of death for male and female mice from a particular strain. Day of death was noted as the day the mice were killed. A mouse was killed when body weight dropped below 14 g, the mouse exhibited a slow response to stimuli, and the mouse had a dehydrated, frail appearance. The Gehan–Breslow–Wilcoxon test was used to evaluate statistical significance between mean lifespans.
Nest-building performance.
A nestlet (Ancare), a 5 × 5 × 0.65 cm square of compressed cotton fiber, was placed on the center of the home cage of 58- to 60-d-old single-housed male mice. Mice were not exposed to nestlets before that age. On the third day, the quality of nest built was scored as described in Figure 7 and based on previous methods (Deacon, 2006).
Figure 7.

Lifespan extended with neuron-specific rescue. A, Graphs of the percentage area of thalamus, cerebellum, and liver occupied by CD68-positive cells are shown for G, P, C, E and R; N; Npc1−/− mice strains at age P60. As in Figure 6E, the dotted line on the graph represents an arbitrary cutoff for low (<10% CD68-positive tissue area) and high (>10% CD68-positive tissue area) microglial activity. B, Weight curves and SD for each strain and sex were plotted and compared with the weight progression of Npc1−/− and Npc1+/− male mice. C, The mean survival age and 95% confidence limits were graphed for each transgenic line with the exception of E; N; Npc1−/− mice (asterisk). E; N; Npc1−/− mice were killed for tissue analysis before signs of severe morbidity. Age of death for this line is not known, but mice with the expected NPC1-YFP expression profile (Figs. 2D,E, 4C) and reduced inflammation (Fig. 6E) readily surpass 100 d (vertical line).
Dystonic features.
To observe hindlimb posture, mice were placed in an open Nalgene 500 ml screw cap container and filmed while rearing. The foot positions in relation to the pelvis and trunk were scored as described in Figure 7. The best score achievable was recorded for each mouse.
Immunoblot analysis.
Whole or half portions of the cerebellum, brainstem, interbrain, and cortex were isolated and snap-frozen. Frozen samples were homogenized in 3 ml per gram of tissue in cold RIPA buffer supplemented with complete Mini, EDTA-free, protease inhibitor cocktail tablets (Roche). After 30 min incubation on ice, samples were centrifuged twice at 10,000 × g for 10 min at 4°C. Supernatants were stored at −20°C in Laemmli protein sample buffer. Before gel electrophoresis in 4–15% or 4–20% Tris-HCl gradient gels (Bio-Rad), protein samples were not heated above 37°C. PBS solution containing 2% BSA+ 0.1% Tween 20 was used for blocking and wash steps. Blots were incubated for 1 h at room temperature or 4°C overnight with primary antibodies: chicken anti-GFP (Aves), rabbit anti-NPC1 (Novus), rabbit anti-Calbindin-D28K (Sigma), rabbit GFAP (Abcam), rat anti-myelin basic protein (MBP) (Abcam), and mouse anti-actin (Millipore). Blots were incubated with secondary HRP-conjugated antibodies (Invitrogen and Aves) for 30 min at room temperature. The HRP chemiluminescence produced by adding SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) was detected and analyzed using a ChemiDoc XRS System (Bio-Rad).
Immunocytochemistry and filipin staining.
Whole brains were fixed in 4% paraformaldehyde:PBS overnight at 4°C. Tissues were washed in PBS and sectioned with a Vibratome. Fifty micrometer sagittal sections were processed free floating in 2% BSA+ 0.2% Triton X-100:PBS solution through all blocking, antibody incubation, and wash steps. Sections were incubated overnight at 4°C with primary antibodies: chicken anti-GFP (Aves), rabbit GFAP (Abcam), rat CD68 (AbD Serotec), rabbit anti-Calbindin-D28K (Sigma), rabbit anti-MAP2 (Millipore), and mouse anti-S100 (Abcam). After washes, sections were incubated with secondary Alexa-conjugated antibodies (Invitrogen) and filipin complex (Sigma) overnight at 4°C. Tissue sections were brush-transferred and mounted on slides with Fluoromount-G (SouthernBiotech). Epifluorescent images were obtained using a Zeiss Axioplan2 fluorescent microscope equipped with an AxioCam HRc CCD camera. A 10% neutral density filter was used to prevent photobleaching of filipin stain at high magnification. Confocal images were obtained with a Leica TCS SP2 laser-scanning microscope. Images were processed using Volocity imaging software.
Microglia concentration and location.
Sagittal vibratome sections through the whole brain of male and female mice of each strain were immunostained with anti-CD68. With ImageJ software, CD68 fluorescence was converted to binary data by manual thresholding so that the space occupied by the resulting black image approximated the area of fluorescence. The percentage area occupied by black pixels was then measured and the SEM reported. To generate a whole-brain volume image, 40 μm coronal cryosections were individually stained with anti-CD68 and Hoechst (Invitrogen). Images were taken using a Leica MZ FLIII scope equipped with a Leica DFC500 color camera. Image sequences were loaded into ImageJ and aligned using MultiStackReg v1.45 plugin (B. L. Busse; http://www.stanford.edu/∼bbusse/work/downloads.html).
Results
Conditional rescue of NPC1 loss
The Tet system is a bitransgenic system requiring the use of reporter and driver transgene combination for gene expression control (Zhu et al., 2002). The bitransgenic nature of the Tet system and the breeding strategy used allowed for a single, newly created, mouse reporter line to be crossed to several previously established mouse driver lines (Fig. 1A,B). We obtained mouse driver lines harboring transgenes that would direct expression of the reporter tetO-Npc1-YFP transgene (N) in specific cell populations in the brain or visceral tissue, either constitutively or when induced with Dox. Mice harboring the driver Pcp2-tTA transgene (P) are known to drive tetO reporter gene expression specifically in PNs of the cerebellum (Zu et al., 2004). The Eno2-tTA transgene (E) drives reporter expression in neurons throughout the CNS (Chen et al., 1998). Camk2a-tTA (C) driven reporter expression is restricted to predominantly forebrain neurons (Mayford et al., 1996). GFAP-tTA (G) drives reporter expression in astrocytes (Wang et al., 2004), and ROSA-rtTA (R), in the presence of Dox, can induce tetO reporter gene expression in a majority of visceral tissues (Hochedlinger et al., 2005).
Figure 1.

Conditional rescue of NPC1 loss. A, Diagram of mating scheme used to generate mice with Tet-inducible NPC1-YFP. The combination of a reporter transgene, N, and a driver transgene, X, allows production of NPC1-YFP in place of NPC1 in a Npc1−/− mouse. B, List of abbreviations for the driver transgenes found in the mouse strains acquired from the Jackson Laboratory and their cell type specificity. Transgenes that drive gene expression in neurons are highlighted. C, Representative immunoblot of brainstem lysate from an Npc1+/−, Npc1−/−, N; Npc1−/−, and G; N; Npc1−/− mouse shows driver-dependent production of NPC1-YFP protein. Blots were probed with an antibody against NPC1 or GFP. The higher molecular weight of NPC1-YFP compared with NPC1 is due to the YFP tag. Anti-actin was used to determine equal loading of total protein.
Ideally, NPC1-YFP expression in N; Npc1−/− mice should be dependent only on the driver transgene. Leaky expression could confound rescue results. To test for baseline expression of NPC1-YFP in the absence of a driver, extensive immunoblot and immunofluorescence analysis was performed. Out of nine tetO-Npc1-YFP reporter lines generated, only one line was chosen. In N; Npc1−/− mice, NPC1-YFP was undetectable by immunoblot (Fig. 1C). Figure 1C shows the immunoblot analysis for brainstem, but a detailed analysis of other tissues was done as well (data not shown). All tissue data show that N was generally silent without a driver transgene. In addition, there was no statistically significant difference (p = 0.437) in the mean survival age of N; Npc1−/− mice (78.5 d, n = 12) compared to Npc1−/− mice (76.0 d, n = 9). Thus, without a driver, N alone did not alter disease progression. As a result, any changes in disease phenotypes of P, G, C, E, or R; N; Npc1−/− mice can be attributed to the effect of NPC1-YFP expression driven by the driver transgenes.
Cell type and tissue-specific production of NPC1-YFP
To accurately observe the location and cell-type specificity of NPC1 within a tissue, NPC1-YFP, a fluorescent protein-tagged version of NPC1, was necessary. The following is a summary of the NPC1-YFP expression profiles for the Tet active bitransgenic mouse lines used in this study.
G; N mice produced NPC1-YFP in S100-positive cells (Fig. 2A). Immunoreactive S100 protein marks the cytoplasm of astrocytes and Bergmann glia, specialized astrocytes that surround PNs. In the cerebellum, NPC1-YFP highlighted Bergmann glia but was absent from PNs (Fig. 2B). Based on immunoblot analysis, NPC1-YFP could be detected throughout the CNS (Fig. 2C). Indeed, astrocytes populate a large fraction of the brain.
Figure 2.

Cell type and tissue-specific expression of NPC1-YFP. A, Immunofluorescent confocal analysis of NPC1-YFP (GFP) localization within astrocytes (S100) or PNs (D28K) in the cerebellum. NPC1-YFP localizes to astrocytes in a G; N mouse and within PN in a P; N mouse (arrows). NPC1-YFP did not localize to other cells. Nuclei were labeled with Hoechst. B, Epifluorescent microscopy showing the distribution of NPC1-YFP relative to PNs. NPC1-YFP colocalizes with PNs in a P; N but not in a G; N mouse cerebellum. C, Immunoblot analysis shows expression of NPC1-YFP throughout the brain in G; N mice. Expression of NPC1-YFP in P; N mice is restricted to the cerebellum. Anti-actin was used as a loading control. D, Immunofluorescent confocal microscopy shows localization of NPC1-YFP (GFP) to MAP2-positive cells in the thalamus of an E; N mouse. E, For contrast, grayscale, inverted images were created using Photoshop. Black pixels are positive for fluorescence, gray is tissue background, and white represents no signal. Immunofluorescent signal from sagittal brain sections showed strong NPC1-YFP accumulation in the striatum (STR) and in the PN layer (arrow) of the cerebellum (CB). Relatively, weak NPC1-YFP immunofluorescence is detectable in the cortex (CTX). The level of NPC1-YFP in the cortex is representative of other CNS regions that are not shown. F, In a C; N mouse, the predominant NPC1-YFP expression is localized to the forebrain areas including hippocampus (HP), but little and sparse NPC1-YFP is found in the thalamus (TH). Also, the majority of cerebellar PNs do not produce NPC1-YFP in this mouse strain. G, NPC1-YFP is detected only in the olfactory bulb (OB) and choroid plexus (CP, arrow) of the brain of R; N mice. Aside from brain, NPC1-YFP is detected in multiple viscera tissue. Sections of liver and intestine are shown.
P; N mice produced NPC1-YFP specifically in PNs (Fig. 2A,B). PN cytoplasm was marked by Calbindin-D28K immunoreactivity (D28K). In these mice, NPC1-YFP was detectable by immunoblot only in the cerebellum (Fig. 2C).
E; N mice produced NPC1-YFP in MAP2-positive cells in the thalamus (Fig. 2D). MAP2 is specific for neurons and NPC1-YFP was detected in the majority of neurons throughout the brain, although at varying levels. Lowest expression occurred in the thalamus and expression was mostly absent from CA3 neurons of the hippocampus (Fig. 3A). Highest expression was detected in the PN layer of the cerebellum and striatum (Fig. 2E). This matched the previous reported expression profile for Eno2-tTA (Chen et al., 1998). For a more complete representation of the expected expression profile for this line, see Figure 4C.
Figure 3.
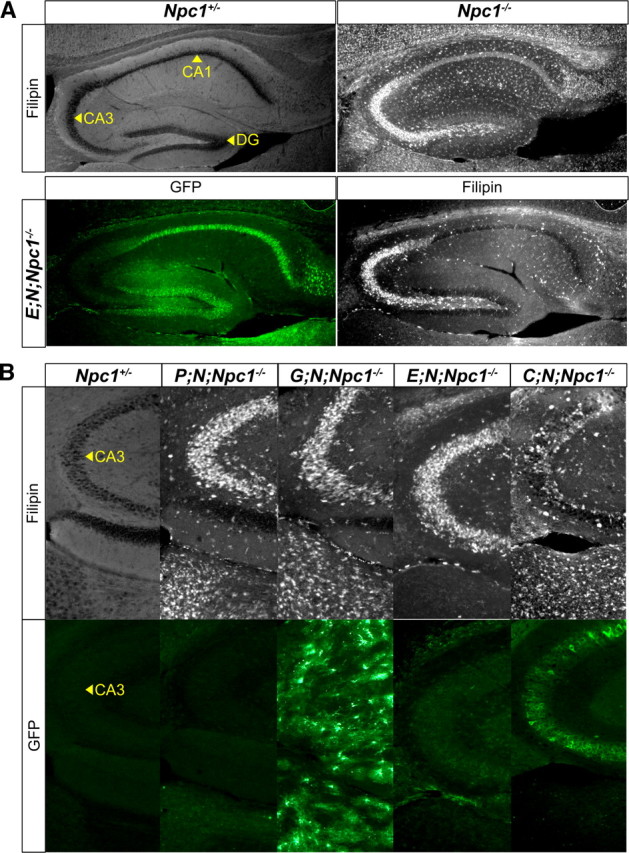
Neuron-autonomous correction of the CNS cholesterol storage defect in Npc1−/− mice. Immunofluorescence with anti-GFP marks neuron or astrocyte regions of the hippocampus that are positive for NPC1-YFP. Filipin marks hippocampus areas that are positive for accumulated free cholesterol. A, Sagittal sections of the hippocampus from an adult Npc1−/− mouse had substantial filipin staining in the CA3 neuron region. In an E; N; Npc1−/− mouse, NPC1-YFP produced in the DG or CA1 neurons did not reduce cholesterol accumulation in CA3 neurons, where NPC1-YFP expression was weak or absent. B, NPC1-YFP present in CA3 neurons (bottom) reduced CA3 cholesterol accumulation (top) in a C; N; Npc1−/− mouse. Despite NPC1-YFP production throughout the hippocampus in a G; N; Npc1−/− mouse, cholesterol accumulation was not reduced in CA3 neurons or elsewhere in the surrounding area.
Figure 4.

CNS region-specific neuron rescue of NPC disease in Npc1−/− mice. Sagittal brain sections of adult Npc1−/− mice were stained with filipin to reveal cholesterol accumulation in specific regions. The brain regions shown are portions of cortex (CTX), striatum (STR), thalamus (TH), hypothalamus (HT), midbrain (MB), cerebellum lobule (CB), pons (PO), and medulla (MD). Shown on the right of the images is an outline of the sagittal cross section of the mouse brain depicting the range of NPC1-YFP protein expression in green. Light green represents weak or variable expression based on the average pattern seen. A, In a representative male P70 N; Npc1−/− mouse, speckled and abundant filipin staining was noticeable in all brain areas. B, In a male P80 C; N; Npc1−/− mouse, decreased filipin staining was noticeable in the CTX, STR, and MB areas. Little to no decrease in filipin staining was noted in HT, PO, and MD. Note that there is a difference in expression profile in mice of the opposite sex. Greater NPC1-YFP coverage and more reduced filipin staining in HT, TH, PO, and MD areas can be seen in female C; N; Npc1−/− mice (data not shown). C, In a male P220 E; N; Npc1−/− mouse, filipin staining was largely absent from most brain regions except the HT, MD, and some of the basal forebrain located to the left of the TH.
C; N mice produced NPC1-YFP predominantly in neurons of the cortex, hippocampus, and striatum (Fig. 2F). This matched the previously reported forebrain expression profile for Camk2a-tTA (Mayford et al., 1996). Expression was mostly absent from the thalamus and cerebellum. For a more complete representation of the expected expression profile for this line, see Figure 4B.
R; N mice, after induction with Dox in the drinking water, produced NPC1-YFP in various visceral tissues, including liver and intestine. Target gene expression with this driver has previously been reported to be detectable in the liver, stomach, intestine, skin, heart, lungs, kidney, and thymus, but not in the brain (Hochedlinger et al., 2005). In the brain, NPC1-YFP can be detected only in the olfactory bulb and choroid plexus (Fig. 2G).
Neuron-specific NPC1-YFP corrects sterol accumulation in the CNS of NPC mice
To determine whether NPC1-YFP produced in astrocytes or neurons can correct the neuronal cholesterol accumulation phenotype of the disease, we used filipin stain of sagittal brain sections, followed by fluorescent microscopy. Filipin, a fluorescent polyene macrolide that binds specifically to unesterified membrane cholesterol and related sterols, is used clinically in the study and diagnosis of NPC disease (Vanier et al., 1991). It has been previously used to reveal cholesterol accumulation in PNs deficient in NPC1 (Elrick et al., 2010). PNs with endogenous NPC1 did not accumulate sterols.
We chose to test the ability of NPC1-YFP to correct cholesterol accumulation in hippocampus for two reasons. Filipin staining is evident in the CA3 neuron region of the Npc1−/− mouse (Fig. 3A), and the feedforward organization of the hippocampus made it feasible to test whether cholesterol accumulation in these neurons can be suppressed by rescue of presynaptic or postsynaptic neuron neighbors—granule cells in the dentate gyrus (DG) make direct contact with CA3 pyramidal cells, which synapse with CA1 pyramidal cells (Yeckel and Berger, 1990).
We found that NPC1-YFP could rescue the NPC disease phenotype of CA3 neurons only when directly expressed in CA3 neurons. In an E; N; Npc1−/− mouse, NPC1-YFP produced by neurons in the CA1 and DG did not reduce the filipin stain in the CA3 region, where NPC1-YFP was, for the most part, not found (Fig. 3A). The few CA3 neurons that produced NPC1-YFP did not reduce the appearance of strong filipin staining (Fig. 3B). CA3 filipin staining was also not reduced in P; N; Npc1−/− mice, which did not produce NPC1-YFP in any CA3 neurons. Reduced filipin staining in the CA3 neuron region was seen only in C; N; Npc1−/− mice that produced NPC1-YFP in a majority of CA3 neurons.
Astrocyte NPC1-YFP did not prevent the cholesterol accumulation in neurons despite the production of NPC1-YFP throughout the CA3 neuron region in G; N; Npc1−/− mice (Fig. 3B). The lack of rescue in G; N; Npc1−/− mice and the different neuronal expression patterns of NPC1-YFP in the brains of P, C, or E; N; Npc1−/− mice demonstrated the neuron-autonomous action of NPC1. This neuron-autonomous rescue of the NPC disease phenotype allowed brain region-specific rescue of sterol accumulation. In C; N; Npc1−/− mice, where NPC1-YFP expression is predominantly found in forebrain neurons of the cortex and striatum, but not the thalamus, filipin staining is generally reduced in the forebrain with the exception of the thalamus (Fig. 4A,B). By comparison, in E; N; Npc1−/− mice, which produced NPC1-YFP in forebrain neurons of the cortex, striatum, and thalamus, filipin staining is reduced in all these brain regions (Fig. 4B,C). Thus, anatomically defined CNS rescue of the disease can be achieved using the tetO-Npc1-YFP transgenic mouse strain to target NPC1-YFP to neurons in specific brain regions.
Cell-autonomous survival of cerebellar Purkinje neurons
To test whether neuron-specific rescue of NPC1 is sufficient to prevent neuron degeneration in an Npc1−/− mouse, we drove NPC1-YFP expression exclusively in cerebellar PNs. In NPC disease, cerebellar PNs are highly susceptible to loss of NPC1 and are among the first neurons to degenerate (German et al., 2001). Thus, PN survival in an NPC disease environment offered a sensitive assay for neuron survival.
PN degeneration occurs in stripes, with the most severe loss beginning in the anterior portion of the cerebellum (Sarna et al., 2003). This patterned loss is detected as loss of D28K staining. With age, almost complete loss of D28K staining is seen throughout the cerebellum with the exception of lobule X (Ko et al., 2005). To compare the amount of PN loss in P; N and G; N; Npc1−/− mice, we stained midline cerebellar sections with D28K.
In P; N; Npc1−/− mice, the patterned loss of PNs was abolished (Fig. 5A). The survival of PNs by PN-specific NPC1-YFP was striking in the context of the ages of the mice and the amount of NPC1-YFP produced in the cerebellum. The cerebella of G; N; Npc1−/− mice, in comparison with P; N; Npc1−/−, exhibited more widespread NPC1-YFP, yet the cerebellum of a postnatal day 90 (P90) P; N; Npc1−/− mouse showed more continuous D28K staining than either a P65 or P80 G; N; Npc1−/− mouse. These results confirm the cell-autonomous survival of PNs in P; N; Npc1−/− mice.
Figure 5.

Cell-autonomous survival of Purkinje neurons. A, Sagittal, midline sections of the cerebellum were stained with anti-D28K to mark remaining PNs and anti-GFP to locate NPC1-YFP expression. At age P65, in a G; N; Npc1−/− mouse, patterned loss of PNs (loss of D28K stain from lobules) can be seen. At age P80, significant loss of PNs is noticeable. At age P90, in a P; N; Npc1−/− mouse, PNs remain and NPC1-YFP is localized to the PN layer (boxed region and zoomed in inset). Note that lobule X was used as a landmark for section comparison since PNs in lobule X are unaffected. B, Representative immunoblot comparing the levels of D28K and MBP in whole cerebellum samples from P; N; Npc1−/− and G; N; Npc1−/− mice to Npc1+/− and Npc1−/− mice. MBP isoforms at 21.5, 18, and 17 kDa are shown. Notice the much larger amount of NPC1-YFP produced in a G; N; Npc1−/− mouse cerebellum compared to an age-matched P; N; Npc1−/− mouse; despite this, there is no obvious rescue of MBP and D28K level in a G; N; Npc1−/− mouse sample, but there is in a P; N; Npc1−/− mouse sample.
In G; N; Npc1−/− mice, despite the more extensive production of NPC1-YFP in the cerebellum in comparison to P; N; Npc1−/− mice, PN loss was not halted. Immunoblots using whole cerebellum protein samples showed no obvious increase in the amount of D28K protein level in samples taken from G; N; Npc1−/− mice as compared to age-matched Npc1−/− mice (Fig. 5B). In addition, MBP levels were assessed. Myelin, which is present on PN axons and is important for neuron survival and function, has been previously shown to decrease in Npc1−/− mice. Neuronal axon injury has been suggested as the most likely explanation for the hypomyelination seen as the disease progresses (Takikita et al., 2004). The reduced MBP level in G; N; Npc1−/− mice cerebella (Fig. 5B) further demonstrated that astrocyte NPC1-YFP provides little benefit to neurons. NPC1-YFP in neurons, even though neurons occupy a smaller fraction of the brain than glial cells, is enough to prevent neuron loss and demyelination.
Autonomous neuron rescue paralleled by nonautonomous glial activity
In Npc1−/− mice but not wild-type mice, reactive astrocytes and microglial are pervasive throughout the CNS during development and before neuron degeneration occurs (Baudry et al., 2003). It has previously been suggested that proinflammatory signals that can trigger glial inflammatory responses originate from astrocytes as a consequence of NPC1 loss in these cells (Suzuki et al., 2007). However, in the cerebella of Npc1−/− mice, gliosis was not seen in areas where PNs were still present (Fig. 6A). In the molecular layer, reactive astrocytes and microglia marked by intense GFAP and CD68 immunoreactivity (Ritz and Hausmann, 2008) were concentrated only in or near sites of PN loss, despite the abundance of reactive astrocytes and microglia in the granule layer, the layer containing PN axon projections. This suggests that glial cell activity remains responsive to neurons and does not occur solely because of NPC1 loss in glia or in response to inflammatory processes nearby.
Figure 6.

Neuronal NPC1-YFP suppresses CNS inflammation. A, Extended focus derived from confocal image stacks showing location of PNs (D28K), with reactive astrocytes (GFAP) and microglia (CD68), in the cerebellum of Npc1−/− mice. CD68-positive microglia were not present in Npc1+/− mice (data not shown), but populated the granule layer (GL) of Npc1−/− mice. Reactive microglia crossed into the molecular layer (ML) in areas of PN loss (PNs are underlined with a yellow bar). In the ML, reactive microglia associated closely with reactive astrocyte processes (arrows). Scale bar, 50 μm. B, Quantitative immunoblot comparisons of GFAP levels in the cerebellum of P; N; Npc1−/− and G; N; Npc1−/− mice at different ages demonstrate decreasing astrocyte reactivity in P; N; Npc1−/− but little change in G; N; Npc1−/− mice. GFAP intensity among samples was normalized against actin on the same blot and compared to the average GFAP levels in cerebella of P64 Npc1+/− mice. C, GFAP immunofluorescence in sagittal sections through thalamus and hippocampus of an N; Npc1−/− mouse compared to an age-matched E; N; Npc1−/− mouse. Notice the reduction in GFAP abundance in the thalamus of an E; N; Npc1−/− mouse, with little change in the hippocampus, which already had low levels of astrocyte activity. D, A fluorescence 3D opacity image was rendered from a sequence of registered coronal sections through the brain of an N; Npc1−/− mouse. Intense CD68-positive regions were located in the thalamus (TH) and cerebellar (CB) regions of the brain (circles). E, Percentage area of brain region that was CD68-positive in P60 N; Npc1−/− mice was quantified by threshold and graphed. The dashed horizontal line at 10% is an arbitrary reference for comparing the extent of inflammation. Anything below 10% is considered low and above high. The highest levels of reactive microglia occurred in the thalamus and cerebellum, while lower levels were observed in the midbrain and remaining forebrain and hindbrain regions. In G; N; Npc1−/− mice, reduction of CD68 was seen throughout the brain at age P60, but these improvements disappeared with age. In contrast, stable reduction of CD68, at least in the thalamus and cerebellum, was seen in E; N; Npc1−/− mice at ages P60 and P125+. The P125+ data were generated by averaging values from P125, P175, and P220 E; N; Npc1−/− mice.
Neuron-specific rescue in Npc1−/− mice allowed us the opportunity to verify that gliosis was a result of neuron degeneration and not because of loss of NPC1 function from the astrocytes themselves. Changes in astrocyte activity were analyzed by protein blots of whole cerebellum lysates from mice of various ages (Fig. 6B). In P64–P70 mice, approximately threefold more GFAP immunoreactivity was detected in Npc1−/− compared to control Npc1+/− cerebellum. Cerebella from P62–P63 P; N; Npc1−/− mice contained less GFAP than age-matched G; N; Npc1−/− mice. Although there was an increase in GFAP immunoreactivity in the cerebella of P100 P; N; Npc1−/− mice as compared to P62 mice, the amount of GFAP remained the same as P62–P63 G; N; Npc1−/− mice. The amount of GFAP in P100 P; N; Npc1−/− mice also remained below the level of GFAP in P64–P70 Npc1−/− cerebella. These results demonstrate that PN survival alone significantly delayed gliosis in the cerebellum. Lowered astrocyte activity in the thalamus was also observed in E; N; Npc1−/− mice (Fig. 6C), which demonstrates that neuron influence on astrocyte activity is not limited to PNs in the cerebellum.
As we observed with astrocytes, microglia in Npc1−/− mice proliferated and occupied areas of neuronal loss or degeneration (Fig. 6A). Unlike GFAP, which was also present in nonactivated astrocytes, CD68 staining was limited to reactive microglia, which facilitated quantitation. To identify brain regions most affected in Npc1−/− mice, we quantified the amount of tissue area occupied by CD68-positive microglia throughout the brain. In P65 Npc1−/− mice, the highest concentration of CD68 immunoreactivity was found in the cerebellum and thalamus as compared to other regions of the CNS (Fig. 6D,E). In comparison to G; N; Npc1−/− mice, fewer CD68-positive microglia occupied the thalamus and cerebellum of E; N; Npc1−/− mice at P60. In contrast to G; N; Npc1−/− mice, microglia activity in E; N; Npc1−/− mice in both the thalamus and cerebellum remained low despite age (Fig. 6E). Microglia activity increased mostly in the hindbrain region of E; N; Npc1−/− mice with age. This is likely a result of the variable, and sparse NPC1-YFP expression observed in this region (Fig. 4C).
Local reduction of inflammation as an indicator of region-specific rescue
The activity of microglia in Npc1−/− mice appeared highly compartmentalized and locally controlled; lowering inflammation in one area of the brain did not alter the level of inflammation in another. For example, in P; N; Npc1−/− mice, we have shown that NPC1-YFP expression is restricted to cerebellar PNs (Figs. 2A–C, 5A). As a result, substantially fewer CD68-positive microglia occupied the cerebellum, but the percentage of CD68 remained unchanged in the thalamus (Fig. 7A). Thus, we used the amount of CD68-positive cells present in a tissue as an indicator of which injured areas of the brain were rescued with NPC1-YFP. The level of reactive macrophages in the liver, also marked by CD68, was used to determine disease rescue in visceral tissue. The liver expresses the highest amounts of NPC1 in the body and, in Npc1−/− mice liver, dysfunction and accumulation of cholesterol is prominent (Garver et al., 2007).
As expected, in both C; N; Npc1−/− and E; N; Npc1−/− mice, reduced liver inflammation was not observed since expression of NPC1-YFP was not identified outside of the brain in these animals. Both mouse strains did have reduced inflammation in the thalamus. In E; N; Npc1−/− mice, however, microglia activity was lowered to levels comparable to areas of brain with the lowest CD68 percentage, such as the midbrain (Figs. 6E, 7A). In R; N; Npc1−/− mice, which produced NPC1-YFP in liver but not in the brain, CD68 levels in the liver were low, but full diseased levels were still seen in the brain.
Neuron rescue ameliorates NPC disease
To determine the effect of neuron rescue on NPC neurological phenotypes, two disease signs were measured that present as clinical symptoms of NPC disease in humans: ataxia and dystonia. Ataxia in mice was assessed by measuring motor incoordination (Kamens et al., 2005), and dystonia was characterized by limb hyperextension and rigidity, plus hypotonia of the lumbosacral trunk region.
To quantify the overall benefit to the health-related quality of life of the animal, mouse weight and nest building were used. Weight monitoring allowed us to plot disease progression and assess improvements in lifespan. Retention of nest-building activity, which requires recognition of bedding material, sufficient motor agility for shredding of nest material, and the desire to organize the bedding into a nest structure for protection and warmth, would imply the continued use of coordinated higher mental and motor functions (Deacon, 2006).
First, production of NPC1-YFP in liver and other tissues of the body, excluding neurons of the CNS (Figs. 2E, 7A), did not prevent weight loss or discernibly delay disease progression of R; N; Npc1−/− mice (Fig. 7B). These mice were fed doxycycline in the drinking water continuously from birth to induce NPC1-YFP production during their entire postnatal lifespan. A similar tetracycline, minocycline, a more effective antibiotic than doxycycline, has been shown to not alter the course of the disease in Npc1−/− mice (Erickson and Bernard, 2002). Skin biopsies were used to check for continuous NPC1-YFP induction (data not shown).
Second, producing NPC1-YFP in astrocytes only slightly modified disease progression, causing a lifespan increase of only 1 week for G; N; Npc1−/− mice (85.5 d, n = 6) in comparison with N; Npc1−/− mice (78.5 d, n = 12). The difference was not statistically significant (p = 0.28) (Fig. 7C). However, G; N; Npc1−/− mice at age P60 did weigh more than N; Npc1−/− mice (21.99 g, n = 8 for G; N; Npc1−/− mice, 17.67 g, n = 10 for N; Npc1−/− mice; p = 0.0005). Despite improved weight gain, G; N; Npc1−/− mice failed to build nests (score 1.1, n = 8), as did N; Npc1−/− mice (score 1.4, n = 7) at age P60. Dystonic features similar to that of C; N; Npc1−/− mice (Fig. 8B) were noticeable at age P70 (date not shown) and mice appeared ataxic although motor coordination was not measured.
Figure 8.

The effect of neuron rescue on neurological signs and behavior. A, The degree of motor coordination deficit, or ataxia, is represented here as the number of leg slips recorded in relation to travel distance on a parallel rod walkway (see Materials and Methods). The progression of ataxia for Npc1+/−, Npc1−/−, and neuron-rescued Npc1−/− mouse lines are shown for comparison. The projected average and range of the SDs are shown from ages P45 to P70. B, The ability of Npc1−/− mice to recognize, shred, and build a nest from nestlets was measured. Mice were given nestlets at P60 when N; Npc1−/− mice consistently began to score a 1. A score of 1 means the nestlet was left untouched. A score of 2 means it was chewed on; 3, the nestlet was partially shredded; 4, the nestlet was fully shredded; 5, the shredded material was shaped into a nest pocket. The graph shows the average (white bar) and range of nest scores for the neuron-rescue lines (black) compared to controls (gray). C, At age P70, hindlimb dystonic postures were readily noticeable in N; Npc1−/− mice. Of all Npc1−/− mice strains, only the E; N; Npc1−/− mouse showed a significant correction of stance while rearing. The extent of dystonia was scored as represented by still images. A score of 0 signifies a normal stance could be achieved. A score of 1 means either one or both limbs would hyperextend, preventing a normal stance; 2, limb hyperextension along with hypotonia of the lumbosacral trunk region was noticeable; 3, severe limb hyperextension and rigidity. Graph shows the average and SD of dystonia scores for the neuron-rescued lines.
Third, despite production of NPC1-YFP in neurons in various areas of the brain in C; N; Npc1−/− mice (Figs. 2D, 4B), weight gain was not improved (Fig. 7B) and increase in lifespan was comparable to P; N; Npc1−/− mice, although it varied between male and female C; N; Npc1−/− mice (Fig. 7C). No improvement in motor coordination and only a slight correction of dystonia was seen (Fig. 8A,C). Surprisingly, despite motor deficits, the nest-building performance of C; N; Npc1−/− mice at P60 (score 3.1, n = 7) was similar to P; N; Npc1−/− mice (score 2.9, n = 14) (Fig. 8B). Both lines approached wild-type nest-building activity (score 4.5, n = 13). Although C; N; Npc1−/− mice appeared slightly more adept at shredding the material, P; N; Npc1−/− mice seemed to more readily build a nest pocket even with nonshredded material.
Considerable extension of lifespan, weight gain, and improved motor coordination was seen in both P; N; Npc1−/− and E; N; Npc1−/− mice (Figs. 7B,C, 8A,B). In comparison to the lifespan and nesting habits of E; N; Npc1−/− mice (>120 d, n = 4 and score 4.0, n = 8), P; N; Npc1−/− mice showed only modest improvements (98 d, n = 12 and score 2.9, n = 14) (Figs. 7B, 8B). P; N; Npc1−/− mice showed no correction of dystonia at P70 (Fig. 8C). The hindlimb dystonic feature was considerably suppressed only in E; N; Npc1−/− mice. E; N; Npc1−/− mice were also the only mice to have a sustained reduction of inflammation in the thalamus (Fig. 7A).
Discussion
We have shown that neuron-targeted gene rescue in a mouse model of NPC disease is sufficient to correct CNS cholesterol accumulation, prevent neurodegeneration, reduce glial activity, and significantly improve the health of the animal. Thus, we conclude, loss of NPC1 function in neurons is predominantly responsible for the CNS pathogenesis in mice. Therapies including enzyme replacement to correct secondary enzyme defects (Devlin et al., 2010) and future genetic interventions that target neurons in an NPC1 disease patient may prove optimal for treatment.
The neuron-mediated progression of neurodegenerative disease is surprising considering that defects in glia-to-neuron interaction may be involved (Rossi and Volterra, 2009). Previous studies on NPC disease have documented glial dysfunction such as defective steroidogenesis (Chen et al., 2007) and abnormal lipid trafficking (Lloyd-Evans et al., 2008) that can negatively affect neurons. Also, it has been reported that an astrocyte-targeted GFAP promoter-driven Npc1 transgene (GFAPNpc1) can triple the lifespan of Npc1−/− mice (Zhang et al., 2008). However, since transgenes are subject to variegating and nontarget expression (Martin and Whitelaw, 1996; Bao-Cutrona and Moral, 2009), neurons in these mice could have produced some NPC1, especially since neuronal cholesterol was reported as reduced; moreover, GFAPNpc1 was not tagged to allow for more precise cellular detection of NPC1 within tissue. Thus, it may not have been astrocyte rescue alone that ameliorated the disease.
To test whether neuron rescue is sufficient to correct the disease, we generated various neuron-specific rescue lines using the Tet system. This system was chosen over other gene induction systems because of the wide commercial and academic availability of driver transgenes for the targeted expression of a single reporter transgene. We chose drivers whose cell type specificity and expression profiles have been previously reported (Fig. 1B). Although we cannot be absolutely certain that NPC1-YFP was absent from all other cell types except neurons, the use of multiple mouse lines with different drivers to induce varied NPC1-YFP expression patterns in the brain allowed the demonstration of full neuron autonomy. In an Npc1−/− mouse, NPC1-YFP produced in a specific neuron population corrected the cholesterol accumulation phenotype only within those neurons (Fig. 3A) and the production of NPC1-YFP in neighboring astrocytes did not change neuronal cholesterol accumulation (Fig. 3B). Along with the reduction of cholesterol in the specific brain areas (Fig. 4), the anatomical location of neuron rescue in Npc1−/− mice could be identified by local reduction of inflammation (Fig. 6). In addition, the lack of improvement in weight and lifespan of the R; N; Npc1−/− mice (Fig. 7), which produced NPC1-YFP in virtually all tissues except the brain (Fig. 2), allowed us to exclude potential confounding effects of non-nervous system rescue.
Our study does not exclude the possibility that glial NPC1 is required for the overall health of neurons, but it appears that the loss of NPC1 does not significantly affect glial function that is critical for neuron survival. Prior work has shown that the secretion of sterols was not inhibited in Npc1−/− astrocytes. Lipoproteins generated by Npc1−/− glia were capable of supporting axon elongation in vitro (Mutka et al., 2004; Karten et al., 2005). Prior work has also demonstrated the cell-autonomous death of PNs by using chimeric mice, mice comprised of a mixture of wild-type and Npc1−/− cells (Ko et al., 2005), and conditional knock-out mice, wild-type mice with Npc1 gene deletion in PNs (Elrick et al., 2010). However, these studies did not address whether PNs can survive alone despite the loss of NPC1 from all other neurons, glia cells, or other cells of the body. Here we show in vivo that despite glial NPC1 deficiency, cerebellar PNs, which are sensitive to many genetic and acquired disorders as well as toxic environmental factors (Sarna and Hawkes, 2003), survived as long as they produced NPC1-YFP (Fig. 5).
The targeted rescue of PNs in an Npc1−/− mouse allowed us to observe the benefit cerebellar improvements alone could have on NPC disease. PNs are the sole neuronal output of the cerebellar cortex, and loss of PNs in an otherwise normal brain has long been known to cause motor abnormalities. Current research has begun to suggest that the cerebellum can regulate nonmotor brain functions as well (Strick et al., 2009), including the early development of the whole brain. Thus, rescuing cerebellar function in a disease that also affects the cerebellum could have broad and significantly beneficial therapeutic outcomes. The benefit PN rescue alone had on weight gain, nest-building activity, motor ability, and lifespan in P; N; Npc1−/− mice supports this view and points to an important cerebellar involvement in the severity of NPC disease progression.
Despite significant benefits, cerebellar PN survival ultimately did not halt disease progression or prevent premature death of P; N; Npc1−/− mice. Improved motor coordination in these mice was temporary as ataxia seemed to eventually increase with age (Fig. 8A), and weight gain was unsustainable (Fig. 7B). E; N; Npc1−/− mice also showed a similar worsening of condition with age, but these mice exhibited an extraordinary increase in lifespan marked by a more delayed and gradual weight loss (Fig. 7B). The broad neuronal rescue in E; N; Npc1−/− mice would suggest that P; N; Npc1−/− mice eventually succumb to the effect of neurological deterioration of other brain regions.
With the given data, we conclude that rescue of thalamic pathways is essential for the prolonged survival of Npc1−/− mice. This deduction is based, in part, on the persistent inflammation in the thalamus of P; N; Npc1−/− and C; N; Npc1−/− mice but not in E; N; Npc1−/− mice, which can survive several weeks longer than either P; N; Npc1−/− or C; N; Npc1−/− mice (Figs. 6E, 7A–C). Therefore, rescue of neurons that are present or synapse in the thalamic region correlated with the most pronounced effect on lifespan.
Caution is necessary in attributing all the survival effect to thalamic rescue. Considering the complexity and interconnectivity of the nervous system, it is likely that other brain regions rescued in E; N; Npc1−/− mice may have contributed to improved lifespan. Nevertheless, it is reasonable to conclude that thalamic rescue would be beneficial. Thalamic atrophy, which occurs in many common neurodegenerative diseases, including Parkinson's and multiple sclerosis (Halliday, 2009; Rocca et al., 2010), can affect motor functions, consciousness, arousal, and sleep. In NPC disease mice, it is known that the sensory thalamus is extremely vulnerable (Yamada et al., 2001), with degeneration of thalamic neurons starting early in life and possibly preceding a dying-back type degeneration of afferent neurons in the brainstem or elsewhere (Ohara et al., 2004). It would be interesting to determine whether thalamus-specific neuron rescue would be sufficient to extend animal lifespan and to document the behavioral consequence of correcting only the thalamus.
Using the tetO-Npc1-YFP transgenic mouse strain generated for this study, future studies can take advantage of gene delivery of a tTA transgene, or other existing mouse driver lines, to target the thalamus or other CNS areas affected in Npc1−/− mice, such as the brainstem (Luan et al., 2008). The targeting of increasingly discrete neuronal networks and the development of assays to measure specific behaviors will be useful to associate rescue of a particular neural circuit with improvements to health-related quality of life, prolonged lifespan, and inhibition of neurological signs. For example, dystonia, which can be caused by damage to multiple brain regions, including the basal ganglia, thalamus, brainstem, and cerebellum (Breakefield et al., 2008), was not greatly suppressed in P; N; Npc1−/− mice, despite improvements in cerebellar pathology (Fig. 5), or in C; N; Npc1−/− mice, even though NPC1-YFP was produced in major areas of the forebrain and other brain regions (Fig. 4B). Thus, it remains unclear which neuronal networks are necessary to reduce dystonia. More precise region-specific control over neuron rescue and more quantitative criteria for measuring dystonia may begin to address this issue.
Our work may be relevant to other lysosomal storage disorders that cause neurological disease. Recent genetic studies on a mouse model of Gaucher disease determined that microglia are not the primary determinant of CNS pathology (Enquist et al., 2007). For NPC disease, we have observed that the inflammatory process is selective and responds to local neuronal dysfunction and degeneration without inevitably harming healthy or functional neurons. This is exemplified in the cerebellum, where the presence of reactive glia in the molecular layer occurs almost exclusively in areas of PN loss, despite accumulation of reactive microglia and astrocytes in the granule layer (Fig. 6). Although correction of NPC1 loss in astrocytes did curtail glial reactivity, increased inflammation and neuron loss was unavoidable unless neuronal NPC1 loss was corrected. In other lysosomal storage diseases and, possibly, more common neurological disorders with lysosomal system dysfunction such as Alzheimer's and Parkinson's (Nixon et al., 2001; Dehay et al., 2010), defects within neurons themselves may be the central cause of neurological decay. Restoring or enhancing the right cellular function in neurons may prove to be the best neuroprotective strategy.
Footnotes
Continuous funding and support from the Ara Parseghian Medical Research Foundation (APMRF) made this work possible. M.E.L. was supported by grants from the National Institutes of Health (Training Grants GM07790 and GM007276) and APMRF, A.D.K. was supported by fellowships from the Pew Foundation and APMRF, and M.P.S. is an Investigator of the Howard Hughes Medical Institute. We thank L. Milenkovic for edits to the manuscript and J. Hong for helpful discussions. We also acknowledge, with admiration, the children and families whose experiences with NPC disease, and perseverance, provided the strongest possible motivation for our research.
The authors declare no competing financial interests.
References
- Bao-Cutrona M, Moral P. Unexpected expression pattern of tetracycline-regulated transgenes in mice. Genetics. 2009;181:1687–1691. doi: 10.1534/genetics.108.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Chen G, Li HM, Chen YR, Gu XS, Duan S. Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann-Pick disease type C. Glia. 2007;55:1509–1518. doi: 10.1002/glia.20563. [DOI] [PubMed] [Google Scholar]
- Chen J, Kelz MB, Zeng G, Sakai N, Steffen C, Shockett PE, Picciotto MR, Duman RS, Nestler EJ. Transgenic animals with inducible, targeted gene expression in brain. Mol Pharmacol. 1998;54:495–503. doi: 10.1124/mol.54.3.495. [DOI] [PubMed] [Google Scholar]
- Deacon RM. Assessing nest building in mice. Nat Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin C, Pipalia NH, Liao X, Schuchman EH, Maxfield FR, Tabas I. Improvement in lipid and protein trafficking in Niemann-Pick C1 cells by correction of a secondary enzyme defect. Traffic. 2010;11:601–615. doi: 10.1111/j.1600-0854.2010.01046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue C, Marion S, Erickson RP. Expression of Npc1 in glial cells corrects sterility in Npc1(−/−) mice. J Appl Genet. 2009;50:385–390. doi: 10.1007/BF03195698. [DOI] [PubMed] [Google Scholar]
- Elrick MJ, Pacheco CD, Yu T, Dadgar N, Shakkottai VG, Ware C, Paulson HL, Lieberman AP. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum Mol Genet. 2010;19:837–847. doi: 10.1093/hmg/ddp552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist IB, Lo Bianco C, Ooka A, Nilsson E, Månsson JE, Ehinger M, Richter J, Brady RO, Kirik D, Karlsson S. Murine models of acute neuronopathic Gaucher disease. Proc Natl Acad Sci U S A. 2007;104:17483–17488. doi: 10.1073/pnas.0708086104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP, Bernard O. Studies on neuronal death in the mouse model of Niemann-Pick C disease. J Neurosci Res. 2002;68:738–744. doi: 10.1002/jnr.10257. [DOI] [PubMed] [Google Scholar]
- Fink JK, Filling-Katz MR, Sokol J, Cogan DG, Pikus A, Sonies B, Soong B, Pentchev PG, Comly ME, Brady RO, Barton NW. Clinical spectrum of Niemann-Pick disease type C. Neurology. 1989;39:1040–1049. doi: 10.1212/wnl.39.8.1040. [DOI] [PubMed] [Google Scholar]
- Garver WS, Jelinek D, Oyarzo JN, Flynn J, Zuckerman M, Krishnan K, Chung BH, Heidenreich RA. Characterization of liver disease and lipid metabolism in the Niemann-Pick C1 mouse. J Cell Biochem. 2007;101:498–516. doi: 10.1002/jcb.21200. [DOI] [PubMed] [Google Scholar]
- German DC, Quintero EM, Liang CL, Ng B, Punia S, Xie C, Dietschy JM. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J Comp Neurol. 2001;433:415–425. doi: 10.1002/cne.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giménez E, Montoliu L. A simple polymerase chain reaction assay for genotyping the retinal degeneration mutation (Pdeb(rd1)) in FVB/N-derived transgenic mice. Lab Anim. 2001;35:153–156. doi: 10.1258/0023677011911525. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM. Thalamic changes in Parkinson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 3):S152–S155. doi: 10.1016/S1353-8020(09)70804-1. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Hol EM, Roelofs RF, Moraal E, Sonnemans MA, Sluijs JA, Proper EA, de Graan PN, Fischer DF, van Leeuwen FW. Neuronal expression of GFAP in patients with Alzheimer pathology and identification of novel GFAP splice forms. Mol Psychiatry. 2003;8:786–796. doi: 10.1038/sj.mp.4001379. [DOI] [PubMed] [Google Scholar]
- Imrie J, Dasgupta S, Besley GT, Harris C, Heptinstall L, Knight S, Vanier MT, Fensom AH, Ward C, Jacklin E, Whitehouse C, Wraith JE. The natural history of Niemann-Pick disease type C in the UK. J Inherit Metab Dis. 2007;30:51–59. doi: 10.1007/s10545-006-0384-7. [DOI] [PubMed] [Google Scholar]
- Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamens HM, Crabbe JC. The parallel rod floor test: a measure of ataxia in mice. Nat Protoc. 2007;2:277–281. doi: 10.1038/nprot.2007.19. [DOI] [PubMed] [Google Scholar]
- Kamens HM, Phillips TJ, Holstein SE, Crabbe JC. Characterization of the parallel rod floor apparatus to test motor incoordination in mice. Genes Brain Behav. 2005;4:253–266. doi: 10.1111/j.1601-183X.2004.00100.x. [DOI] [PubMed] [Google Scholar]
- Kapur R, Donohue C, Jelinek D, Erickson RP. Amelioration of enteric neuropathology in a mouse model of Niemann-Pick C by Npc1 expression in enteric glia. J Neurosci Res. 2009;87:2994–3001. doi: 10.1002/jnr.22126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karten B, Hayashi H, Francis GA, Campenot RB, Vance DE, Vance JE. Generation and function of astroglial lipoproteins from Niemann-Pick type C1-deficient mice. Biochem J. 2005;387:779–788. doi: 10.1042/BJ20041694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, Gordon MD, Jin JY, Scott MP. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol Biol Cell. 2001;12:601–614. doi: 10.1091/mbc.12.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP. Cell-autonomous death of cerebellar Purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56:481–493. doi: 10.1002/glia.20622. [DOI] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Erickson RP, Walkley SU, Bryant MA, Incao A, Heidenreich RA, Pavan WJ. Rescue of neurodegeneration in Niemann-Pick C mice by a prion-promoter-driven Npc1 cDNA transgene. Hum Mol Genet. 2002;11:3107–3114. doi: 10.1093/hmg/11.24.3107. [DOI] [PubMed] [Google Scholar]
- Luan Z, Saito Y, Miyata H, Ohama E, Ninomiya H, Ohno K. Brainstem neuropathology in a mouse model of Niemann-Pick disease type C. J Neurol Sci. 2008;268:108–116. doi: 10.1016/j.jns.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Martin DI, Whitelaw E. The vagaries of variegating transgenes. Bioessays. 1996;18:919–923. doi: 10.1002/bies.950181111. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Mutka AL, Lusa S, Linder MD, Jokitalo E, Kopra O, Jauhiainen M, Ikonen E. Secretion of sterols and the NPC2 protein from primary astrocytes. J Biol Chem. 2004;279:48654–48662. doi: 10.1074/jbc.M405345200. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Mathews PM, Cataldo AM. The neuronal endosomal-lysosomal system in Alzheimer's disease. J Alzheimers Dis. 2001;3:97–107. doi: 10.3233/jad-2001-3114. [DOI] [PubMed] [Google Scholar]
- Ohara S, Ukita Y, Ninomiya H, Ohno K. Degeneration of cholecystokinin-immunoreactive afferents to the VPL thalamus in a mouse model of Niemann-Pick disease type C. Brain Res. 2004;1022:244–246. doi: 10.1016/j.brainres.2004.06.037. [DOI] [PubMed] [Google Scholar]
- Park WD, O'Brien JF, Lundquist PA, Kraft DL, Vockley CW, Karnes PS, Patterson MC, Snow K. Identification of 58 novel mutations in Niemann-Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum Mutat. 2003;22:313–325. doi: 10.1002/humu.10255. [DOI] [PubMed] [Google Scholar]
- Patel SC, Suresh S, Kumar U, Hu CY, Cooney A, Blanchette-Mackie EJ, Neufeld EB, Patel RC, Brady RO, Patel YC, Pentchev PG, Ong WY. Localization of Niemann-Pick C1 protein in astrocytes: implications for neuronal degeneration in Niemann-Pick type C disease. Proc Natl Acad Sci U S A. 1999;96:1657–1662. doi: 10.1073/pnas.96.4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz MF, Hausmann ON. Effect of 17beta-estradiol on functional outcome, release of cytokines, astrocyte reactivity and inflammatory spreading after spinal cord injury in male rats. Brain Res. 2008;1203:177–188. doi: 10.1016/j.brainres.2008.01.091. [DOI] [PubMed] [Google Scholar]
- Rocca MA, Mesaros S, Pagani E, Sormani MP, Comi G, Filippi M. Thalamic damage and long-term progression of disability in multiple sclerosis. Radiology. 2010;257:463–469. doi: 10.1148/radiol.10100326. [DOI] [PubMed] [Google Scholar]
- Rossi D, Volterra A. Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res Bull. 2009;80:224–232. doi: 10.1016/j.brainresbull.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Hawkes R. Patterned Purkinje cell death in the cerebellum. Prog Neurobiol. 2003;70:473–507. doi: 10.1016/s0301-0082(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol. 2003;456:279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- Strick PL, Dum RP, Fiez JA. Cerebellum and nonmotor function. Annu Rev Neurosci. 2009;32:413–434. doi: 10.1146/annurev.neuro.31.060407.125606. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Sugimoto Y, Ohsaki Y, Ueno M, Kato S, Kitamura Y, Hosokawa H, Davies JP, Ioannou YA, Vanier MT, Ohno K, Ninomiya H. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: a potential basis for glial cell activation in the NPC brain. J Neurosci. 2007;27:1879–1891. doi: 10.1523/JNEUROSCI.5282-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikita S, Fukuda T, Mohri I, Yagi T, Suzuki K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J Neuropathol Exp Neurol. 2004;63:660–673. doi: 10.1093/jnen/63.6.660. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Rodriguez-Lafrasse C, Rousson R, Duthel S, Harzer K, Pentchev PG, Revol A, Louisot P. Type C Niemann-Pick disease: biochemical aspects and phenotypic heterogeneity. Dev Neurosci. 1991;13:307–314. doi: 10.1159/000112178. [DOI] [PubMed] [Google Scholar]
- Wang J, Lin W, Popko B, Campbell IL. Inducible production of interferon-gamma in the developing brain causes cerebellar dysplasia with activation of the Sonic hedgehog pathway. Mol Cell Neurosci. 2004;27:489–496. doi: 10.1016/j.mcn.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Yamada A, Saji M, Ukita Y, Shinoda Y, Taniguchi M, Higaki K, Ninomiya H, Ohno K. Progressive neuronal loss in the ventral posterior lateral and medial nuclei of thalamus in Niemann-Pick disease type C mouse brain. Brain Dev. 2001;23:288–297. doi: 10.1016/s0387-7604(01)00209-1. [DOI] [PubMed] [Google Scholar]
- Yeckel MF, Berger TW. Feedforward excitation of the hippocampus by afferents from the entorhinal cortex: redefinition of the role of the trisynaptic pathway. Proc Natl Acad Sci U S A. 1990;87:5832–5836. doi: 10.1073/pnas.87.15.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Strnatka D, Donohue C, Hallows JL, Vincent I, Erickson RP. Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1-/- mice. J Neurosci Res. 2008;86:2848–2856. doi: 10.1002/jnr.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zheng T, Lee CG, Homer RJ, Elias JA. Tetracycline-controlled transcriptional regulation systems: advances and application in transgenic animal modeling. Semin Cell Dev Biol. 2002;13:121–128. doi: 10.1016/s1084-9521(02)00018-6. [DOI] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]