

Abstract
Presynaptic NMDA receptors (NMDARs) modulate release and plasticity at many glutamatergic synapses, but the specificity of their expression across synapse classes has not been examined. We found that non-postsynaptic, likely presynaptic NR2B-containing NMDARs enhanced AMPA receptor-mediated synaptic transmission at layer 4 (L4) to L2/3 (L4–L2/3) synapses in juvenile rat barrel cortex. This modulation was apparent at room temperature when presynaptic NMDARs were activated by elevation of extracellular glutamate or application of exogenous NMDAR agonists. At near physiological temperatures, modulation of transmission by presynaptic NMDARs occurred naturally, without the need for external activation. Blockade of presynaptic NMDARs depressed unitary and extracellularly evoked EPSCs at L4–L2/3 synapses, accompanied by increases in paired-pulse ratio and coefficient of variation, indicative of a decrease in presynaptic release probability. NMDAR agonists increased the frequency of miniature EPSCs in L2/3 neurons, without altering their amplitude or kinetics. Focal application of NMDAR antagonist revealed that the NMDARs that modulate L4–L2/3 transmission are located in L2/3, not L4, consistent with localization on terminals or axons of L4–L2/3 synapses, rather than on the somatodendritic compartment of presynaptic L4 neurons. In contrast, presynaptic NMDARs did not modulate L4–L4 synapses, which originate from the same presynaptic neurons as L4–L2/3 synapses, or cross-columnar L2/3–L2/3 horizontal projections, which synapse onto the same postsynaptic target neurons. Thus, presynaptic NMDARs selectively modulate L4–L2/3 synapses, relative to other synapses made by the same neurons. Existence of these receptors may support specialized processing or plasticity by L4–L2/3 synapses.
Keywords: synaptic release, cortical layer, presynaptic modulation, release probability, glutamate, barrel cortex
Introduction
Synapse efficacy, dynamics, and plasticity vary markedly across classes of excitatory synapses, greatly increasing the computing power of neural circuits (Murthy et al., 1997; Reyes et al., 1998; Tóth and McBain, 2000; Koester and Johnston, 2005). This functional heterogeneity reflects, in part, differential expression of presynaptic neurotransmitter receptors, which modulate release and plasticity. Classically, presynaptic receptors are metabotropic, including GABAB, endocannabinoid, and metabotropic glutamate receptors; the latter of which modulates release in a target-cell selective manner (Scanziani et al., 1998). Previously, functional presynaptic ionotropic receptors have also been identified (MacDermott et al., 1999; Schmitz et al., 2000; Contractor et al., 2001; Engelman and MacDermott, 2004), including presynaptic kainate receptors, which are differentially expressed at synapses onto interneurons and contribute to target-specific differences in release (Sun and Dobrunz, 2006). Whether other presynaptic ionotropic receptors are also differentially expressed and modulate function of specific synapse classes remains unknown.
NMDA receptors (NMDARs) classically exist postsynaptically at excitatory synapses, and contribute to postsynaptic depolarization and calcium influx (Dingledine et al., 1999). Presynaptic NMDARs (PreNMDARs) have been detected physiologically at many cortical synapses, including synapses onto layer 2 (L2) and L5 cells in entorhinal cortex (Berretta and Jones, 1996; Woodhall et al., 2001; Yang et al., 2006), synapses between L5 pyramidal cells in visual cortex (Sjöström et al., 2003), ascending inputs onto L2/3 pyramidal cells (presumptive L4–L2/3 synapses) in somatosensory cortex (S1) (V. A. Bender et al., 2006), and synapses onto L2/3, L4, and L5 cells in visual cortex (Corlew et al., 2007). Cortical PreNMDARs modulate transmitter release and long-term synaptic depression (LTD) (Sjöström et al., 2003; V. A. Bender et al., 2006; Corlew et al., 2007). In contrast to this abundance of synapses showing functional regulation by PreNMDARs, electron microscopy studies have typically found presynaptic NMDARs at only a small subset of cortical excitatory terminals (Aoki et al., 1994; Conti et al., 1997; Charton et al., 1999), although prevalence may be greater in early postnatal development [earlier than postnatal day 23 (P23)] (Corlew et al., 2007). Two models for PreNMDAR expression could explain this discrepancy. First, PreNMDARs may be expressed at many cortical excitatory synapse classes, but at low levels within each class. Alternatively, PreNMDARs may be selectively expressed at a specific subset of synapse classes, and be absent from other classes. If PreNMDAR expression is synapse class specific, it would indicate that neurons selectively form and traffic these receptors to govern functional properties of specific synapses.
We showed previously that non-postsynaptic NMDARs are required for LTD at presumptive L4–L2/3 synapses in S1. At room temperature, these receptors acutely regulated release probability only in the presence of elevated extracellular glutamate (during high-frequency burst firing or when glutamate transporters were partially blocked) (V. A. Bender et al., 2006). Here, we show that PreNMDARs selectively modulate L4–L2/3 synapses, but not L4–L4 synapses, or cross-columnar synapses onto the same postsynaptic L2/3 pyramidal cells. At near-physiological temperatures, this modulation occurs during standard, low-frequency transmission, without artificial elevation of glutamate. Thus, PreNMDARs are selectively localized to L4–L2/3 synapses, and are likely to regulate synapse function during normal, low-frequency synaptic transmission.
Materials and Methods
All procedures were approved by the University of California, San Diego, Institutional Animal Care and Use Committee. Long–Evans rats (P14–P22, either sex; Harlan Sprague Dawley, Indianapolis, IN) were anesthetized with isoflurane and decapitated, and the brain was rapidly removed in ice-cold oxygenated Ringer's solution [containing (in mm) 119 NaCl, 26 NaHCO3, 11 d-(+)-glucose, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, and 2.5 CaCl2, pH 7.20–7.23, osmolarity 292 mOsm]. Acute brain slices (350–400 μm) containing the posteromedial barrel subfield of S1 were cut on a vibrating microtome (VT1000S; Leica, Nussloch, Germany) at an oblique angle, 50° toward coronal from the midsagittal plane (Allen et al., 2003). In some experiments, the rostral pole of the brain was tilted 10° up during cutting (Feldmeyer et al., 2002), which increased the rate of finding synaptically connected L4–L2/3 neuron pairs (from 2.2 to 4.3%). After sectioning, slices were preincubated in Ringer's solution at 30°C for 30 min, and then incubated at room temperature (22–24°C) until use (0.5–8.5 h). S1 was identified by the presence of three to five large (250–450 μm) barrels in L4, visible under transillumination (Feldman, 2000). All recordings were made at room temperature, except for Figure 8, for which data were recorded at 30–32°C using a feedback-controlled automatic heater (model TC344B; Warner Instruments, Hamden, CT).
Figure 8.

PreNMDARs are active, but not saturated, at 30–32°C in the absence of TBOA. A1, Single example of depression of extracellularly evoked L4–L2/3 synaptic responses by 50 μm d-APV at 30–32°C, without TBOA or exogenous NMDAR agonist. Focal BMI and iMK-801 were included to isolate AMPA-EPSCs. The first EPSC amplitude is shown throughout the recording. Inset, Pairs of AMPA-EPSCs (30 Hz) during baseline (black), d-APV application (gray), and after d-APV washout (dashed black). Each trace is mean of the last 10 sweeps in each condition. A2, Mean effect of d-APV on amplitude of first AMPA-EPSC at L4–L2/3 synapses (n = 7; filled symbols) and at extracellularly evoked L2/3–L2/3 cross-columnar synapses (n = 7; open symbols). Bars are SEM. A3, Effects of d-APV on PPR for cells in (A2). B1, d-APV (50 μm) decreased mEPSC frequency in L2/3 pyramidal cells at 30–32°C. B2, Cumulative probability histogram for inter-mEPSC interval during baseline (solid black) and d-APV (gray; p < 0.01, Kolmogorov–Smirnov test). Inset, Mean mEPSC before (black) and during (gray) d-APV application. C1, Effects of HQA (20 μm) and subsequent APV (50 μm) on mEPSC frequency in L2/3 pyramidal cells. C2, Cumulative probability histogram for inter-mEPSC interval during baseline (solid black), HQA (gray), and HQA plus APV (dashed black); (p < 0.01, Kolmogorov–Smirnov test for all pairwise comparisons). Inset, Mean mEPSC during baseline (black), HQA (gray), and HQA plus APV (dashed black). D1, HQA (20 μm) and HQA plus d-APV (50 μm) do not affect mEPSC frequency in L4. D2, Cumulative probability histogram for inter-mEPSC interval during baseline (solid black), HQA (gray), and HQA plus APV (dashed black); (p > 0.05, Kolmogorov–Smirnov test for all pairwise comparisons). Inset, Mean mEPSC during baseline (black), HQA (gray), and HQA plus APV (dashed black). *p < 0.05; **p < 0.01.
Whole-cell recording
Whole-cell recordings were made with 3–8 MΩ pipettes using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Recordings were filtered at 2 kHz and digitized at 5 kHz using a 12 bit data acquisition board (National Instruments, Austin, TX) and custom data acquisition and analysis routines running in Igor (Wavemetrics, Lake Oswego, OR). Neurons with pyramidal shaped somata were selected for recording using infrared differential interference contrast optics. A glass pipette (3–5 μm tip diameter) containing 5 mm bicuculline methiodide (BMI; Sigma, St. Louis, MO) in Ringer's solution was placed in L2/3 within 100 μm of the recording electrode to block GABAA receptors focally (Castro-Alamancos et al., 1995; Feldman, 2000). For extracellular stimulation of the L4–L2/3 projection, a concentric bipolar stimulating electrode (FHC, Bowdoinham, ME) was placed at the base of an L4 barrel of the same column as the recorded pyramidal neuron. For extracellular stimulation of the horizontal L2/3–L2/3 projection, an identical stimulating electrode was placed in L2/3 of the neighboring column centered above the neighboring barrel and at the same subpial depth as the recorded neuron.
For voltage-clamp experiments, the internal solution contained (in mm) 108 d-gluconic acid, 108 CsOH, 20 HEPES, 5 tetraethylammonium Cl, 2.8 NaCl, 0.4 EGTA, 0.3 GTP, 4 ATP, and 10 phosphocreatine, adjusted to pH 7.2 with CsOH (290 mOsm). Membrane potential of L2/3 pyramidal cells (measured immediately after break-in) was −82 ± 3.4 mV (SD; n = 28 cells). Membrane potentials and holding potentials were corrected for the measured liquid junction potential of −12 mV. Initial holding current to achieve a holding potential of −90 mV was −48 ± 34 pA (SD) and increased by an average of −29 ± 57 pA (SD) during 50 min of recording. Series resistance and input resistance were calculated from the response to a 50–150 ms long −5 mV step at the end of each sweep (Feldman, 2000). The mean series resistance was 15 ± 4 MΩ (SD) and input resistance was 333 ± 176 MΩ (SD), calculated using the method described previously (Isaac et al., 1995). After 50 min of recording, holding current had changed by −29 ± 57 pA (SD), series resistance had changed by 0.2 ± 3.9 MΩ (SD), and input resistance had changed by −33 ± 97 MΩ (SD). Cells were excluded if holding current increased by more that −200 pA, if input resistance changed by >200 MΩ, or if series resistance changed by >6 MΩ.
EPSCs were evoked by extracellular stimulation using a 200-μs-long pulse through an ISO-flex stimulus isolation unit (A.M.P.I., Jerusalem, Israel). Stimulus intensity was 5.17 ± 2.59 μA (SD; range, 2.1–10.8 μA). Pairs of stimuli (30 Hz) were delivered every 30 s. Responses were either single-component EPSCs or multicomponent EPSCs with well isolated initial components in both EPSCs (in which case only the first component was analyzed). The amplitude of each EPSC was measured relative to a 2 ms long baseline period starting 3 ms before stimulation. The amplitude of the first EPSC was 151 ± 88 pA (SD).
All experiments were performed by recording isolated AMPA receptor-mediated EPSCs (AMPA-EPSCs), except where noted in Results. AMPA-EPSCs were isolated by holding the postsynaptic cell at −90 mV to block voltage-dependent postsynaptic NMDARs. In addition, postsynaptic NMDARs were blocked pharmacologically in most experiments by loading the postsynaptic cell internally with the NMDA channel blocker (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801; 1 mm) via the recording pipette (Berretta and Jones, 1996; Woodhall et al., 2001; Humeau et al., 2003; Mameli et al., 2005; Samson and Pare, 2005; V. A. Bender et al., 2006; Corlew et al., 2007), which selectively blocks postsynaptic NMDA currents without affecting NMDA currents in neighboring cells (V. A. Bender et al., 2006). No attempt was made to block kainate receptor-mediated EPSCs, which may account for a small component of the primarily AMPA-EPSCs that were studied.
Drugs
Ifenprodil, d-2-amino-5-phosphonovaleric acid (d-APV), tetrodotoxin citrate (TTX), 6,7-dinitroquinoxaline-2,3-dione (DNQX), homoquinolinic acid (HQA), dl-threo-β-benzyloxyaspartic acid (TBOA), and picrotoxin (all from Tocris Bioscience, Ellisville, MO) were dissolved in Ringer's and stored as stock solutions (100× to 1000×) at −20°C. The concentration of TBOA used here would be predicted to block most activity of presynaptic and postsynaptic neuronal excitatory amino acid transporters (EAATs), but only affect a subset of glial EAATs (Shigeri et al., 2004), as such, it should increase the concentration of ambient glutamate, but presumably less than the ∼10-fold increase that has been observed for application of 200 μm TBOA (Cavelier and Attwell, 2005). MK-801 (1 mm; Tocris Bioscience) and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA; 5 mm, Sigma) were dissolved directly into the internal solution.
Focal d-APV puffing
For experiments in which d-APV was applied via focal puffing in L2/3, a glass pipette (3–5 μm tip diameter) containing 2.5 mm d-APV dissolved in Ringer's solution was positioned near the recorded neuron, at a distance of 106 ± 28 μm (SD; range, 70–160 μm). For focal puffing of d-APV in L4, the puffer pipette was positioned in the center of the L4 barrel ∼100 μm apical to the L4 stimulating electrode. Sweeps were collected at a 30 s intersweep interval and d-APV was puffed 15 s before each sweep by applying a 20 ms burst pressure (5 psi, compressed nitrogen) using a PV830 Pneumatic PicoPump (World Precision Instruments, Sarasota, FL).
Synaptically connected pairs
L4–L2/3 pairs.
For synaptically coupled L4 to L2/3 pairs, we made a whole-cell voltage-clamp recording from a postsynaptic L2/3 pyramidal cell using Cs+-internal-containing MK-801 (iMK-801). A 6–8 MΩ glass recording electrode containing (in mm) 116 potassium gluconate, 20 HEPES, 6 KCl, 2 NaCl, 0.5 EGTA, 0.3 GTP, 4 ATP, and 10 phosphocreatine, adjusted to pH 7.2 with KOH (290 mOsm), was then positioned just over the surface of the slice in L4. The potassium-based internal solution was puffed manually at random points throughout the L4 barrel while whole-cell currents in the L2/3 cell were visually monitored using an oscilloscope. “Hot spots” of connectivity were identified by a dramatic increase in synaptic current frequency in the L2/3 cell.
Once a hot spot was found, a whole-cell recording was established with a regular spiking L4 neuron. In 3/119 cases, this cell was synaptically connected to the L2/3 neuron. In the remaining cases, the electrode was withdrawn from the cell and reused to form a loose seal (< 1 GΩ) on another L4 spiny neuron which was stimulated in cell-attached mode as described previously (Feldmeyer et al., 1999, 2002). Briefly, the presynaptic loose patch was hyperpolarized with −700 pA (in current clamp, the resulting potential was approximately −50 mV, indicating a seal resistance of 70–100 MΩ) and a 10 ms current pulse (1–6 nA) was applied to evoke one or more action potentials, which were usually visible as small deflections in the voltage trace. If the L4 neuron elicited an EPSC, indicating a connected pair, the loose seal electrode was withdrawn and the cell repatched in whole-cell mode with a new electrode. Once a connected L4–L2/3 pair was found, presynaptic action potentials were evoked by 10 ms presynaptic current injection to evoke unitary AMPA-EPSCs (AMPA-uEPSCs). Pairs of AMPA-uEPSCs were evoked at 30 Hz with 30 s between sweeps.
Connectivity rate.
One hundred and thirty-two postsynaptic L2/3 pyramidal cells were recorded. In 119 neurons, an L4 hot spot was identified in which 968 potential presynaptic L4 cells were tested for connectivity. Of L4 cells within hot spots, 5.9% appeared connected during loose-patch stimulation; however, only 59.6% (n = 28) of those that survived repatching were found to be actually connected with the postsynaptic neuron. Thus, we estimate the overall connectivity rate within a hot spot to be 3.6% (5.9% apparent connectivity during loose-patch stimulation × 59.6% of apparently connected cells actually connected). This connectivity rate may underestimate true connectivity because it assumes that every cell which did not appear connected by loose-patch stimulation was not actually connected. An additional 117 L4 cells outside of hot spots were tested for connectivity, but no pairs were found, suggesting that L4 cells projecting to a single L2/3 target neuron exist in clusters; such clusters have been observed for intralaminar connections in L5 of visual cortex (Song et al., 2005) and in L4 and L2/3 of S1 (Feldmeyer et al., 1999, 2006) and for L4–L2/3 connections in S1 (Feldmeyer et al., 2002). Of 28 connected pairs, 17 were discarded because one cell was lost before the experiment was completed or because of significant run-down in baseline transmission.
L4–L4 pairs.
Sixty-eight simultaneous recordings were made between adjacent L4 excitatory cells. Twelve were synaptically coupled [connectivity rate, 18%; consistent with the data of Feldmeyer et al. (1999)], of which five were discarded because one cell died before completion of the experiment. Ten coupled cell pairs were tested for reciprocal connectivity, of which one was found to be reciprocally connected; in that pair, the experiment was performed on the stronger connection. All six pairs that were tested for sensitivity to 1 μm DNQX showed significant blockade of the unitary EPSC (mean blockade, 51 ± 6%), confirming that these were excitatory connections.
For all L4 cells included in the study, initial membrane potential (V m) was −84 ± 7 mV (SD) and input resistance was 299 ± 96 MΩ (SD). For L4 current-clamp recordings, series resistance was measured by manual compensation using the whole-cell amplifier; the initial series resistance was 18.8 ± 7.7 MΩ (SD), and when measured again at the end of the experiment (35–55 min later) it had increased by 0.8 ± 1.8 MΩ (SD). No difference was found in initial V m, V m stability, input resistance, or input resistance stability, between L4 neurons in the L4–L2/3 pairs experiment (which were patched after searching for hot spots and loose patching) and in the L4–L4 pairs experiment (which were patched directly). Thus, identification of hot spots and loose patch stimulation did not appear to compromise cell health.
Miniature EPSCs
Spontaneous AMPA-mediated miniature EPSCs (mEPSCs) were recorded using the whole-cell voltage-clamp methods described above, except that 500 nm TTX and 100 μm picrotoxin were included in the Ringer's solution, and focal BMI was omitted. All mEPSCs were measured using iMK-801. mEPSC frequency, amplitude, and other parameters were analyzed off-line using MiniAnalysis software (Synaptosoft, Decatur, GA) with a mEPSC threshold set at 4.5 pA (∼2.5 times root-mean-square noise). Automated detection of mEPSCs was verified by visual inspection, with the experimenter blind to the experimental condition during analysis. Input and series resistance were monitored by applying −5 mV holding potential steps (3–5 pulses at 0.14 Hz every 5 min during recording). mEPSCs were recorded during an initial 10 min baseline period, followed by application of NMDAR agonists or antagonists for 10 min each. Only data from the last 5 min of each 10 min recording epoch was analyzed to ensure that drugs had fully equilibrated.
Average mEPSC shape was determined from individual mEPSCs aligned to the most positive sample point before mEPSC onset (Synaptosoft). This alignment method produced an apparent upward deflection before the average mEPSC waveform (see Fig. 2 B, inset), and also tended to temporally align any rhythmic noise present in the recording (as apparent in the tail of the average mEPSC in Fig. 2 B).
Figure 2.

Activation of PreNMDARs increases mEPSC frequency in L2/3 pyramidal neurons. A, Representative mEPSCs from a L2/3 pyramidal neuron during baseline (top), 15 μm NMDA application (middle), and 15 μm NMDA plus 50 μm d-APV application (bottom). Recordings were made in 500 nm TTX, 100 μm picrotoxin, and iMK-801. B, Cumulative probability histogram for inter-mEPSC interval during baseline (solid black), NMDA (gray), and NMDA plus APV (dashed black) conditions. The histogram is a mean of individual histograms from six cells (p < 0.01 for baseline vs NMDA and NMDA vs NMDA plus APV; p > 0.05 for baseline vs NMDA plus APV, Kolmogorov–Smirnov test). Inset, Average mEPSC waveform across the population (black, baseline; gray, NMDA; dashed, NMDA plus APV). C, Mean mEPSC frequency for each cell. Error bars represent the population mean. **p < 0.01. D, Mean mEPSC amplitude for each cell.
Statistics
All comparisons were made by using a two-tailed paired Student's t test (for single comparisons) or a repeated-measures ANOVA (for multiple comparisons), unless otherwise noted. The critical level of significance was p < 0.05. All data are presented as mean ± SE, unless noted as SD.
Results
PreNMDARs regulate evoked release at L4–L2/3 synapses
To study L4–L2/3 excitatory synaptic responses, we made whole-cell recordings from single L2/3 pyramidal neurons in acute slices of S1 cortex from P14–P22 Long–Evans rats, while stimulating with just suprathreshold extracellular stimulation in the underlying L4 barrel. In a previous study, we showed that bath application of d-APV (50 μm) reduced the amplitude of EPSCs recorded at −90 mV, and increased paired-pulse ratio (PPR), suggesting that NMDARs modulate evoked presynaptic release at L4–L2/3 synapses (V. A. Bender et al., 2006). This effect was only observed during high-frequency presynaptic bursts, or when EAATs were partially blocked with a subsaturating concentration of TBOA (25 μm), suggesting that an increase in glutamate concentration, caused transporter blockade (Cavelier and Attwell, 2005; Lien et al., 2006) or presynaptic bursting, was required to activate the relevant NMDARs (V. A. Bender et al., 2006).
To confirm that the NMDARs that modulate release at L4–L2/3 synapses were non-postsynaptic, we recorded isolated AMPA-EPSCs with internal iMK-801 in the postsynaptic cell (1 mm) to block postsynaptic NMDA receptors (see Materials and Methods). Cells were held at −90 mV and recordings were made at room temperature. TBOA (15 μm) was applied to elevate extracellular glutamate (Cavelier and Attwell, 2005), and BMI was applied locally to block GABAA-mediated inhibition (Castro-Alamancos et al., 1995; Feldman, 2000). Two pulses at 30 Hz were applied every 30 s. We asked whether d-APV reduced AMPA-EPSCs and/or changed PPR under these conditions, when postsynaptic NMDA currents were already blocked with iMK-801. In an example cell (Fig. 1 A), bath application of 50 μm d-APV caused a decrease in the amplitude of evoked AMPA currents and an increase in PPR, without coincident changes in holding current, series resistance, or input resistance. This decrease in AMPA-EPSC and increase in PPR is consistent with a decrease in the probability of evoked vesicle release per presynaptic action potential (Dobrunz and Stevens, 1997; Zucker and Regehr, 2002; Sun et al., 2005). Both of these effects recovered with washout of d-APV.
Figure 1.

Non-postsynaptic NMDARs modulate synaptic transmission at L4–L2/3 synapses in S1. A, Representative recording showing that bath application of d-APV reduces the amplitude of AMPA-EPSCs at presumptive L4–L2/3 synapses activated by extracellular L4 stimulation. This recording was made in 15 μm TBOA, focal BMI, and internal MK-801 to block postsynaptic NMDA receptors. Top, Amplitude of the first AMPA-EPSC throughout the recording for this cell. Inset, Pair of AMPA-EPSCs (30 Hz) during baseline (black), d-APV application (gray) and after d-APV washout (dashed black). Each trace in this and other figures is the mean of the last 10 sweeps in each condition. Bottom, Holding current, series resistance, and input resistance. B, Mean effect of d-APV on amplitude of first AMPA-EPSC for five cells. Error bars indicate SEM. d-APV (50 μm) was applied for 14 min. C, Mean effect of 30 min d-APV application on first AMPA-EPSC amplitude (50 μm d-APV, n = 5 cells, filled squares; control experiments, n = 5 cells, open squares). D, Effect of d-APV on PPR for 14 min (filled diamonds) and 30 min (filled squares) applications. Open symbols, PPR for corresponding time points during control experiments in which d-APV was not applied. **p < 0.01; *p < 0.05.
Across the population of neurons, d-APV application for 14 min (n = 5 cells) reversibly decreased AMPA-EPSCs and increased PPR (Fig. 1 B,D, filled diamonds). Baseline PPR was slightly, but not significantly, <1.0 (PPR, 0.920 ± 0.056; n = 15; p = 0.17, t test), consistent with previous studies of extracellular stimulation at this projection (K. J. Bender et al., 2006). Longer duration d-APV application (30 min) (n = 5 cells) produced a similar magnitude effect, indicating that 14 min d-APV exposure was sufficient to produce the maximal effect (Fig. 1 C,D, filled circles). Interleaved control experiments confirmed that the presence of TBOA itself, without d-APV application, did not cause a decrease in AMPA-EPSC amplitude or a change in PPR (n = 5) (Fig. 1 C,D, open circles). These data indicate that in the presence of TBOA (which increases extracellular glutamate concentration), blockade of non-postsynaptic, putatively presynaptic NMDARs decreases release probability.
PreNMDARs regulate mEPSC frequency
To test whether PreNMDARs modulate spontaneous transmitter release, we tested whether activation of PreNMDARs altered the frequency, but not amplitude or kinetics, of AMPA-mediated mEPSCs. mEPSCs were measured at −90 mV, at room temperature, from L2/3 pyramidal neurons in TTX (500 nm) and picrotoxin (100 μm), with iMK-801 (1 mm) to block postsynaptic NMDARs (no TBOA was present in mEPSC experiments). mEPSCs were recorded under baseline conditions (10 min), after which 15 μm NMDA was bath applied to activate NMDARs (10 min), and then d-APV was added to antagonize the effect (10 min) (Fig. 2 A). NMDA caused a small but significant increase in mean mEPSC frequency (Fig. 2 C), which was evident as a leftward shift in the cumulative probability histogram of inter-mEPSC interval compiled across six cells (Fig. 2 B) (25,291 total individual mEPSCs). Addition of d-APV returned mEPSC frequency to baseline levels (Fig. 2 C) (baseline, 6.6 ± 0.9 Hz; NMDA, 7.6 ± 1.0 Hz; NMDA plus APV, 6.4 ± 0.9 Hz; p < 0.0001, repeated-measures ANOVA; post hoc pairwise-comparison p values in Fig. 2). In contrast, NMDA and d-APV did not alter mEPSC amplitude (Fig. 2 D) (baseline, 9.5 ± 0.4 pA; NMDA, 9.7 ± 0.2; NMDA plus APV, 9.4 ± 0.2; p = 0.58). NMDA and d-APV also did not alter mEPSC kinetics, consistent with the interpretation that mEPSCs at −90 mV represent AMPA, but not NMDA currents (Fig. 2 B, inset) (rise time: baseline, 2.1 ± 0.1 ms; NMDA, 2.2 ± 0.2 ms; NMDA plus APV, 2.2 ± 0.2 ms; p = 0.34; decay time: baseline, 4.9 ± 0.4 ms; NMDA, 5.2 ± 0.5 ms; NMDA plus APV, 5.2 ± 0.6 ms; p = 0.24). These data indicate that activation of PreNMDARs by exogenous agonist modestly, but significantly, increases spontaneous release probability.
To test whether PreNMDARs actively promote release under normal baseline conditions in the slice, we applied d-APV without NMDA in 6 cells, also at room temperature. Under these conditions, d-APV had no effect on mEPSC frequency (baseline, 5.90 ± 1.22 Hz; d-APV, 5.89 ± 1.23 Hz; p = 0.96) or amplitude (baseline, 8.8 ± 0.3 pA; d-APV, 8.7 ± 0.3 pA; p = 0.56). This indicates that, at least at room temperature, PreNMDARs promote spontaneous release only under conditions of exogenous NMDAR agonist. Taken with the results from Figure 1 (with TBOA) this confirms that, at room temperature, PreNMDAR activation depends on enhanced extracellular glutamate.
Localization of PreNMDARs on L4–L2/3 terminals
Presynaptic ionotropic receptors could influence release by being located on the presynaptic terminal [e.g., by locally influencing membrane potential (Turecek and Trussell 2001, 2002; Trussell 2002), or directly supplying calcium for release (Chen et al., 2000)]. Alternatively, they could be located on the presynaptic somatodendritic compartment, and generate somatic depolarization that spreads down the axon to influence release, as has been shown to occur with subthreshold depolarization at some terminals (Alle and Geiger, 2006; Shu et al., 2006). To determine the location of the PreNMDARs that modulate evoked release at L4–L2/3 synapses, we focally puffed d-APV (2.5 mm via a picospritzer pipette) in L2/3. We first verified that the focal d-APV puffing protocol blocked local NMDARs on neural elements in L2/3, but not in L4, by recording pharmacologically isolated postsynaptic NMDAR currents at +30 mV from L2/3 and L4 neurons (in 10 μm DNQX and 100 μm picrotoxin, with 5 mm internal BAPTA). Focal d-APV puffing in L2/3 blocked 77.4 ± 5.4% (n = 4) of synaptically evoked, postsynaptic NMDA currents recorded in L2/3 neurons 103 ± 30 μm (SD) away from the puffer pipette, but only 16.7 ± 2.6% (n = 4) of synaptically evoked NMDA currents recorded in L4 neurons in the underlying barrel (Fig. 3 A, insets). Thus, this protocol blocked NMDARs located in L2/3 (presumably including the presynaptic axons and terminals of L4 cells that extended into L2/3), relatively selectively compared with L4.
Figure 3.
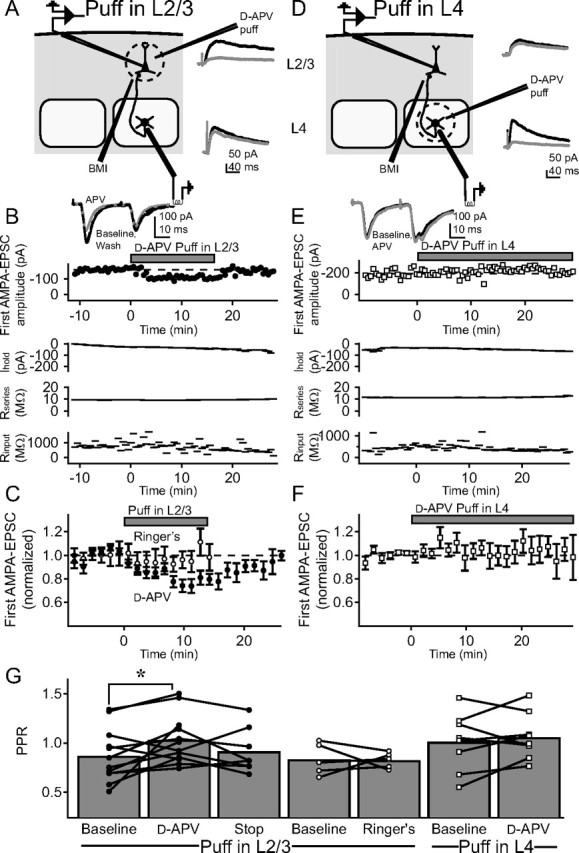
Focal application of d-APV reveals that PreNMDARs at the L4–L2/3 projection are located in L2/3, not in L4. A–F, AMPA-EPSCs were measured in L2/3 pyramidal cells in response to stimulation in L4, while d-APV was focally applied via a puffer pipette in either L2/3 (A–C), or in L4 (D–F). A, Experiment schematic for experiments in which d-APV was focally applied via a puffer pipette in L2/3. Inset, Postsynaptic NMDA-EPSCs measured from a representative L2/3 neuron (top trace pair) and L4 neuron (bottom trace pair) before (black) and during (gray) puffer application of 2.5 mm d-APV in L2/3. Each trace shown is the average of the last 10 sweeps of each condition. Focal d-APV application blocked NMDA currents strongly in L2/3, but only modestly in L4. B, Example cell showing the effect of focal puffing of d-APV in L2/3 on AMPA-EPSCs on the L4–L2/3 projection. Top, Amplitude of the first of a pair of AMPA-EPSCs. Inset, Pairs of AMPA-EPSCs during baseline (black), d-APV puffing (gray), and after recovery (black, dashed). Each trace shown is the average of the last 10 sweeps of each condition. Bottom, Holding current, series resistance, and input resistance for this recording. C, Mean effect of d-APV puffing in L2/3 on AMPA-EPSCs (n = 12 cells, filled circles), and effect of puffing Ringer's solution in L2/3 instead of d-APV (n = 5, open circles). D–F, Equivalent experiments on nine cells in which d-APV was puffed in L4, rather than in L2/3. G, Effect on PPR of focal puffing of DAPV in L2/3 (filled circles), Ringer's in L2/3 (open circles), and d-APV in L4 (open squares). Error bars represent population means. *p < 0.05.
Next, we tested whether focal d-APV puffing in L2/3 modulated L4–L2/3 AMPA-EPSCs. We stimulated in L4 while recording AMPA-EPSCs in 15 μm TBOA, focal BMI, and iMK-801. After a stable baseline in which two AMPA-EPSCs were evoked at 30 Hz every 30 s, 2.5 mm d-APV was puffed focally in L2/3, 108 ± 29 μm (SD) away from the postsynaptic cell. One puff was delivered 15 s before each sweep (Fig. 3 A). Figure 3 B shows an example cell in which this protocol caused a rapid decrease in AMPA-EPSC amplitude and an increase in PPR, which recovered after puffing was stopped. This effect was consistent across a population of 12 cells (Fig. 3 C,G, filled circles). In interleaved control experiments (n = 5 cells), we focally puffed Ringer's solution using the same protocol, and observed no change in evoked EPSC amplitude or PPR (Fig. 3 C,G, open circles).
To ensure that this effect was not caused by the modest blockade of L4 NMDARs produced by d-APV puffing in L2/3, we performed the converse experiment, focally puffing d-APV in the center of the L4 barrel (∼100 μm apical to the site of stimulation), to block NMDARs in the somatodendritic compartment of L4 neurons (Fig. 3 D). d-APV puffing in L4 blocked 80.6 ± 1.2% (n = 4) of pharmacologically isolated postsynaptic NMDA currents in local L4 neurons, but only 20.5 ± 6.8% (n = 3) of postsynaptic NMDA currents in overlying L2/3 neurons (Fig. 3 D, insets). Thus, puffing in L4 preferentially blocked NMDARs located in L4. d-APV puffing in L4 did not affect AMPA-EPSCs at L4–L2/3 synapses (n = 10 cells) (Fig. 3 E,F), and did not change PPR (Fig. 3 G, open squares). Together, these results indicate that d-APV modulates release by acting on non-postsynaptic NMDARs that are located in L2/3, rather than in L4, consistent with localization on L4 axons or terminals in L2/3, rather than on L4 cell bodies or dendrites.
PreNMDARs contain NR2B subunits
NMDARs are heterotetrameric channels made up of two NMDA receptor 1 (NR1) subunits and two NR2 subunits (Dingledine et al., 1999). Of the four types of NR2 subunits, cortical neurons preferentially express NR2A and/or NR2B (Monyer et al., 1994). In cortex, postsynaptic NMDARs undergo an early postnatal developmental switch from NR2B-containing to NR2A-containing receptors (Monyer et al., 1994; Flint et al., 1997; Kew et al., 1998; Stocca and Vicini, 1998; Tovar and Westbrook, 1999; Liu et al., 2004), whereas PreNMDARs are thought to preferentially retain NR2B subunits (Woodhall et al., 2001; Sjöström et al., 2003; Yang et al., 2006). To determine the subunit composition of PreNMDARs at L4–L2/3 synapses, we applied the antagonist ifenprodil, which blocks NR2B-containing NMDARs (Williams, 1993). We first tested whether NR2B receptors were largely absent from postsynaptic NMDARs, as expected by this age (Flint et al., 1997), by measuring L4-evoked postsynaptic NMDA currents in L2/3 pyramidal cells (+30 mV holding potential, in 10 μm DNQX and 100 μm picrotoxin, 5 mm internal BAPTA), and applying increasing concentrations of ifenprodil. Postsynaptic NMDA currents were almost completely insensitive to 3 μm ifenprodil (4.3 ± 0.1% block, n = 3), but were progressively blocked by higher ifenprodil concentrations, consistent with the weak affinity of ifenprodil for postsynaptic NR2A-containing NMDARs (Fig. 4 A) (Williams, 1993).
Figure 4.

PreNMDARs preferentially contain NR2B subunits. A, Blockade of pharmacologically isolated postsynaptic NMDA-mediated EPSCs by increasing ifenprodil concentration. Inset, Representative traces from a single cell during baseline (solid black), 3 μm (solid gray), 4 μm (dashed black), and 6 μm ifenprodil (dashed gray). B, Effect of 3 μm ifenprodil on amplitude of the first EPSC for a representative cell. Inset, Pairs of EPSCs before (black) and during (gray) 3 μm ifenprodil. Each trace shown is the average of the last 10 sweeps of each condition. C, Group data for cells with 3 μm ifenprodil application (n = 10, filled circles) and for control experiments without ifenprodil (n = 8; open circles). D, Change in PPR for 3 μm ifenprodil application (filled circles) and control (open circles). Error bars represent population means. **p < 0.01. E, Representative mEPSCs from a single L2/3 pyramidal neuron during baseline, 20 μm HQA application, and 20 μm HQA plus 3 μm ifenprodil. F, Mean cumulative probability histogram for inter-mEPSC interval during baseline (solid black), HQA (gray), and HQA plus ifenprodil (dashed black; p < 0.01 for baseline vs HQA and for HQA vs HQA plus ifenprodil; p > 0.05 for baseline vs HQA plus ifenprodil, Kolmogorov–Smirnov test). Inset, Mean frequency for each cell during each condition. Error bars represent population mean. *p < 0.05; **p < 0.01.
To test whether NR2B-containing receptors regulated release at L4–L2/3 synapses, we applied 3 μm ifenprodil while measuring L4–L2/3 EPSCs recorded at −90 mV in the presence of 15 μm TBOA and focal BMI. iMK-801 was not present in these experiments. Pairs of EPSCs (30 Hz) were evoked every 30 s. After a stable baseline period, 3 μm ifenprodil was applied. Ifenprodil caused a decrease in EPSC amplitude and an increase in PPR, indicating a decrease in release probability (Fig. 4 B–D, filled symbols). Interleaved control experiments confirmed that no change in EPSC amplitude or PPR was observed without ifenprodil (Fig. 4 C,D, open symbols).
As an additional test of the subunit composition of PreNMDARs, we measured the effect of the NR2A- and NR2B-preferring NMDAR agonist HQA (20 μm) on mEPSC frequency (de Carvalho et al., 1996; Woodhall et al., 2001). mEPSCs were measured (500 nm TTX, 100 μm picrotoxin and 1 mm iMK-801) during a baseline period (10 min), after addition of HQA (20 μm, 10 min), and after subsequent addition of ifenprodil (3 μm, 10 min, Fig. 4 E). HQA caused an increase in mEPSC frequency, which was reversed by ifenprodil (Fig. 4 F) (baseline, 7.4 ± 0.9 Hz; HQA, 8.7 ± 1.1 Hz; HQA plus ifenprodil, 7.3 ± 1.0 Hz; p < 0.0001). There was no effect on mEPSC amplitude (baseline, 9.1 ± 0.4 pA; HQA, 8.8 ± 0.3 pA; HQA plus ifenprodil, 9.0 ± 0.4 pA; p = 0.68), rise time (baseline, 1.93 ± 0.06 ms; HQA, 2.04 ± 0.04 ms; HQA plus ifenprodil, 2.06 ± 0.09 ms; p = 0.16), or decay time (baseline, 5.0 ± 0.3 ms; HQA, 4.8 ± 0.3 ms; HQA plus ifenprodil, 4.9 ± 0.3 ms; p = 0.68). Thus, these results are consistent with the hypothesis that the PreNMDARs that modulate spontaneous release onto L2/3 pyramidal cells preferentially contain NR2B subunits, relative to synaptically activated postsynaptic receptors, which lack NR2B. Presynaptic receptors may be NR1/NR2B or NR1/NR2A/NR2B, both of which are blocked by ifenprodil (Neyton and Paoletti, 2006). Involvement of NR2C and 2D could not be assessed because of inavailability of selective antagonists.
Synapse specificity of PreNMDARs
We examined the presence of functional PreNMDARs with respect to two types of synapse specificity: first, input specificity, in which PreNMDARs are not localized on all excitatory synaptic inputs to a neuron, but are instead localized to one class of inputs; and second, target specificity, in which PreNMDARs are not expressed on all axonal terminals of a neuron, but only those terminals that contact a specific target neuron class.
Input specificity
We first tested whether PreNMDARs are selectively expressed at L4–L2/3 inputs onto L2/3 pyramidal cells, relative to other excitatory inputs onto these same cells, by testing for functional PreNMDARs at horizontal, cross-columnar L2/3 inputs onto L2/3 neurons. We recorded AMPA-EPSCs from a single L2/3 pyramidal cell in one barrel column in 15 μm TBOA and iMK-801, with focal BMI. We placed one extracellular stimulating electrode in L4 of the same column to activate the L4–L2/3 pathway (as above), and another in L2/3 of a neighboring column to activate the cross-columnar L2/3–L2/3 pathway (Fig. 5 B). We alternately stimulated each pathway with two pulses (30 Hz), with 15 s between pathways, and tested whether bath-applied d-APV (50 μm) modulated release on either pathway. In an example cell, d-APV decreased AMPA-EPSC amplitude and increased PPR on the L4–L2/3 pathway, but caused no change in either AMPA-EPSC amplitude or PPR on the simultaneously recorded L2/3 cross-columnar pathway (Fig. 5 A). Similar results were obtained across eight cells (Fig. 5 C,D). Thus, PreNMDARs functionally regulate L4 inputs onto L2/3 pyramidal cells, but not cross-columnar L2/3 inputs onto L2/3 pyramidal cells, indicating that the presence of PreNMDARs depends on the origin of presynaptic input.
Figure 5.

PreNMDARs are located on the L4–L2/3 projection but not on other cortical inputs to L2/3 pyramidal cells. A, Representative experiment testing the effect of d-APV on AMPA-EPSCs evoked on the L2/3 cross-columnar pathway (L2/3–L2/3) and on L4–L2/3 inputs to the same postsynaptic cell. Responses to the two pathways were measured in alternation. Top plots, Amplitude of the first AMPA-EPSC on each pathway. Insets, Pairs of AMPA-EPSCs before (black) and during (gray) d-APV application. Each trace shown is the average of the last 10 sweeps of each condition. Bottom plots, Holding current, series resistance, and input resistance for this cell. B, Recording set-up for these experiments. C, Mean effect of d-APV on first AMPA-EPSC amplitude for L2/3–L2/3 inputs (triangles) and simultaneously measured L4–L2/3 inputs (circles) (n = 8 cells). D, PPR changes in this experiment. Error bars represent population means. *p < 0.05.
Target specificity
To test whether the presence of functional PreNMDARs is dependent on the postsynaptic cell that different axon collaterals target, we asked whether other synapses made by L4 neurons, in addition to L4–L2/3 synapses, also express functional PreNMDARs. In addition to projecting to L2/3, L4 excitatory neurons are extensively interconnected in their home barrels (Feldmeyer et al., 1999; Petersen and Sakmann, 2000). We therefore tested whether unitary connections between L4 neurons are regulated by PreNMDARs (Fig. 6 A). We recorded uEPSCs at −90 mV from 12 synaptically connected L4–L4 pairs in the presence of 15 μm TBOA. Seven pairs survived long enough to assess the effects of d-APV application. Two presynaptic action potentials (30 Hz) were evoked every 30 s. After a stable baseline, 50 μm d-APV was applied. d-APV application caused no change in the amplitude of the first uEPSC or PPR as seen in a single, representative cell (Fig. 6 B) and across a population of seven cells (Fig. 6 C,D). These results indicate that PreNMDARs functionally regulate L4–L2/3 synapses, but not L4–L4 synapses.
Figure 6.
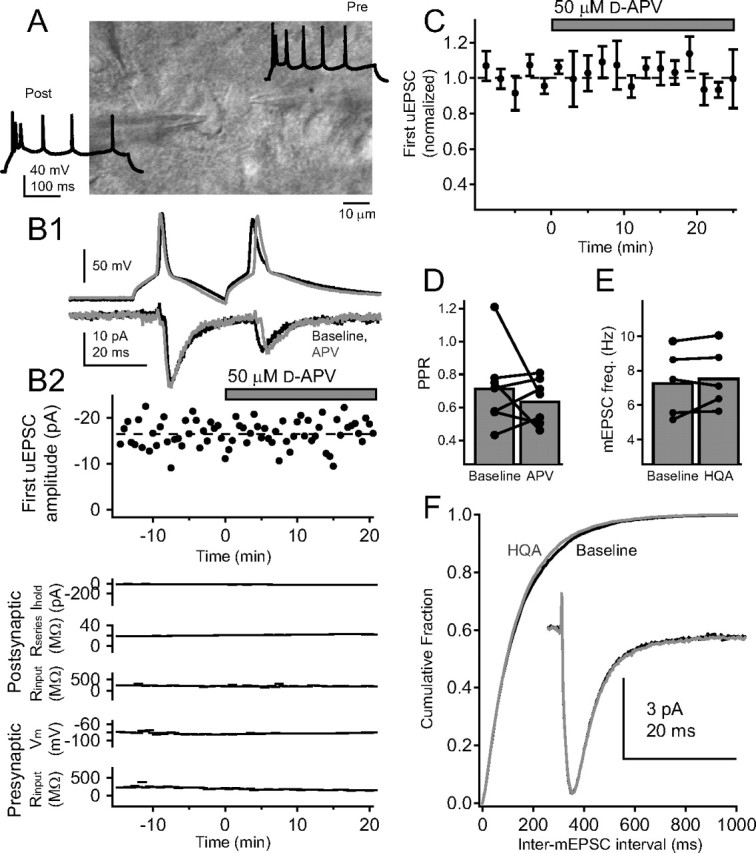
Functional PreNMDARs are absent from local synapses between L4 excitatory cells. A, Differential interference contrast image of example synaptically coupled L4 excitatory cells, with regular-spiking pattern for these cells. B1, Postsynaptic uEPSCs elicited by a pair of presynaptic spikes before (black) and after (gray) 50 μm d-APV application for the regular-spiking pair above. Each trace shown is the average of the last 10 sweeps of each condition. B2, Top, Lack of effect of d-APV on amplitude of the first uEPSC for one representative cell pair. Bottom: Postsynaptic holding current, postsynaptic series resistance, postsynaptic input resistance, presynaptic membrane potential, and presynaptic input resistance for this pair. C, Mean effect of d-APV application on first uEPSC amplitude (n = 7 pairs). D, Effect of d-APV on PPR. Error bars show population means. E, Mean mEPSC frequency for five L4 cells before and after 20 μm HQA application. F, Cumulative probability histogram of mEPSC interval before (black) and after (gray) HQA application (p > 0.1, Kolmogorov–Smirnov test). Inset, Mean mEPSC before (black) and during (gray) HQA application.
Because L4 neurons form many intralaminar connections with each other (Feldmeyer et al., 1999), spontaneous mEPSCs on L4 neurons will reflect, to a large degree, inputs from other L4 cells. Therefore, as a second test of whether functional PreNMDARs regulate release at L4–L4 synapses, we measured the effects of the NMDAR agonist HQA on mEPSCs in L4 excitatory neurons in 500 nm TTX, 100 μm picrotoxin, and 1 mm iMK-801 (holding potential, −90 mV). Bath application of HQA (20 μm) did not affect mEPSC frequency (Fig. 6 E) (baseline, 7.3 ± 0.9 Hz; HQA, 7.6 ± 0.8 Hz; p = 0.35), amplitude (baseline, 7.8 ± 0.1 pA; HQA, 7.9 ± 0.1 pA; p = 0.30), rise time (baseline, 2.03 ± 0.04 ms; HQA, 2.08 ± 0.05 ms; p = 0.14), or decay time (baseline, 5.0 ± 0.2 ms; HQA, 5.0 ± 0.1 ms; p = 0.60). Together, these results indicate that PreNMDARs regulate spontaneous and evoked release on L4–L2/3 synapses, but not at L4–L4 synapses or horizontal L2/3–L2/3 synapses. Thus, the expression of functional PreNMDARs exhibits both target specificity and input specificity.
Confirmation of functional PreNMDARs at unitary L4–L2/3 synapses
To verify that PreNMDAR regulation of synaptic transmission occurs specifically at L4–L2/3 synapses, as opposed to other synapses that may be activated by extracellular L4 stimulation, we tested whether PreNMDARs regulated release probability at synaptically coupled pairs of presynaptic L4 excitatory cells and postsynaptic L2/3 pyramidal cells (Feldmeyer et al., 2002). To obtain dual recordings, the postsynaptic L2/3 pyramidal cell was whole-cell voltage-clamped at −90 mV with iMK-801, and the presynaptic L4 cell was identified by loose-patch stimulation and repatched in whole-cell current-clamp mode using K+-gluconate internal (see Materials and Methods). Recordings were made at room temperature. We tested 1085 pairs, of which 28 were connected, and 11 remained stable long enough for analysis. PPR was measured in eight pairs (before TBOA addition) to be 0.95 ± 0.15 at 20 ms interstimulus interval (ISI) and 0.65 ± 0.06 at 200 ms ISI, which were not different from PPR previously measured at L4–L2/3 inputs using extracellular L4 stimulation (p = 0.87 for 20 ms, p = 0.15 for 200 ms) (K. J. Bender et al., 2006) (K. Bender and D. Feldman, unpublished observations). In two cells, AMPA-uEPSC reversal potential was measured to be +7 mV, consistent with glutamatergic transmission. All L4 cells exhibited a regular-spiking pattern consistent with excitatory spiny stellate or star pyramidal cells (Fig. 7 A, inset) (Connors and Gutnick, 1990; Feldmeyer et al., 2002)
Figure 7.

PreNMDARs regulate transmission at synaptically coupled L4–L2/3 pairs. A1, Example AMPA-uEPSCs from one coupled presynaptic L4 cell and postsynaptic L2/3 cell in response to a pair of presynaptic spikes during baseline (solid black) and 50 μm d-APV (gray). Each trace shown is the average of the last 10 sweeps of each condition. Inset, Regular-spiking pattern for presynaptic L4 cell. A2, Top, Effect of d-APV on first AMPA-uEPSC amplitude for this cell pair. Bottom, Postsynaptic holding current, postsynaptic series resistance, postsynaptic input resistance, presynaptic membrane potential, and presynaptic input resistance for this pair. B, Mean effect of d-APV on first AMPA-uEPSC amplitude across seven cell pairs (filled circles) and across four control cell pairs without d-APV (open circles). C, Change in PPR caused by d-APV application (filled circles) and for control pairs (open circles). Error bars represent population means. *p < 0.05. D, CV analysis of effects of d-APV on first AMPA-uEPSC amplitude. Small filled circles, CV−2 versus mean EPSC amplitude for each pair in which 50 μm d-APV was applied. Large filled circle, Population average and SE for these pairs. Large open circle, Average effect on control pairs with no d-APV application. All values are normalized to the baseline period for each pair.
We tested the effects of d-APV application on pairs of AMPA-uEPSCs (30 Hz; 30 s intersweep interval; n = 7 pairs) in the presence of 15 μm TBOA and postsynaptic iMK-801 (postsynaptic holding potential, −90 mV). In an example pair, d-APV application caused a reduction in amplitude of the first AMPA-uEPSC, and an increase in PPR, without any coincident changes in presynaptic membrane potential or input resistance, or postsynaptic holding current, input resistance, or series resistance (Fig. 7 A). Across the population, d-APV consistently decreased first AMPA-uEPSC amplitude (Fig. 7 B) and increased PPR (Fig. 7 C). To ensure that this effect did not represent nonspecific synaptic rundown, we performed interleaved control experiments in which we recorded unitary L4–L2/3 connections in the presence of TBOA, but did not apply d-APV. In these experiments (n = 4 pairs), no change was observed in the amplitude of the first AMPA-uEPSC, and no consistent change in PPR (Fig. 7 B,C).
As an additional test to determine whether d-APV affected presynaptic release probability, we calculated the coefficient of variation (CV) for the first AMPA-uEPSC amplitude before and after d-APV application. Manipulations that cause postsynaptic depression are associated with a decrease in mean EPSC amplitude without change in CV−2, whereas depression of presynaptic release is expected to cause a decrease in CV−2 greater than the decrease in mean EPSC amplitude (Malinow and Tsien, 1990; Faber and Korn, 1991; Larkman et al., 1992; Sjöström et al., 2003). We found that d-APV treatment caused CV−2 to decrease significantly more than mean EPSC amplitude (Fig. 7 D) (CV−2 APV/CV−2 baseline, 0.42 ± 0.08; meanAPV/meanbaseline, 0.61 ± 0.07; p = 0.03), consistent with a presynaptic locus of depression. In contrast, control cell pairs in which d-APV was not applied exhibited no change in mean (meanAPV/meanbaseline, 0.98 ± 0.06; p = 0.81, t test) or CV−2 (CV−2 APV/CV−2 baseline, 1.08 ± 0.33; p = 0.83, t test) (Fig. 7 D). These results indicate that PreNMDARs regulate release probability at unitary L4–L2/3 synaptic connections.
PreNMDARs are active at physiological temperatures without external activation by TBOA or applied agonists
The data shown above demonstrate that at room temperature, PreNMDARs at L4–L2/3 synapses are active only when extracellular glutamate is elevated by TBOA, or when exogenous NMDAR agonists are applied (Figs. 1 –7) (V. A. Bender et al., 2006). In contrast, other studies report that at physiological temperatures, PreNMDARs modulate spontaneous and evoked transmission without any external activation of these receptors (Berretta and Jones, 1996; Woodhall et al., 2001; Sjöström et al., 2003; Corlew et al., 2007). To test if PreNMDARs are endogenously active at physiological temperatures, we reassayed for PreNMDAR activity at 30−32°C, without TBOA or exogenous agonists (Fig. 8). We first tested whether d-APV application reduced evoked release at extracellularly stimulated L4–L2/3 synapses. Pairs of AMPA-EPSCs (30 Hz, 30 s intersweep interval, focal BMI, iMK-801) were measured. 50 μm d-APV reversibly and significantly decreased the amplitude of the first EPSC to 73.3 ± 5.3% of baseline (p < 0.01), and significantly increased PPR (p < 0.05) (Fig. 8 A1, example recording, A2,A3, population of seven cells). Input specificity of PreNMDAR function remained intact, because d-APV failed to affect AMPA-EPSCs evoked on L2/3–L2/3 cross-columnar inputs onto L2/3 pyramidal cells (Fig. 8 A2,A3).
To test whether PreNMDARs were activated by baseline glutamate in the absence of evoked synaptic release, we measured the effect of d-APV on AMPA-mEPSCs in L2/3 pyramidal neurons (−90 mV holding potential, 30–32°C, 500 nm TTX, 100 μm picrotoxin, iMK-801, no TBOA). d-APV (50 μm) applied after a 10 min baseline period significantly decreased mEPSC frequency (Fig. 8 B1), evident as a rightward shift in the cumulative histogram of inter-mEPSC interval (Fig. 8 B2) (p < 0.01, Kolmogorov–Smirnov test). There was no significant change in average mEPSC amplitude (baseline, 9.9 ± 0.7 pA; APV, 10.4 ± 0.6 pA; p = 0.13), rise time (baseline, 1.86 ± 0.11 ms; APV, 1.89 ± 0.12 ms; p = 0.42), or decay time (baseline, 4.51 ± 0.44 ms; APV, 4.46 ± 0.40 ms; p = 0.50). Thus, PreNMDARs were active even in the absence of evoked synaptic transmission.
To determine whether PreNMDAR activity was saturated under these baseline, nonspiking conditions, we recorded AMPA-mEPSCs in L2/3 pyramidal cells as above, and after a 10 min baseline period, applied 20 μm HQA for 10 min. HQA caused a significant increase in mEPSC frequency, and subsequent addition of 50 μm d-APV for 10 min reduced mEPSC frequency below baseline levels (Fig. 8 C1). These effects were evident in the cumulative probability histogram for inter-mEPSC interval (Fig. 8 C2) (p < 0.01 for all pairwise comparisons, Kolmogorov–Smirnov test). There was no change in mEPSC amplitude (baseline, 10.2 ± 0.7 pA; HQA, 9.9 ± 0.6 pA; HQA plus APV, 10.1 ± 0.9 pA; p = 0.64), rise time (baseline, 1.94 ± 0.09 ms; HQA, 1.99 ± 0.09 ms; HQA plus APV, 1.91 ± 0.09 ms; p = 0.10), or decay time (baseline, 4.7 ± 0.3 ms; HQA, 4.6 ± 0.4 ms; HQA plus APV, 4.6 ± 0.3 ms; p = 0.57). These findings suggest that PreNMDARs are active, but not saturated, by endogenous baseline glutamate in S1 slices at 30–32°C.
To determine whether functional PreNMDARs were absent from synapses onto L4 neurons at 30–32°C, we recorded AMPA-mEPSCs from L4 neurons (V hold, −90 mV, 500 nm TTX, 100 μm picrotoxin, iMK-801, no TBOA). Neither HQA nor subsequent application of d-APV altered mEPSC frequency (Fig. 8 D1) (baseline, 11.9 ± 2.4 Hz; HQA, 12.3 ± 2.7 Hz; HQA plus APV, 11.8 ± 2.1 Hz; p = 0.87) or cumulative probability histogram for inter-mEPSC interval (Fig. 8 D2) (p > 0.05, Kolmogorov–Smirnov test for all pairwise comparisons). No changes occurred in mESPC amplitude (baseline, 10.7 ± 0.8 pA; HQA, 10.4 ± 0.9 pA; HQA plus APV, 10.2 ± 1.0 pA; p = 0.50), rise time (baseline, 1.76 ± 0.04 ms; HQA, 1.89 ± 0.07 ms; HQA plus APV, 1.92 ± 0.14 ms; p = 0.26), or decay time (baseline, 4.0 ± 0.2 ms; HQA, 4.0 ± 0.3 ms; HQA plus APV, 4.0 ± 0.3 ms, p = 0.98). These results indicate that PreNMDARs are endogenously active, but not saturated, at L4–L2/3 synapses at 30–32°C, but that functional expression remains input and synapse specific, as observed at room temperature.
Discussion
PreNMDARs modulate synaptic transmission at many excitatory cortical synapses (Berretta and Jones, 1996; Woodhall et al., 2001; Sjöström et al., 2003; Yang et al., 2006; Corlew et al., 2007), particularly during the first 3 postnatal weeks (Fiszman et al., 2005; Mameli et al., 2005; Corlew et al., 2007). Our results demonstrate that functional, NR2B-containing PreNMDARs are present in L2/3 of S1 at P14–P22, but are restricted to a specific subset of synapses: they are present at L4–L2/3 excitatory synapses, but not at other synapses made by L4 cells (L4–L4 synapses) or at other inputs onto L2/3 cells (synapses of the cross-columnar L2/3–L2/3 projection). Thus, expression of functional PreNMDARs depends both on the origin and the target of synaptic inputs. These results indicate that functional expression of PreNMDARs is not a general property of developing cortical synapses, but is highly targeted to specific synapses within and across neurons. These results also demonstrate, for the first time to our knowledge, that synapses of single cortical excitatory neurons onto different excitatory postsynaptic targets can have distinct, target-specific properties.
Presynaptic localization and function of PreNMDARs
The NMDARs that modulate transmission at L4–L2/3 synapses are not located on the postsynaptic neuron because their function was not blocked by postsynaptic hyperpolarization or internal MK-801, manipulations that block postsynaptic NMDAR currents in L2/3 neurons (V. A. Bender et al., 2006). Our iMK-801 data also rule out the possibility that the relevant NMDARs are extrasynaptic postsynaptic receptors which, like PreNMDARs, are also enriched for NR2B (Tovar and Westbrook, 1999) (but see Thomas et al., 2006). d-APV does not reduce AMPA-mediated transmission by a nonselective effect on L2/3 neurons, because d-APV selectively reduced L4–L2/3 synaptic responses, but not horizontal L2/3–L2/3 synaptic responses, recorded simultaneously onto the same postsynaptic target.
Given that the relevant receptors are not postsynaptic, they could be located on the somatodendric compartment of the presynaptic cell, the presynaptic terminal itself, or on a third cell. We were able to exclude the somatodendritic compartment of the presynaptic L4 cell, because focal puffing of d-APV in L2/3 reduced release at the L4–L2/3 projection, whereas focal puffing in L4, where presynaptic somata and dendrites are located, did not. Thus, the relevant NMDARs must be located in L2/3, either on the axons or terminals of presynaptic L4 cells, or on an unknown third cell, such as a synaptically associated glial cell (Conti et al., 1996, 1999; DeBiasi et al., 1996; Lalo et al., 2006; Verkhratsky and Kirchhoff, 2007). Because NMDAR blockade decreases the probability of evoked transmitter release (evidenced by increased PPR and decreased CV−2 of AMPA-uEPSCs) and because NMDAR agonists and antagonists increase and decrease the probability of spontaneous release, respectively (evidenced by effects on mEPSC frequency), the simplest possible location for these NMDARs is on the presynaptic L4–L2/3 terminal, where release probability can be directly modulated. This hypothesis is supported by electron microscopic evidence that places NMDAR protein on axon terminals in L2/3 of visual cortex (Aoki et al., 1994; Conti et al., 1997; Charton et al., 1999; Corlew et al., 2007).
Our results indicate that PreNMDAR activation serves to enhance the probability of spontaneous and evoked transmitter release. If located on presynaptic terminals, PreNMDAR activation could enhance release via depolarization of the terminal (e.g., by activating voltage-sensitive calcium channels to increase resting [Ca2+]) (Turecek and Trussell, 2001, 2002; Trussell, 2002), via calcium influx through the NMDA receptor channel (Chen et al., 2000), or via an unknown (calcium- and voltage-independent) signaling process. It is possible that the mechanism for increasing spontaneous mEPSC frequency may be distinct from the mechanism for enhancing evoked release. Other presynaptic receptors use a variety of calcium-dependent and -independent signaling pathways to modulate release (Trussell, 2002).
Activation of PreNMDARs
When are PreNMDARs active, and what is the source of glutamate for activation? At near-physiological temperatures, d-APV reduced evoked transmission and probability of spontaneous release, without the need for artificial elevation of glutamate or application of exogenous agonists, indicating that PreNMDARs are active under these conditions (Fig. 8). Glutamate for PreNMDAR activation could arise from (1) basal, ambient glutamate that exists in the absence of evoked synaptic transmission (Sah et al., 1989; Cavelier and Attwell, 2005; Herman and Jahr, 2007; Le Meur et al., 2007), (2) glutamate released by action potential-evoked transmission at the same synapse that expresses the PreNMDARs, and/or (3) activity-dependent glutamate release from nearby synapses, neuronal cell bodies and dendrites, or glia (Isaacson and Strowbridge, 1998; Schoppa et al., 1998; Jourdain et al., 2007).
We found that APV decreased AMPA-mEPSC frequency (measured at 30–32°C in the presence of TTX), indicating that PreNMDARs are activated by basal, ambient glutamate, without evoked release. However, PreNMDARs were not maximally activated under these conditions, because NMDAR agonist application further increased mEPSC frequency (Fig. 8 D). This implies that additional glutamate, potentially from activity-dependent sources, i.e., sources (2) and (3), may further increase PreNMDAR activation beyond that achieved by ambient glutamate. That both ambient and activity-dependent glutamate release contribute to PreNMDAR activation is consistent with previous reports showing that PreNMDARs modulate spontaneous, action potential-independent release (Berretta and Jones, 1996; Woodhall et al., 2001; Sjöström et al., 2003; Corlew et al., 2007), and that high-frequency bursting is required for PreNMDAR modulation of evoked release at L5–L5 synapses in visual cortex (Sjöström et al., 2003), and at L4–L2/3 synapses in S1 at room temperature (V. A. Bender et al., 2006). At L4–L2/3 synapses at near physiological temperatures, PreNMDAR modulation of evoked release did not require sustained bursts (Fig. 8 A). The finding that PreNMDARs modulate spontaneous and low-frequency evoked release at near physiological temperatures predicts that PreNMDARs will be active at L4–L2/3 synapses in vivo.
Blockade of PreNMDARs decreased the amplitude of the first AMPA-EPSC in a pair or a train (Fig. 8 A) (Sjöström et al., 2003), indicating that PreNMDARs are active during the first action potential to invade the presynaptic terminal. In contrast, synaptic modulation by presynaptic kainate receptors is absent on the first spike of a train, and develops as the train progresses (Sun and Dobrunz, 2006). This suggests that kainate receptors, which have a low affinity for glutamate (Pinheiro et al., 2007), require build-up of local synaptic glutamate released during trains, whereas PreNMDARs, which contain NR2B subunits with a higher affinity for glutamate (Laurie and Seeburg, 1994; Priestley et al., 1995), are effectively activated by ambient glutamate, or by slow activity-dependent release of glutamate, for example by glia, that may build up over multiple trains (Cavelier and Attwell, 2005; Jourdain et al., 2007).
Synapse specificity of PreNMDARs
Our data demonstrate that in juvenile S1, functional PreNMDARs are not broadly expressed, but instead regulate release at a highly specific subset of excitatory synapses within L4 and L2/3. This specificity is apparent at the cellular level, with functional PreNMDARs evident at L4–L2/3 synapses, but not at horizontal, cross-columnar synapses, or at L4–L4 synapses. Thus, the molecular targeting or functional regulation of PreNMDARs must be tightly spatially controlled in L4 neurons. Differences between our results in somatosensory cortex and the previous findings of Corlew et al. (2007) showing PreNMDAR expression in L4 of visual cortex may reflect different cellular specializations or developmental programs in these two areas.
Excitatory synapses made by collaterals from the same presynaptic axon can have markedly different release properties, dynamics, and expression of presynaptic metabotropic and kainate receptors (Reyes et al., 1998; Scanziani et al., 1998; Tóth and McBain, 2000; Koester and Johnston, 2005; Sun and Dobrunz, 2006). Our results indicate that PreNMDARs can also be expressed in a target-specific manner. Thus, differential expression of presynaptic receptors (including ionotropic receptors) may be a general mechanism by which a single presynaptic cell regulates release dynamics and other synapse properties in a target-cell-specific manner.
Potential functional role of PreNMDARs
PreNMDARs appear to function to enhance release probability at L4–L2/3 synapses. This suggests that one role of PreNMDARs may be to dynamically offset the synaptic depression that occurs with modest, sustained activity at this synapse (Feldmeyer et al., 2002). Selective expression of PreNMDARs at feedforward L4–L2/3 synapses, rather than at cross-columnar L2/3–L2/3 synapses or at L4–L4 synapses, suggests that PreNMDARs preferentially promote ascending, feedforward activation of single S1 columns, relative to lateral spread of excitation across columns. Thus, activation of PreNMDARs could serve to sharpen the representation of single whiskers, especially during trains of sensory-evoked responses or cortical up states (McCormick, 2005).
In addition to acutely regulating synapse function, PreNMDARs are also important in activity-dependent plasticity, particularly in endocannabinoid-mediated, presynaptically expressed LTD (Sjöström et al., 2003; V. A. Bender et al., 2006; Corlew et al., 2007). It has been suggested that these receptors participate in a novel presynaptic coincidence detection mechanism for this form of LTD (Duguid and Sjöström, 2006) (but see V. A. Bender et al., 2006). Synapse-specific and developmentally regulated expression of PreNMDARs (Corlew et al., 2007; present study) suggests synapse-specific and developmentally regulated expression of this form of LTD. Thus, synapse-specific expression of PreNMDARs provides a potentially important regulatory mechanism for synapse-specific modulation of excitatory synaptic transmission and plasticity.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS046652-04. D.J.B. was supported by a predoctoral fellowship from Howard Hughes Medical Institute. We thank Per Jesper Sjöström for technical advice, Jeffrey Isaacson and Massimo Scanziani for helpful discussions, and Kevin Bender and Renna Stevens for substantial technical assistance and comments on this manuscript.
References
- Alle H, Geiger JR. Combined analog and action potential coding in hippocampal mossy fibers. Science. 2006;311:1290–1293. doi: 10.1126/science.1119055. [DOI] [PubMed] [Google Scholar]
- Allen CB, Celikel T, Feldman DE. Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat Neurosci. 2003;6:291–299. doi: 10.1038/nn1012. [DOI] [PubMed] [Google Scholar]
- Aoki C, Venkatesan C, Go CG, Mong JA, Dawson TM. Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci. 1994;14:5202–5222. doi: 10.1523/JNEUROSCI.14-09-05202.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Allen CB, Bender VA, Feldman DE. Synaptic basis for whisker deprivation-induced synaptic depression in rat somatosensory cortex. J Neurosci. 2006;26:4155–4165. doi: 10.1523/JNEUROSCI.0175-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Donoghue JP, Connors BW. Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J Neurosci. 1995;15:5324–5333. doi: 10.1523/JNEUROSCI.15-07-05324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol (Lond) 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charton JP, Herkert M, Becker CM, Schroder H. Cellular and subcellular localization of the 2B-subunit of the NMDA receptor in the adult rat telencephalon. Brain Res. 1999;816:609–617. doi: 10.1016/s0006-8993(98)01243-8. [DOI] [PubMed] [Google Scholar]
- Chen WR, Xiong W, Shepherd GM. Analysis of relations between NMDA receptors and GABA release at olfactory bulb reciprocal synapses. Neuron. 2000;25:625–633. doi: 10.1016/s0896-6273(00)81065-x. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Conti F, DeBiasi S, Minelli A, Melone M. Expression of NR1 and NR2A/B subunits of the NMDA receptor in cortical astrocytes. Glia. 1996;17:254–258. doi: 10.1002/(SICI)1098-1136(199607)17:3<254::AID-GLIA7>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Conti F, Minelli A, DeBiasi S, Melone M. Neuronal and glial localization of NMDA receptors in the cerebral cortex. Mol Neurobiol. 1997;14:1–18. doi: 10.1007/BF02740618. [DOI] [PubMed] [Google Scholar]
- Conti F, Barbaresi P, Melone M, Ducati A. Neuronal and glial localization of NR1 and NR2A/B subunits of the NMDA receptor in the human cerebral cortex. Cereb Cortex. 1999;9:110–120. doi: 10.1093/cercor/9.2.110. [DOI] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci. 2007;27:9835–9845. doi: 10.1523/JNEUROSCI.5494-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBiasi S, Minelli A, Melone M, Conti F. Presynaptic NMDA receptors in the neocortex are both auto- and heteroreceptors. NeuroReport. 1996;7:2773–2776. doi: 10.1097/00001756-199611040-00073. [DOI] [PubMed] [Google Scholar]
- de Carvalho LP, Bochet P, Rossier J. The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem Int. 1996;28:445–452. doi: 10.1016/0197-0186(95)00091-7. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Duguid I, Sjöström PJ. Novel presynaptic mechanisms for coincidence detection in synaptic plasticity. Curr Opin Neurobiol. 2006;16:312–322. doi: 10.1016/j.conb.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Engelman HS, MacDermott AB. Presynaptic ionotropic receptors and control of transmitter release. Nat Rev Neurosci. 2004;5:135–145. doi: 10.1038/nrn1297. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Egger V, Lubke J, Sakmann B. Reliable synaptic connections between pairs of excitatory layer 4 neurones within a single “barrel” of developing rat somatosensory cortex. J Physiol (Lond) 1999;521:169–190. doi: 10.1111/j.1469-7793.1999.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Lubke J, Silver RA, Sakmann B. Synaptic connections between layer 4 spiny neurone-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: physiology and anatomy of interlaminar signaling within a cortical column. J Physiol (Lond) 2002;538:803–822. doi: 10.1113/jphysiol.2001.012959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Lübke J, Sakmann B. Efficacy and connectivity of intracolumnar pairs of layer 2/3 pyramidal cells in the barrel cortex of juvenile rats. J Physiol (Lond) 2006;575:583–602. doi: 10.1113/jphysiol.2006.105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszman ML, Barberis A, Lu C, Fu Z, Erdelyi F, Szabo G, Vicini S. NMDA receptors increase the size of GABAergic terminals and enhance GABA release. J Neurosci. 2005;25:2024–20231. doi: 10.1523/JNEUROSCI.4980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissiere S, Luthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- Kew JN, Richards JG, Mutel V, Kemp JA. Developmental changes in NMDA receptor glycine affinity and ifenprodil sensitivity reveal three distinct populations of NMDA receptors in individual rat cortical neurons. J Neurosci. 1998;18:1935–1943. doi: 10.1523/JNEUROSCI.18-06-01935.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Johnston D. Target cell-dependent normalization of transmitter release at neocortical synapses. Science. 2005;308:863–866. doi: 10.1126/science.1100815. [DOI] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci. 2006;26:2673–2683. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkman A, Hannay T, Stratford K, Jack J. Presynaptic release probability influences the locus of long-term potentiation. Nature. 1992;360:70–73. doi: 10.1038/360070a0. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH. Ligand affinities at recombinant N-methyl-d-aspartate receptors depend on subunit composition. Eur J Pharmacol. 1994;268:335–345. doi: 10.1016/0922-4106(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol (Lond) 2007;580:373–383. doi: 10.1113/jphysiol.2006.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Mu Y, Vargas-Caballero M, Poo MM. Visual stimuli-induced LTD of GABAergic synapses mediated by presynaptic NMDA receptors. Nat Neurosci. 2006;9:372–380. doi: 10.1038/nn1649. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci. 2004;24:8885–8895. doi: 10.1523/JNEUROSCI.2476-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Mameli M, Carta M, Partridge LD, Valenzuela CF. Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDA receptors. J Neurosci. 2005;25:2285–2294. doi: 10.1523/JNEUROSCI.3877-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA. Neuronal networks: flip-flops in the brain. Curr Biol. 2005;15:R294–R296. doi: 10.1016/j.cub.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC, Sakmann B. The excitatory neuronal network of rat layer 4 barrel cortex. J Neurosci. 2000;20:7579–7586. doi: 10.1523/JNEUROSCI.20-20-07579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Perrais D, Coussen F, Barhanin J, Bettler B, Mann JR, Malva JO, Heinemann SF, Mulle C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci USA. 2007;104:12181–12186. doi: 10.1073/pnas.0608891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestley T, Laughton P, Myers J, Le Bourdelles B, Kerby J, Whiting PJ. Pharmacological properties of recombinant human N-methyl-d-aspartate receptors comprising NR1a/NR2A and NR1a/NR2B subunit assemblies expressed in permanently transfected mouse fibroblast cells. Mol Pharmacol. 1995;48:841–848. [PubMed] [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science. 1989;246:815–818. doi: 10.1126/science.2573153. [DOI] [PubMed] [Google Scholar]
- Samson RD, Pare D. Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci. 2005;25:1847–1855. doi: 10.1523/JNEUROSCI.3713-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Charpak S. Target cell-specific modulation of transmitter release at terminals from a single axon. Proc Natl Acad Sci USA. 1998;95:12004–12009. doi: 10.1073/pnas.95.20.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Kinzie JM, Sahara Y, Segerson TP, Westbrook GL. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci. 1998;18:6790–6802. doi: 10.1523/JNEUROSCI.18-17-06790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev. 2004;45:250–265. doi: 10.1016/j.brainresrev.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Song S, Sjostrom PJ, Reigl M, Nelson S, Chklovskii DB. Highly nonrandom features of synaptic connectivity in local cortical circuits. PLoS Biol. 2005;3:507–519. doi: 10.1371/journal.pbio.0030068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol (Lond) 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HY, Dobrunz LE. Presynaptic kainate receptor activation is a novel mechanism for target cell-specific short-term facilitation at Schaffer collateral synapses. J Neurosci. 2006;26:10796–10807. doi: 10.1523/JNEUROSCI.2746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HY, Lyons SA, Dobrunz LE. Mechanisms of target-cell specific short-term plasticity at Schaffer collateral synapses onto interneurones versus pyramidal cells in juvenile rats. J Physiol (Lond) 2005;568:815–840. doi: 10.1113/jphysiol.2005.093948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Tóth K, McBain CJ. Target-specific expression of pre- and postsynaptic mechanisms. J Physiol (Lond) 2000;525:41–51. doi: 10.1111/j.1469-7793.2000.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO. Modulation of transmitter release at giant synapses of the auditory system. Curr Opin Neurobiol. 2002;12:400–404. doi: 10.1016/s0959-4388(02)00335-5. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Reciprocal developmental regulation of presynaptic ionotropic receptors. Proc Natl Acad Sci USA. 2002;99:13884–13889. doi: 10.1073/pnas.212419699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Kirchhoff F. NMDA Receptors in glia. Neuroscientist. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Woodhall G, Evans DI, Cunningham MO, Jones RSG. NR2B-containing NMDA autoreceptors at synapses on entorhinal cortical neurons. J Neurophysiol. 2001;86:1644–1651. doi: 10.1152/jn.2001.86.4.1644. [DOI] [PubMed] [Google Scholar]
- Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci. 2006;26:406–410. doi: 10.1523/JNEUROSCI.4413-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]