

Abstract
Previously our laboratory has shown that ketamine exposure (24 hours of clinically relevant anesthesia) causes significant increases in neuronal cell death in perinatal rhesus monkeys. Sensitivity to this ketamine-induced neurotoxicity was observed on gestational days 120-123 (in utero exposure via maternal anesthesia) and on postnatal days (PNDs) 5-6, but not on PNDs 35-37. In the present study, six monkeys were exposed on PND 5 or 6 to intravenous ketamine anesthesia to maintain a light surgical plane for 24 hrs and six control animals were unexposed. At 7 months of age all animals were weaned and began training to perform a series of cognitive function tasks as part of the National Center for Toxicological Research (NCTR) Operant Test Battery (OTB). The OTB tasks used here included those for assessing aspects of learning, motivation, color discrimination, and short-term memory. Subjects responded for banana-flavored food pellets by pressing response levers and press-plates during daily (M-F) test sessions (50 min) and were assigned training scores based upon their individual performance. As reported earlier [47] beginning around 10 months of age, control animals significantly outperformed (had higher training scores than) ketamine-exposed animals for approximately the next 10 months. For animals now over 3 and one-half years of age, the cognitive impairments continue to manifest in the ketamine-exposed group as poorer performance in the OTB learning and color and position discrimination tasks, as deficits in accuracy of task performance, but also in response speed. There are also apparent differences in the motivation of these animals which may be impacting OTB performance. These observations demonstrate that a single 24-hr episode of ketamine anesthesia, occurring during a sensitive period of brain development, results in very long-lasting deficits in brain function in primates and provide proof-of-concept that general anesthesia during critical periods of brain development can result in subsequent functional deficits. Supported by NICHD, CDER/FDA and NCTR/FDA.
Keywords: developmental neurotoxicity, brain function, ketamine anesthesia, cognition, nonhuman primate
1. Introduction
Our increasing ability to keep premature infants alive is resulting in an ever-increasing population in our nation’s neonatal intensive care units: over 12% of US births are preterm [81]. Part of this success lies in the increased number of complicated surgical and other interventions that are brought to bear in this already-at-risk population. Many of these procedures, as well as those carried out in term babies, infants and toddlers are performed under various forms of anesthesia and sedation, often in combination with other potent central nervous system therapeutics. Ketamine, like its congener phencyclidine (PCP), is a dissociative anesthetic that acts primarily through blockade of N-methyl-D-aspartate (NMDA)-type glutamate receptors. Unlike PCP, ketamine has been commonly used for a variety of pediatric procedures, albeit of generally short duration [12,17,26]. Concerns over the potential adverse effects of exposures to ketamine were piqued by the findings that blockade of NMDA receptors by ketamine and related compounds causes robust increases in apoptotic cell death in the rat during the brain growth spurt period [23]. These findings were subsequently replicated and extended by others and in our own laboratories [18,60,63,85] where it has been repeatedly demonstrated that multiple doses of ketamine given to neonatal rat pups on Postnatal Day (PND) 7, the peak of the brain growth spurt in rats, leads to a massive increase in apoptotic neuronal degeneration. Similar findings have also been demonstrated in mice and current reports indicate that the use of ketamine is under increasing scrutiny [3,14,44,79].
It is known that NMDA receptors play important roles during development in that excitatory amino acids play an important role by regulating neuronal survival and migration, axonal and dendritic structure, and synaptogenesis and plasticity [27,34,55]. It has long been known that modulation of NMDA receptor function during development affects vertebrate neural development [61] and that there are functional consequences associated with changes in NMDA subunit expression during development [13]. There is also evidence to suggest that the infant brain is more responsive to agents that affect NMDA receptor function than are adult brains (e.g., [4,10]) and the pharmacology of the NMDA receptor in the developing rat (up to PND 14) is markedly different from that of the adult-like receptor [66].
NMDA receptors are also important for learning and memory processes. As important targets for excitatory amino acid neurotransmitters they serve to mediate long term potentiation (LTP) [7], an increase in synaptic efficiency that is generally believed to be important for processes associated with learning and memory [8,21,72].
Additional studies in rats extended the ketamine observations to demonstrate that another NMDA receptor antagonist (nitrous oxide) given in combination with the γ-aminobutyric acid (GABA) agonists midazolam and isoflurane also cause similar increases in neuroapoptosis [24,78]. In these publications, the clinically-relevant anesthetic cocktail was given to rodents in a single episode during the brain growth spurt and assessments of brain function in exposed animals as young adults demonstrated clear deficits in learning behaviors [24]. While concerns about adverse functional consequences resulting from developmental exposure to PCP and related compounds had been raised earlier [11], this was a clear demonstration in the rat model that a single bout of anesthesia during a period of rapid brain development could result in very long-term, perhaps permanent, deficits in brain function. Subsequently, similar adverse behavioral outcomes have been demonstrated in rats after 4 or 6 hour exposures (PND 7) to the inhalation anesthetic, isoflurane, a GABA agonist [58,69-71], in mice exposed (PND 10) to ketamine with and without diazepam [14], and in mice exposed to six hours of sevoflurane on PND 6 [59].
It has now been demonstrated that exposure to a host of compounds having NMDA receptor antagonist or GABA receptor agonist properties, many of which are used in the pediatric setting, may also be associated with marked increases in apoptosis when given during the brain growth spurt (reviewed in [22,37,38,42,43]). Indeed, it has been postulated that ethanol, which acts as both an NMDA antagonist and a GABA agonist may cause the fetal alcohol syndrome via these actions [39-41].
Given the findings in the rodent models, it was important to determine if the phenomenon also occurred in primates. Initial in vitro studies using PND 3 monkey cortical neurons in culture demonstrated that ketamine was as effective in killing monkey neurons [76] as it was in killing rat cortical neurons in culture [75]. Additional studies demonstrated that the phenomenon also occurs in vivo in nonhuman primates [67]. In those studies, ketamine exposures were conducted in the developing rhesus monkey, a model that more closely mimics the developing pediatric population [16,76]. A single 24-hour anesthetic episode of intravenous ketamine was initially employed to explore the sensitivity of the monkey at various stages of development. Exposures were carried out on gestation days 120-123 or about three quarters of the way through gestation (term is 165 days), on postnatal days 5-6 and on PNDs 35-37. Marked increases in neuronal cell death were observed when exposures occurred in utero or on PNDs 5 or 6, but not on PNDs 35-37. In this and subsequent studies [84] it was determined that exposures lasting 9 hours or more are sufficient to cause the effect but that exposures lasting only 3 hours are not. Importantly, isoflurane and ketamine have recently been shown to induce neuroapoptosis and oligoapoptosis in the neonatal rhesus monkey after exposures lasting only 5 hours [5,6,36]. In the monkey ketamine-induced cell death appears to be both apoptotic and excitotoxic whereas in the rat it seems to be primarily apoptotic. Given that ketamine was also shown to cause significant abnormal cell death in the primate as it does in rodents, there was heightened interest in the phenomenon and its relevance for the pediatric clinic [3,15,25,30,31]. In addition, a description of the steps that the Food and Drug Administration was taking to address the issue was published [35]. Those steps included developing, in collaboration with the anesthesia community and regulated industry, strategies for further assessing the safety of the pediatric use of anesthetics and for providing data to guide clinicians to make informed decisions when selecting treatment regimens.
The current study was designed to test the hypothesis that, in rhesus monkeys, there are subsequent deficits in brain function associated with a single 24 hr bout of ketamine-induced general anesthesia during the neonatal period. Originally described when used for assessing the acute effects of delta-9-THC on cognition in monkeys [62], the NCTR OTB has been used in our animal and human research laboratories for a number of years in translational studies of cognitive function [48,49]. The OTB contains several complex positively-reinforced tasks, in which correct performance is thought to depend on relatively specific and important brain functions which include learning, color and position discrimination, motivation and short-term memory. Previous experiments from this laboratory have shown that the tasks in the OTB are differentially sensitive to the acute effects of a variety of drugs from different pharmacological classes and that OTB performance by children is not generally distinguishable from that of well-trained rhesus monkeys [48,49]. The similarity in OTB performance between monkeys and children [51,52] is of particular importance with regard to extrapolating to humans the neurobehavioral (and possibly neurotoxic) effects of drugs and toxicants as determined in the monkey model. Additionally, the demonstration that several measures of OTB performance correlate highly with measures of intelligence in children [50] serves to highlight the relevance of such measures.
2. Materials and Methods
2.1 Drugs
Ketamine hydrochloride (Ketaset®, Fort Dodge Animal Health, Fort Dodge, IA) was diluted in lactated Ringer’s solution for intravenous infusion. Ketamine was identified and its purity confirmed (>99%) using high-performance liquid chromatography and mass spectrometry (LC/MS).
2.2 Animals and Exposure Procedure
Twelve postnatal day (PND) 5 (2 controls) or PND 6 (4 controls, 6 ketamine treated) rhesus monkeys served as subjects. All were born and housed at the FDA’s National Center for Toxicological Research (NCTR) nonhuman primate facility. Four male and 8 female monkeys were randomly assigned to ketamine treated (n = 6; one male, five females) and control (n = 6; 3 males and 3 females) groups. Animals were treated as described in detail previously [20,67]. Immediately prior to the initiation of anesthesia or sequestration, monkeys were separated from their anesthetized mothers, removed from their home cage and hand carried to a procedure room. Control monkeys were maintained in a temperature controlled infant incubator (Ohio Care Plus, Ohmeda Medical, Laurel, MD) with access to water but no food and were not sedated for physiological measurements or blood sample collections. Blood samples and physiological measurements were collected from the control animals while they were hand restrained. For treated monkeys, ketamine was given as an initial intramuscular injection (20 mg/kg) to induce anesthesia followed by continuous intravenous infusion at a rate of 20-50 mg/kg/hr to maintain a light surgical plane of anesthesia (as evidenced by lack of voluntary movement, decreased muscle tone, and minimal reaction to physical stimulation with maintenance of an intact palpebral reflex) for 24 hours. Throughout anesthesia, monkeys were also kept in a neonatal incubator (Ohio Care Plus, Ohmeda Medical, Laurel, MD) on a circulating water heating pad and under a nearby heat lamp to maintain body temperature. Dextrose (5% and 0.9% Sodium Chloride Injection USP; B. Braun Medical Inc. Irvine, CA) was administered via intravenous drip (10 ml/kg/hr) to anesthetized animals and by stomach tube (10 ml) every 2 hours to control animals to maintain blood glucose levels. Glycopyrrolate (0.01 mg/kg, American Reagent, Shirley, NY) was administered intramuscularly prior to anesthesia and every six hours to both control and treated monkeys to reduce airway secretions. Control and treated animals were returned to their natural mothers approximately 27 hours after separation and reared by them until weaning.
All animal procedures were approved by the National Center for Toxicological Research (NCTR) Institutional Animal Care and Use Committee and conducted in full accordance with the PHS Policy on the Humane Care and Use of Laboratory Animals.
2.3 Physiological Measurements
The procedures followed for the maintenance and monitoring of experimental subjects during anesthesia have been detailed in an earlier publication [20,67]. Briefly, pulse oximetry (N-395 Pulse Oximeter, Nellcor, Pleasanton, CA), capnography (Tidal Wave Hand-held Capnograph, Respironics, Murrysville, PA), non-invasive sphygmomanometry (Critikon Dynamap Vital Signs Monitor, GE Healthcare, Waukesha, WI), and a rectal temperature probe were used to monitor physiological conditions. Heart rates, respiratory rates, oxygen saturation of hemoglobin, expired CO2 concentrations, and rectal temperatures were recorded every 15 minutes in treated monkeys and every one to two hours in control monkeys. Systolic, diastolic, and mean arterial blood pressures were recorded every 30 minutes in treated monkeys and every two to four hours in controls. Blood (0.25 ml) was collected at one- to four-hour intervals for the measurement of plasma glucose (Ascensia Elite XL Blood Glucose Meter, Bayer Diagnostics, Tarrytown, NY), and determination of venous blood gases (Rapidlab, East Walpole, MA).
2.4 Animal weaning, housing and maintenance
At about six months of age, all subjects were fitted with permanent collars [on PND 182.0 +/- 0.0 (mean +/- SD) for controls and 185.7 +/- 12.2 for ketamine treated subjects] for use in pole and collar restraint/capture/transport procedures (Primate Products, Miami, Florida). Catch poles were attached to individual collars to secure and remove animals from their home cages to transport chairs (Primate Products, Miami, Florida). Animals were weaned [(PND 192.8 +/- 12.2 for controls and 190.7 +/- 13.0 for ketamine treated subjects) and pair-housed with a similar-age companion for approximately 3 months after which they were singly housed (at PND 268.9 +/- 12.2 for controls and 266.7 +/- 13.0 for ketamine treated animals). All subjects were acclimated to the transport chairs used for subsequent behavioral testing approximately two weeks prior to the beginning of training in the NCTR Operant Test Battery (OTB) which began at approximately 7.4 months of age on PND 224.2 +/- 3.7 for controls and 223.8 +/- 3.1 for the ketamine exposed animals. During acclimation, subjects were placed in transport chairs for approximately 1 hr/day on weekdays during which they were proffered banana-flavored food pellets (190 mg dustless, Bio-Serv, Frenchtown, New Jersey) that were subsequently used as reinforcers during operant behavioral testing and exposed to the daily laboratory routine. Daily (M-F) body weights were obtained while animals were chaired. Standard nonhuman primate chow (Purina Mills, Richmond, IN) was given once a day following behavioral or acclimation sessions (M-F) and on weekends at approximately the same time of day and adjusted weekly to insure that animals gained approximately 0.1 kg per month. Fresh fruit and multivitamins (USA Drug, Little Rock, AR) were provided 5 days a week and water was available ad libitum in their home cages.
2.5 OTB Testing
The behavioral tasks and apparatus used in the study (see below) have been previously described [49,62]. All behavioral tasks were conducted in primate operant test chambers using behavior panels that included press-plates and response levers. Banana-flavored food pellets served as positive reinforcers for all tasks. Testing occurred daily (M-F) at the same time of day (+/- 1 hour). OTB training was accomplished via the method of successive approximations and was very similar to that reported in detail elsewhere [56]. During the testing sequence, all animals (in transport chairs) were rotated through one of nine behavior chambers such that no animal was tested in the same chamber for 2 consecutive test days.
2.5.1 Learning Task
OTB training began with a learning task called an Incremental Repeated Acquisition (IRA) task which ultimately required subjects to learn a new sequence of lever presses during every IRA test session. This task and the behavior or intelligence panel--which have been described in detail elsewhere [62] -- utilizes four horizontally-aligned retractable levers and a series of lights to indicate when correct or incorrect responses (lever presses) have been made and how many more correct responses are required to obtain the next reinforcer. During the initial phase of IRA training (50 minute sessions), a press to any one of the four levers resulted in reinforcer delivery. After 40 reinforcers had been earned, training level one was considered complete and one of the four levers was inactivated, but remained extended. Thus, during training level 2, a response to one of the three active levers resulted in reinforcer delivery. After 40 reinforcers had been earned at training level 2, a second lever was de-activated (training level 3) and so on until only one lever was active (training level 4): responses to inactivated levers had no programmed consequences. After 40 reinforcers had been earned at training level 4, the ‘full’ IRA task was presented the next test day and the subject was considered to be at OTB training level 5 (training score = 5; see Figure 1).
Figure 1.
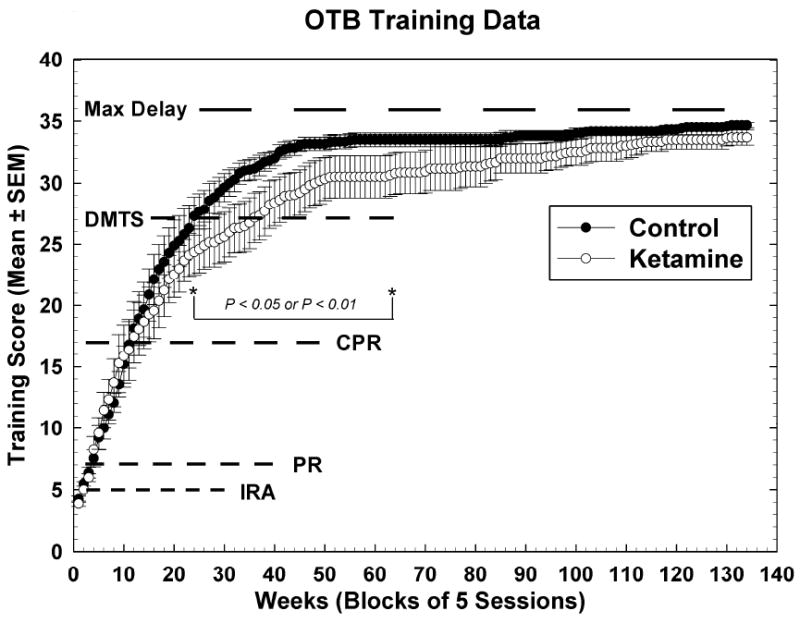
Training scores for OTB task performance. Data are raw means +/- SEMs for 5 test sessions (one week’s worth of data). Dotted lines indicate the scores at which animals began performing under the final rules of reinforcement for the indicated tasks: IRA = incremental repeated acquisition (learning) task; PR = progressive ratio (motivation) task; CPR = conditioned position responding (color and position discrimination) task; DMTS = delayed matching-to-sample (short-term memory) task with no recall delays in place; Max Delay = point at which all DMTS delay sets are attained. Note that the separation between the groups began during the time subjects had to learn the concept associated with correct performance of the DMTS task. Bracket indicates sessions over which training scores for control subjects were significantly higher than those for ketamine-exposed subjects.
During the initial component of the full IRA task (IRA1), subjects had to press the single correct (active) lever (randomly chosen) out of the four. Responses to the correct lever resulted in reinforcer delivery and illumination of a ‘correct response’ indicator light (1-sec) after which the subject could continue the task; incorrect responses (pressing on any of one the three incorrect levers) resulted in illumination of an ‘incorrect response’ indicator light (2-sec) and presentation/continuation of the same lever-pressing ‘problem.’ After completion of 20 correct responses (criterion performance) at IRA1, a 30 second time-out followed after which the task difficulty was ‘incremented’ to a 2-lever sequence (IRA2). Subjects had to add a lever press to the previously learned lever ‘sequence’. Thus, a press to a lever that was different from the correct lever at IRA1 was required, followed by responding to the lever that was correct during IRA1. Errors did not ‘reset’ the response requirement: error correction was permitted. After criterion performance at IRA2 (completion of 20 errorless sequences—not necessarily consecutive), a 30 second time-out followed after which the task was increment to a three lever sequence (IRA3) and so on up to a six lever sequence (IRA6) or the task timed out (50 minutes). Thus, for each IRA session, subjects had to learn a new sequence of lever presses, with task difficulty increasing from a 1 lever sequence to a 6 lever sequence and the percent of the task completed dependant upon the subjects’ performance. The dependent measures assessed for the IRA task were number of sessions to reach OTB training level 5 (final IRA rules), percent task completed [PTC; (number of errorless sequences completed/maximum number of errorless sequences attainable) × 100]; response rate (RR; lever presses per second), and accuracy [ACC; (correct lever presses/total lever presses) × 100].
2.5.2 Color and position discrimination task
After three consecutive IRA sessions (50 min/session maximum) during which subjects completed IRA1, subjects began training to perform a color and position discrimination or Conditioned Position Responding (CPR) task on alternate test days. The ultimate performance of this task required subjects to make right or left choice responses depending upon which color was presented to them at the initiation of a given trial. Here, the response manipulanda were three horizontally aligned press-plates that were illuminated from behind with the color red, yellow, blue, green or white. Training for this task consisted of 10 levels and, as for the IRA and all other OTB tasks, scores were assigned to each level of training and served as metrics that accumulated as animals mastered task performance.
For training in this task, subjects earned 100 reinforcers at each of the first four training levels and 200 reinforcers at each of the last 5. At CPR training level one, all three press-plates were illuminated the same with red, yellow, green or blue color (chosen randomly) and a press to any of them resulted in reinforcer delivery. At CPR training level two, only two of the press-plates (the center press-plate plus only one side press-plate) were illuminated with the same color (chosen randomly) and a press to either of them resulted in reinforcer delivery. Responses to darkened press-plates had no programmed consequences during any part of CPR training or performance. When the left press-plate plus the center press-plate were illuminated, the color was always red or yellow; when the right press-plate plus the center press-plate were illuminated, the color was always blue or green. At CPR training level 3, only the center press-plate was initially illuminated with red, yellow, blue or green (selected randomly). Presses to the illuminated center press-plate immediately extinguished it and resulted in the immediate illumination of the appropriate side plate with the same color (left for red or yellow; right for blue or green). A response to the illuminated side plate resulted in reinforcer delivery. At training level four the center press-plate was randomly illuminated red, yellow, blue or green and a single press to the illuminated center press-plate resulted in the darkening of that plate and the illumination of a single side-press plate with white. If the center press-plate color was red or yellow, the left press-plate was illuminated white: if the center press-plate was blue or green, then the right press-plate was illuminated white. A single response to the illuminated side press-plate resulted in reinforcer delivery. At training level five the center press-plate was randomly illuminated with one of the four colors (red, yellow, blue or green). In contrast to CPR training level four, a press to the center press-plate resulted in the darkening of the center-plate and the illumination of both side keys with white. A response to the left press-plate was reinforced only if the center color had been red or yellow: a response to the right press-plate was reinforced only if the center color had been blue or green. Responding to the incorrect side press-plate resulted in a 10-second time out (all press-plates dark) and re-presentation of the same color on the center press-plate (i.e., the ‘problem’ was repeated). The same problem was presented to the subject until it was solved correctly, after which the center press-plate was randomly illuminated with red, yellow, blue or green (new problem). After 200 reinforcers had been earned at level 5, subjects were considered to have moved to training level 6; after 200 more reinforcers subjects were considered to have moved to training level 7, and so on up to training level 10. At CPR level 10, the CPR session length was reduced to 10 minutes, the maximum number of pellets available was set at 60 and the final rules of the task were in place: the center press-plate was randomly illuminated red, yellow, blue or green and after a response to it, it was immediately extinguished and the two side press-plates were illuminated white. If the center press-plate had been illuminated red or yellow then a left choice response was reinforced; if the center press-plate had been illuminated green or blue then a right choice response was reinforced. Incorrect choice responses resulted in a 10-sec timeout (all plates dark) and the random presentation of a new problem. At this same time training in a short-term memory [Delayed Matching-to-Sample (DMTS)] task (see below) was added to the operant testing schedule. The dependent measures for the CPR task were number of sessions to reach training level 10 (final rules), PTC (correct trials/maximum correct trials allowed), RR and ACC (correct choice responses/total choice responses).
2.5.3 Motivation task
After the presentation of three training CPR sessions (regardless of subject performance) a task for assessing appetitive motivation [Progressive Ratio (PR)] was added to the IRA sessions. PR sessions were 10 minutes in length and the session length of the IRA task, which started one minute after the end of the PR task, was set at 40 minutes. During performance of the PR task (designated PR 1+1), only the far right of the four retractable response levers was used. Here, the first reinforcer of each PR session was obtained after a single lever press (PR 1) and the response requirement for each subsequent reinforcer was increased by one (+ 1). Thus, the second reinforcer required 2 lever presses, the third reinforcer required three lever presses, and so on until the maximum number of reinforcers (100) had been earned or the session timed out (10 minutes). PR training began at level one, and was scored as level two after 10 reinforcers were earned in a single session. The dependent measures for the PR task were number of sessions to reach PR training level 2 and response rate (RR). Earlier analyses of these data and data from other studies has demonstrated that PR response rate is highly correlated with number or reinforcers earned, size of the last ratio completed and percent task completed for this task. Thus, RR is the only endpoint reported here.
2.5.4 Short-term memory task
Training DMTS sessions (40 minutes) were added to CPR sessions as indicated above and began one minute after their termination. Only the three press-plate manipulanda were used for this task. For the first four levels of DMTS training, subjects had to earn 100 reinforcers and at the 5th level, 1000 reinforcers. At training level 1 all three press-plates were illuminated each with an identical geometric symbol (white on black) chosen randomly from one of seven possibilities. A press to any of the press-plates resulted in reinforcer delivery. At level 2, the center press-plate and one of the side press-plates (chosen randomly) were illuminated, each with the same symbol. A press to either of the illuminated press-plates results in reinforcer delivery. At DMTS training level 3 the center press-plate was randomly illuminated with one of the seven possible geometric symbols (‘sample’ stimuli). A press to the center press-plate caused it to extinguish and one of the three press-plates (location random) was immediately illuminated with the same symbol. A response to this illuminated press-plate resulted in reinforcer delivery. At training level 4 the center press-plate was illuminated with one of the seven possible geometric symbols (random selection) as a sample stimulus. A single response to that sample stimulus extinguished it and immediately, two of the three press-plates (locations random) were illuminated: one plate was illuminated with a matching symbol and the other plate with a non-matching symbol. A single response to the press-plate illuminated with the ‘match’ resulted in reinforcer delivery followed by presentation of a new matching problem. A response to the non-matching symbol resulted in a 10-sec time out, followed by re-presentation of the same matching problem. At training level 5 the center press-plate was illuminated as in level 4 but upon responding to the center press-plate, all three of the press-plates were illuminated, one with the ‘matching’ symbol, the others with non-matching symbols. Responses to the correct (matching) symbol were reinforced and responses to incorrect symbols were followed by a 10 second time-out during which all press-plates were dark and after which the same problem was presented. For every 200 reinforcers earned under these criteria, one additional training level/score was attained. Thus, after 1,000 reinforcers subjects were considered to be at DMTS training level 10 and the final task rules were put into effect, but without the addition of recall delays. Here (DMTS training level 10) the maximum number of reinforcers available was 120 and responses to incorrect symbols (non-matching choices) were followed by a 10 second time-out after which a new problem (different sample symbol) could be presented (random selection). At this point in training, there were no programmed delays between observing responses (presses to the center press-plate to indicate observance of the sample stimuli) and choice responses. Delays were inserted after observing responses at subsequent levels of training, with the maximum number of possible delay values being six. Delays were presented in a pseudo random fashion with a maximum of 20 reinforcers being available for each of the six delays.
At DMTS training level ten, the six delay values were set to virtually zero seconds (0.01, 0.01, 0.01, 0.01, 0.01, and 0.01 seconds). After three consecutive DMTS test sessions during which at least 50 reinforcers were earned, the time delay configuration was changed for training level eleven to 0.01, 0.01, 0.01, 1, 1, 1 (i.e., 3 values near zero and three values at 1 second). When performance at this delay configuration met the above criteria, the time delay configuration was changed to 0.01, 0.01, 1, 1, 2, 2 (DMTS training level twelve): two of the six delays were set to 0.01 seconds, two to 1 second and two to 2 seconds. In similar fashion the following delay configurations were incorporated into DMTS performance: 0.01, 1, 1, 2, 2, 4 for DMTS training level thirteen; 0.01, 1, 1, 2, 4, 8 for training level fourteen; 0.01, 1, 2, 4, 8, 16 for training level fifteen; 0.01, 2, 4, 8, 16, 32 for training level sixteen; 0.01, 4, 8, 16, 32, 48 for training level seventeen 0.01, 8, 16, 32, 48, 64 for training level eighteen; 0.01, 16, 32, 48, 64, 80 for training level nineteen. The dependent measures for the DMTS task were number of sessions to reach training level 10 (final rules) and each training level (delay set) thereafter.
2.5.5 Behavioral testing schedule
Typically, animals took quickly to IRA training, usually beginning to lever press for food in a single session, after which they progressed rapidly to IRA training level 5 (final rules). At this point, training for the CPR task began but was often much slower (press-plate behavior is much more difficult to shape) such that the next training point accrual actually came with the addition of the PR task: subjects usually attained the final training level for the PR task within a few sessions. Acquisition of CPR responding followed and that was followed by acquisition of DMTS responding. The final testing schedule was PR (10 minutes)/IRA (40 minutes) on one test day and CPR (10 minutes)/DMTS (40 minutes) on alternate test days. Note that a training score was assigned for every day of testing, even though the specific tasks assessed may have alternated every other test day. Thus, for the OTB training scores, 5 sessions equate to one full week (M-F) of testing. For tasks that alternate test days, 5 sessions equate to two full weeks of testing and 10 sessions equate to four weeks or one full month of testing.
2.6 Statistical Analyses
Statistical analyses of physiological measures were carried out using Student’s t-test with statistical significance defined as p < 0.05: values used are means averaged across 24 hours (all time points). Statistical analyses of OTB performance measures and body weight were effected using a repeated measures model fit by use of the mixed procedure, using SAS software [29,65,74]. The model included terms for Dose, Sex and Weeks/Sessions and their interactions. Since the ketamine group was composed, albeit randomly, of mostly females, a Sex term was also included in the model. The covariance structure on the repeated observations within each animal that was fit was first-order autoregressive. For data to be included in the Accuracy analyses for the IRA and CPR tasks, subjects must have earned at least 5 reinforcers or completed a minimum of 10 trials, respectively.
3. Results
3.1 Physiologic responses to ketamine anesthesia
All monkeys tolerated the procedures well, recovered from anesthesia or sequestration uneventfully and were returned to and accepted by their mothers without incident. Some of the physiologic parameters changed throughout the 24-h experimental period in the ketamine-treated group; however, all important physiological values such as body temperature, blood glucose, and O2 saturation remained within normal ranges for both control and ketamine treated animals [20]. These data indicate that, as previously reported [20,67], ketamine induced neurotoxicity was not due to hypothermia, hypoxia or hypoglycemia. Table 1 summarizes the physiologic parameters for both groups, showing replication of earlier findings [20,67]. The respiratory rate was lower in the ketamine-treated monkeys than in the control animals. Consistent with differences in respiratory rate, expired CO2 was higher in ketamine-treated infants than in control infants. As in the previous study, heart rate in ketamine treated animals was decreased throughout anesthesia compared to that in control animals, as were blood pressures. Body temperatures and venous pH did not vary between groups and venous pO2 and O2 saturation were higher in ketamine-treated animals than in control animals. Oxygen saturation of hemoglobin measured by pulse oximetry averaged 94% or above for both groups.
Table 1.
Physiological parameters for infants infused with ketamine for 24 hours and control monkeys (6/group).
| PND 5/6 infants |
||
|---|---|---|
| Control | Ketamine | |
| Respiratory rate (breaths per min) | 65±10.56 | 47±7.04* |
| Expired CO2 | 23±1.36 | 28±4.49* |
| Heart rate (beats per min) | 200±25.04 | 151±25.98* |
| O2 saturation (%) | 97±1.22 | 94±1.42* |
| Temperature (°F) | 98±1.17 | 99±0.50 |
| Mean Arterial Pressure (mmHg) | 62±8.10 | 52±5.68* |
| Systolic blood pressure | 81±6.46 | 70±8.64* |
| Diastolic blood pressure | 51±6.56 | 43±4.81* |
| Glucose (mg/dl) | 48±17.03 | 55±17.85 |
| Venous pCO2 | 44±4.81 | 43±5.95 |
| Venous pO2 | 22±7.04 | 39±4.01* |
| Venous pH | 7.3±0.05 | 7.3±0.03 |
| Venous O2 saturation | 30±9.72 | 66±5.17* |
Values are means ± SD averaged across 24 hours (all time points).
Indicates significant difference (p<0.05) between control and ketamine-treated monkeys.
3.2 Operant Behavioral Assessments
3.2.1 OTB Training Scores
The acquisition of task performance over time as evidenced by overall OTB training scores is shown in Figure 1. A training score of five was attained once subjects were performing under the final rules of the IRA task (5 points). A training score of seven indicated attainment of targeted PR responding (2 points); the addition of ten more points indicated that CPR performance under the final rules had been attained (training score 17); the training score of 27 indicated attainment of performance under the final rules of the DMTS task with no delays (10 points); and the maximum training score of 36, which indicates performance at the longest DMTS recall delays has yet to be attained. There were no significant differences in the attainment of training scores until subjects were learning to perform the DMTS task (training score >17); during and after this time, ketamine treated animals performed significantly worse than controls from week 24 of training until week 63, a period of about 10 months. This discrepancy appears to be primarily due to the additional time (~10 weeks or over two months) it took the ketamine treated animals to master the ‘matching’ concept necessary for completion of DMTS training. The results of the repeated measures model analysis are: Group [F (1, 21.3) = 8.14, P = 0.0094]; Sex [F (1, 21.3) = 1.75, P = 0.20]; Group*Sex [F (1, 21.3) = 2.26, P = 0.15]; Session [F (133, 1556) = 16.42, P < 0.0001]; Group*Session [F (133, 1556) = 1.81, P < 0.0001]; Sex*Session [F (133, 1556) = 1.43, P = 0.0013]; Group*Sex*Session [F (133, 1556) = 1.62, P < 0.0001].
3.2.2 Learning (IRA) Task Performance
Figure 2 shows acquisition data for the IRA task with the first data point corresponding to test sessions that began when the subjects attained an OTB training score of 5. At this point, the final rules of reinforcement were in effect for this task and henceforth behavior was indicative of subjects continuing to learn how best to solve the lever-pressing problems presented. Figure 2A shows that the PTC for both groups increased in tandem up to about the 36th week of testing, after which the control subjects outperformed the ketamine treated subjects, significantly so over most of the nearly 2 year period following week 36. for IRA PTC: Group [F (1, 12.1) = 4.34, P = 0.06]; Sex [F (1, 12.1) = 0.27, P = 0.61]; Group*Sex [F (1, 12.1) = 0.27, P = 0.61]; Session [F (33, 354) = 6.97, P < 0.0001]; Group*Session [F (33, 354) = 1.47, P = 0.049]; Sex*Session [F (33, 354) = 1.40, P = 0.08]; Group*Sex*Session [F (33, 354) = 1.34, P = 0.11]. Figures 2B and 2C indicate that this effect of ketamine exposure to decrease IRA PTC was due to both a decrease in response speed and accuracy, respectively, since both of these metrics were affected similarly, albeit it could be argued that response rate was affected to a greater degree (more points significantly different from those of the control animals) than accuracy. For IRA response rate: Group [F (1, 12) = 6.51, P = 0.025]; Sex [F (1, 12) = 0.43, P = 0.52]; Group*Sex [F (1, 12) = 0.03, P = 0.86]; Session [F (33, 354) = 5.55, P < 0.0001]; Group*Session [F (33, 354) = 1.88, P = 0.003]; Sex*Session [F (33, 354) = 1.17, P = 0.24]; Group*Sex*Session [F (33, 354) = 1.57, P = 0.026]. For IRA accuracy: Group [F (1, 12) = 3.33, P = 0.09]; Sex [F (1, 12) = 0.08, P = 0.78]; Group*Sex [F (1, 12) = 0.74, P = 0.41]; Session [F (31, 330) = 13.95, P < 0.0001]; Group*Session [F (31, 330) = 2.00, P = 0.0016]; Sex*Session [F (33, 330) = 1.17, P = 0.25]; Group*Sex*Session [F (33, 330) = 1.44, P = 0.06].
Figure 2.
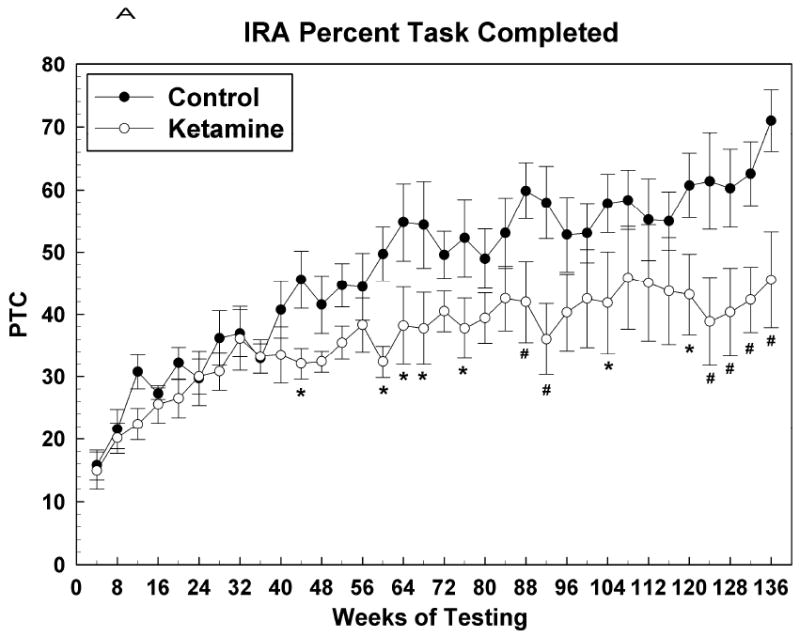
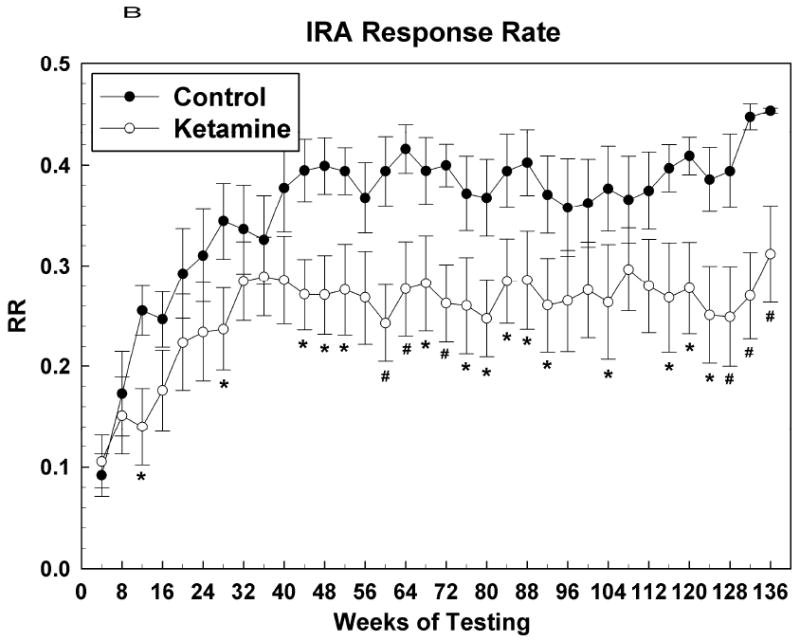
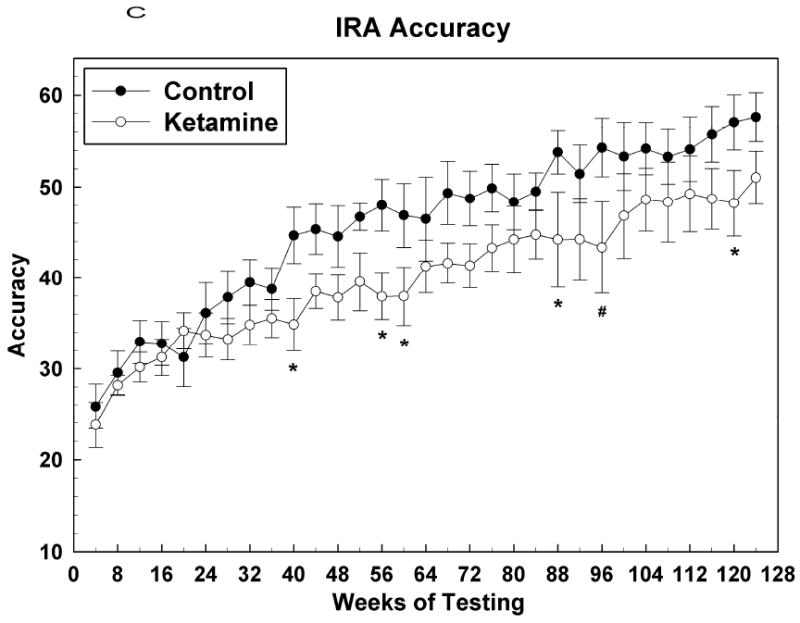
Learning (IRA) task raw data presented as averages of over 10 sessions (means +/- SEMs of 4 weeks worth of data). (A) Percent task completed; (B) Response rate; (C) Accuracy. * = P <0.05; # = p < 0.01.
3.2.3 Motivation (PR) Task Performance
Figure 3 shows the development of response rate for this task where it can be seen that, for both groups, response rate increases rather steeply over about the first 9 months (36 weeks), after which it seems to plateau at about 2.4 responses per second for control subjects and about 1.8 responses per second for the ketamine treated subjects. This difference was only sporadically statistically significant (see Figure 3). For PR response rate: Group [F (1, 12.1) = 2.66, P = 0.13]; Sex [F (1, 12.1) = 0.33, P = 0.57]; Group*Sex [F (1, 12.1) = 0.09, P = 0.77]; Session [F (31, 334) = 7.27, P < 0.0001]; Group*Session [F (31, 334) = 0.92, P = 0.59]; Sex*Session [F (31, 334) = 1.63, P = 0.02]; Group*Sex*Session [F (31, 334) = 0.76, P = 0.82].
Figure 3.
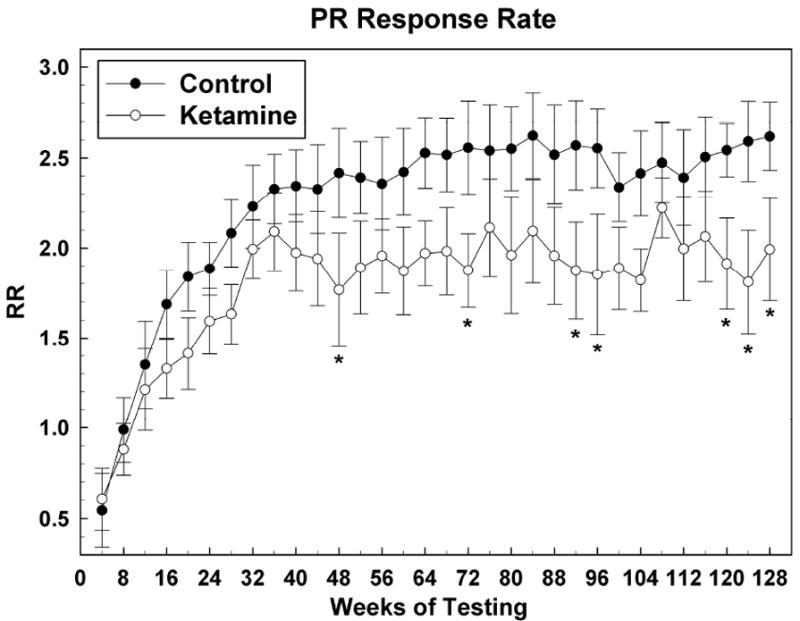
Motivation (PR) task response rate (RR) data presented as in Figure 2. * = P <0.05; # = p < 0.01.
3.2.4 Color and Position Discrimination (CPR) Task Performance
Figure 4 shows acquisition data for the CPR task with the first data point corresponding to the test session during which the subject attained an OTB training score of 17. At this point, the final rules of reinforcement were in effect for this task and henceforth behavior was indicative of subjects continuing to learn how best to solve the color-position choice problems presented. Figure 4A shows that the PTC for both groups increased in tandem up to about the 40th week of CPR testing, after which the control subjects outperformed the ketamine treated subjects, albeit significantly so for only a few months during the period between weeks 40 and 80. After the 80th week PTC measures for both groups tracked together. For CPR PTC: Group [F (1, 12.7) = 4.51, P = 0.0539]; Sex [F (1, 12.7) = 1.47, P = 0.25]; Group*Sex [F (1, 12.7) = 2.27, P = 0.16]; Session [F (31, 336) = 2.13, P = 0.0005]; Group*Session [F (31, 336) = 1.06, P = 0.39]; Sex*Session [F (31, 336) = 0.61, P = 0.95]; Group*Sex*Session [F (31, 336) = 1.19, P = 0.23]. Figures 4B and 4C indicate that this effect of ketamine exposure to decrease CPR PTC was most closely associated with a similar effect on accuracy: response rate was not significantly affected during this time. As testing in this task continued into the end of year two and into year three, the response rate for the control group became significantly faster than that for the ketamine group, which seemed to plateau at around the 48th week of testing. Thus, over the last year or so of testing, ketamine subjects were just as accurate as control subjects in solving this simple color discrimination task but they were significantly slower in doing so. For CPR response rate: Group [F (1, 12.6) = 3.16, P = 0.10]; Sex [F (1, 12.6) = 0.53, P = 0.48]; Group*Sex [F (1, 12.6) = 0.02, P = 0.89]; Session [F (31, 335) = 2.86, P < 0.0001]; Group*Session [F (31, 335) = 1.04, P = 0.42]; Sex*Session [F (31, 335) = 1.28, P = 0.15]; Group*Sex*Session [F (31, 335) = 0.88, P = 0.65]. For CPR accuracy: Group [F (1, 12.5) = 5.41, P = 0.0375]; Sex [F (1, 12.5) = 6.67, P = 0.02]; Group*Sex [F (1, 12.5) = 6.87, P = 0.02]; Session [F (26, 276) = 4.15, P < 0.0001]; Group*Session [F (26, 276) = 1.02, P = 0.45]; Sex*Session [F (26, 276) = 1.18, P = 0.25]; Group*Sex*Session [F (26, 276) = 1.46, P = 0.07].
Figure 4.
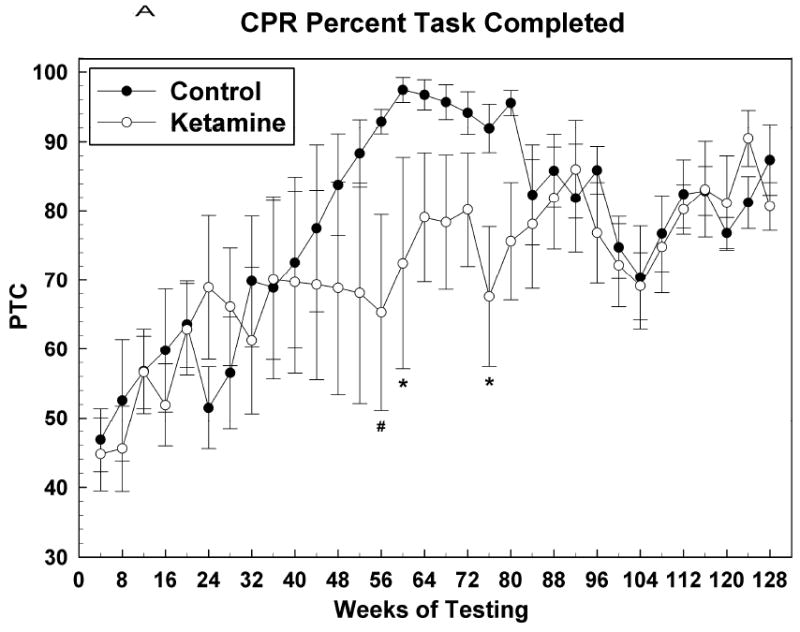
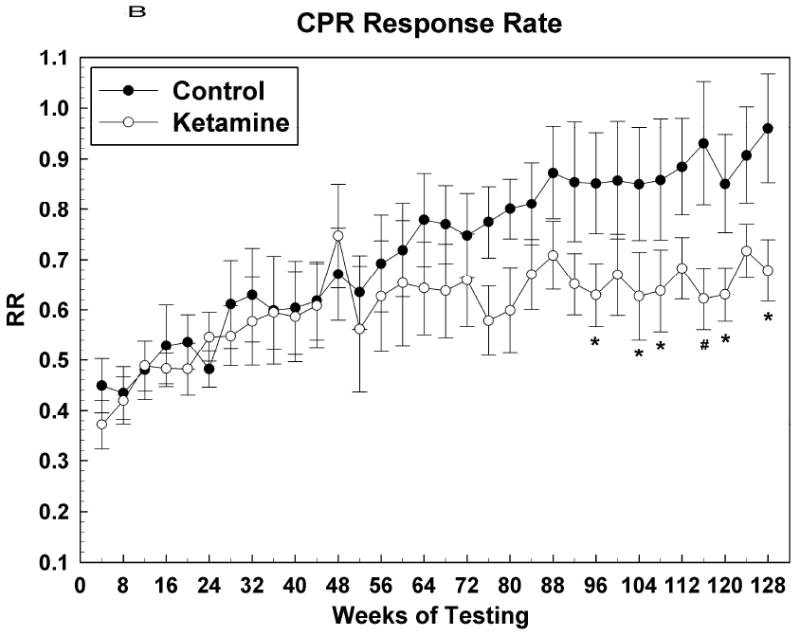
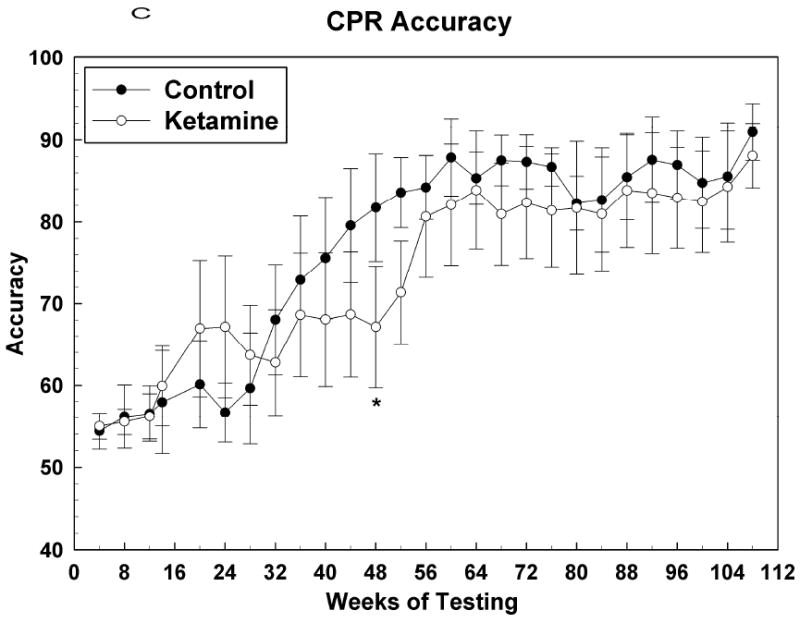
Color and position discrimination (CPR) task data presented as in Figure 2. (A) Percent task completed; (B) Response rate; (C) Accuracy. * = P <0.05; # = p < 0.01.
3.2.5 Short-term memory (DMTS) Task Performance
A telling effect of ketamine treatment on performance of this task was apparent in the OTB Training Score data (Figure 1) where it was shown that ketamine treated subjects took significantly more sessions (~ 10 weeks worth) to master the matching concept than did controls. Data presented in Figure 5 were obtained after subjects had learned the matching-to-sample concept and the final rules of reinforcement for the DMTS task were in effect. The data show the acquisition of the differing memory recall delay levels (difficulties) for this task. It can be seen here that there are no differences between the two groups, demonstrating that once the ketamine treated subjects learned the matching concept they performed no differently that did control subjects, an observation suggesting that aspects of short-term memory were not affected by treatment. For DMTS delay level acquisition: Group [F (1, 14.9) = 0.12, P = 0.73]; Sex [F (1, 14.9) = 0.11, P = 0.74[; Group*Sex [F (1, 14.9) = 0.09, P = 0.77]; Session [F (28, 336) = 17.00, P < 0.0001]; Group*Session [F (28, 336) = 2.46, P < 0.0001]; Sex*Session [F (28, 336) = 3.14, P < 0.0001]; Group*Sex*Session [F (28, 336) = 2.07, P = 0.0015].
Figure 5.
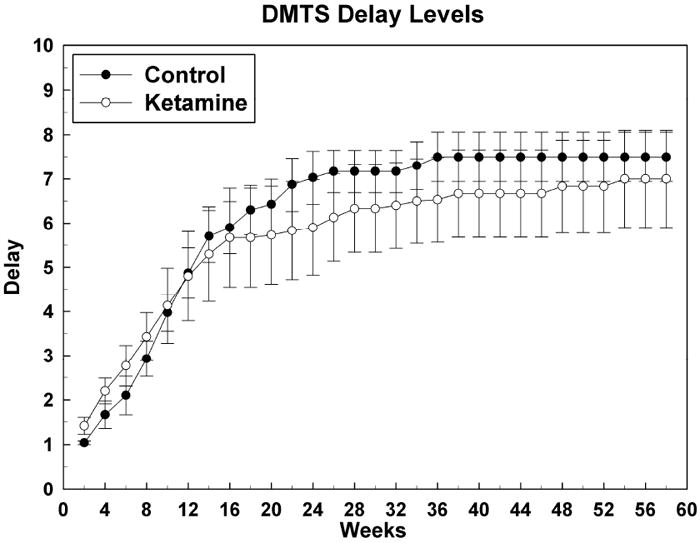
Attainment of short-term memory (DMTS) task delay intervals after mastery of DMTS task performance with no delays: delay sets versus number of DMTS sessions. Data are raw means for 5 test sessions (2 weeks) +/- SEMs. There were no significant differences between the groups at any week of testing.
3.3 Body Weight
Figure 6 shows body weights for both groups for the 138 weeks of testing. Overall the body weights were similar, evidence that our controlled feeding regimen was, with the exception of a few weeks (12, 20, 23, 46 and 117), quite successful in keeping body weights the same for each group and allowing animals to gain weight at approximately a tenth of a kilogram per month.
Figure 6.
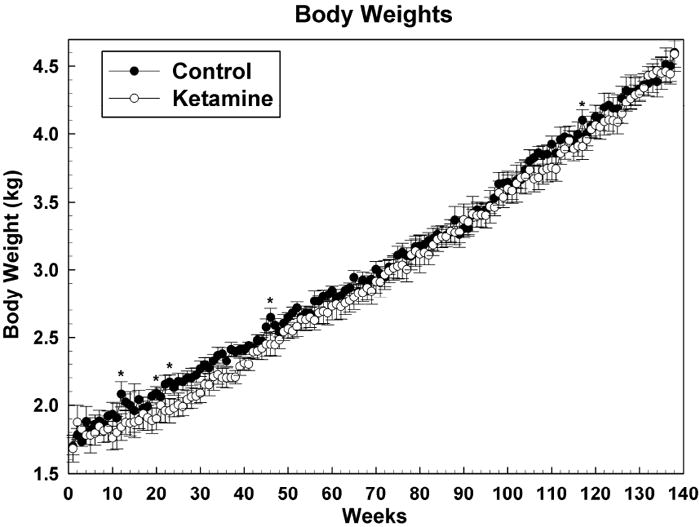
Weekly body weights as means +/- SEMs. * = P <0.05
4. Discussion
The data presented here provide proof of the concept that exposure to ketamine during a period of rapid brain growth in a primate species can result in long-term, perhaps permanent, derangements in important aspects of brain function as evidenced by significant disruptions in OTB performance. Since OTB performance by monkeys is often indistinguishable from that of children [52] and many of the metrics of OTB performance, particularly those for the IRA task, correlate positively and significantly with IQ scores in children [50], it is reasonable to interpret the present findings in monkeys as indications that similar ketamine-induced effects in children could lead to detrimental effects on aspects of intelligence. It has been suggested previously that chronic drug exposures during development in nonhuman primates can provide models of brain dysfunction in humans [45,46] and the studies presented here provide yet another example of that possibility.
It is of interest to note that the most sensitive aspects of OTB behavior in the present study involve concept formation, intuitively an incredibly important brain function. This was evidenced as significant delays in the ability of ketamine-treated animals to master the matching-to-sample concept and their apparent continued inability to master correct performance of the repeated acquisition (learning) task. These behaviors are thought to be subserved by the frontal cortex and hippocampus and the frontal cortex is an area of the monkey brain known to be perhaps the most sensitive to the induction of apoptosis associated with a 24-hr bout of ketamine anesthesia [67,84]. While treated subjects were clearly able to eventually master the performance of the comparatively simple color discriminate task, at least in terms of problem solving accuracy, they continued to underperform in this task as evidenced by continuing deficits in response rate, an indication of diminished psychomotor speed. While there were also significant deficiencies in the performance of ketamine treated animals in the progressive ratio task used to assess motivation, the timing of those effects does not often overlap with effects in the other more cognitively-loaded tasks. An effect of ketamine exposure on motivation does seem likely given the diminished response rate in the progressive ratio task, but that effect could also indicate diminished motoric capabilities.
In other studies on the effects of developmental exposures to psychoactive drugs in monkeys, the OTB learning task was also the most sensitive to disruption [53,54]. In those studies, subjects were treated daily for a full year beginning at about 2 years of age with the prototypic NMDA receptor antagonist, MK-801, or the compound remacemide, an effective sodium channel blocker with limited activity at the NMDA receptor. MK-801 was virtually without effect on acquisition and performance of OTB responding whereas remacemide had effects on learning task performance not unlike those in the present study [53]. Those observations support the hypothesis that NMDA receptor antagonism is ineffective in causing toxicity if given after the sensitive period during early development is over and before the onset of adult sensitivity.
While the data presented here are striking, several very important questions remain unanswered. For example, the precise thresholds for dose and duration of exposure that are needed to cause the functional deficits reported here are not yet known. While it may seem logical that they would be the same as or similar to those that cause frank abnormal cell death, that is not a given. And while it is now known that the period of sensitivity to ketamine-induced cell death in the monkey spans from gestation day 120 (at about 75% of gestation) through the first week of life [67], it is not known how much before gestation day 120 or how much after the first week of life the sensitivity is manifest, nor is it known exactly how these periods of sensitivity will translate to the human situation. In the monkey model it seems that ketamine-induced anesthetic episodes lasting less than three hours are not sufficient to cause abnormal levels of apoptotic cell death whereas exposures of five hours [5] and longer are sufficient [67,84]. In addition, it has recently been demonstrated that 5 hours of isoflurane anesthesia causes substantially more apoptotic cell death (12-fold increase) than ketamine (3-fold) in neonatal monkeys [5], suggesting that isoflurane is even more toxic than ketamine. Behavioral assessments of monkeys exposed to less than 24 hours of ketamine anesthesia or to isoflurane anesthesia have not been reported.
Ideally, the increases in cell death caused by ketamine (and a variety of other NMDA antagonists and GABA agonists) should be monitorable in vivo using specific markers of cell death processes and newer imaging techniques (e.g., microPET) that could provide the opportunity for minimally-invasive and repeated assessments of the phenomenon and, thus, be of use clinically. Recently, [18-F]-annexin-v has been used with microPET imaging to monitor events in rat brain following treatment with ketamine on PND 7 [82]. Annexin-v is thought to bind to phosphatidyl serine residues on the surface of cell membranes of dying cells: in healthy cells the phosphatidyl serine residues remain intracellular and inaccessible to annexin-v. While not specific for apoptosis, the concentration of the annexin-v tracer was shown to be significantly higher on PND 35 in rats previously treated with a neurotoxic regimen of ketamine on PND 7 than in controls [82]. More recently, [18-F]-DFNSH, a dansyl compound thought to label intracellular elements of dying cells, produced results in the rat similar to those of annexin-v [83]. If similar compounds can be found for use in humans, it will be possible to determine in clinical populations whether cell-death phenomena occur as they do in our animal models.
While we have shown in the current rhesus model that the functional deficits resulting from early ketamine exposure are very long lasting, we do not yet know for certain how long they will last. Nor is it known what will happen during or after puberty, another period of rapid and important brain restructuring. Might the noted deficiencies increase or decrease in severity? During old age will these subjects be predisposed to a more rapid onset of the typical cognitive decline associated with aging? As part of the plan for the animals used in the current study, follow-on behavioral challenges are being developed to allow for continued assessments of current and additional functional domains and the testing of agents that might improve cognitive function. Assessments of social interactions are also being contemplated for future studies.
It is also important to pursue strategies for preventing or ameliorating the adverse effects associated with the use of anesthetic agents. Some agents may be less likely to cause toxicity than others. For example isoflurane has been reported to cause greater neurodegeneration than sevoflurane in a neonatal mouse model [28] and greater damage than ketamine in a rhesus monkey model [5]. Recent findings with agents such as L-carnitine [86], 7-nitroindazole [77], lithium [9,68], dexmedetomidine [58], erythropoietin[80], neurotrophin receptor antagonists [19], calcium channel agonists [73], and hypothermia [9] suggest that these agents may protect against the neurodegeneration caused by ketamine and other anesthetic agents. In addition, some compounds for example, xenon and dexmedetomidine seem to be capable of inducing anesthesia and sedation, respectively, without causing cell death and/or protecting against cell death induced by anesthetics or hypoxia [32,33,58,64]. And, very importantly, we do not yet understand how the findings of the present study relate to the use of anesthetics in actual surgical or other painful/stressful circumstances. It is known that painful stimuli (needle sticks, subcutaneous formalin injections) if untreated, can themselves lead to increased cortical and subcortical cell death [2,57] and long-term behavioral changes in the rat pup [1,2,57]. Concomitant short-duration ketamine treatment in these pain/trauma models actually mitigates the associated adverse effects of noxious stimuli. It will be very important going forward to determine the effects of ketamine and related compounds in animal models that closely approximate the clinical situations for which they are used. It will also be necessary to assess the effects of the typical drug cocktails used clinically because it is the rule rather than the exception that human subjects also receive other agents such as benzodiazepines, opiates and/or anticholinergics in combination with general anesthetics.
Acknowledgments
This work was supported by the National Center for Toxicological Research (NCTR)/U.S. Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER)/FDA and the National Institute of Child Health and Human Development (NICHD).
Abbreviations
- CPR
Conditioned Position Responding
- DMTS
Delayed Matching-to-Sample
- IRA
Incremental Repeated Acquisition
- LTP
long term potentiation
- NMDA
N-methyl-D-aspartate
- OTB
Operant Test Battery
- PCP
phencyclidine
- PND
postnatal day
- PR
Progressive Ratio
Footnotes
Disclaimer This document has been reviewed in accordance with United States Food and Drug Administration (FDA) policy and approved for publication. Approval does not signify that the contents necessarily reflect the position or opinions of the FDA nor does mention of trade names or commercial products constitute endorsement or recommendation for use. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the FDA.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anand KJ, Coskun V, Thrivikraman KV, Nemeroff CB, Plotsky PM. Long-term behavioral effects of repetitive pain in neonatal rat pups. Physiol Behav. 1999;66:627–37. doi: 10.1016/s0031-9384(98)00338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anand KJ, Garg S, Rovnaghi CR, Narsinghani U, Bhutta AT, Hall RW. Ketamine reduces the cell death following inflammatory pain in newborn rat brain. Pediatr Res. 2007;62:283–90. doi: 10.1203/PDR.0b013e3180986d2f. [DOI] [PubMed] [Google Scholar]
- 3.Anand KJ, Soriano SG. Anesthetic agents and the immature brain: are these toxic or therapeutic? Anesthesiology. 2004;101:527–30. doi: 10.1097/00000542-200408000-00033. [DOI] [PubMed] [Google Scholar]
- 4.Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H, Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol. 1999;45:724–35. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 5.Brambrink AM, Back SA, Avidan MS, Creeley CE, Olney JE. Ketamine and isoflurane anesthesia triggers neuronal and glial apoptosis in the neonatal Macaque. Proc Am Soc Anesth Abs #A375. 2010 [Google Scholar]
- 6.Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, Dissen GA, Creeley CE, Olney JW. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–41. doi: 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collingridge GL, Lester RA. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol Rev. 1989;41:143–210. [PubMed] [Google Scholar]
- 9.Creeley CE, Olney JW. The young: neuroapoptosis induced by anesthetics and what to do about it. Anesth Analg. 2010;110:442–8. doi: 10.1213/ANE.0b013e3181c6b9ca. [DOI] [PubMed] [Google Scholar]
- 10.D’Souza SW, McConnell SE, Slater P, Barson AJ. N-methyl-D-aspartate binding sites in neonatal and adult brain. Lancet. 1992;339:1240. doi: 10.1016/0140-6736(92)91188-e. [DOI] [PubMed] [Google Scholar]
- 11.Deutsch SI, Mastropaolo J, Rosse RB. Neurodevelopmental consequences of early exposure to phencyclidine and related drugs. Clin Neuropharmacol. 1998;21:320–32. [PubMed] [Google Scholar]
- 12.Durrmeyer X, Vutskits L, Anand KJ, Rimensberger PC. Use of analgesic and sedative drugs in the NICU: integrating clinical trials and laboratory data. Pediatr Res. 2010;67:117–27. doi: 10.1203/PDR.0b013e3181c8eef3. [DOI] [PubMed] [Google Scholar]
- 13.Feldmeyer D, Cull-Candy S. Functional consequences of changes in NMDA receptor subunit expression during development. J Neurocytol. 1996;25:857–67. doi: 10.1007/BF02284847. [DOI] [PubMed] [Google Scholar]
- 14.Fredriksson A, Archer T, Alm H, Gordh T, Eriksson P. Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res. 2004;153:367–76. doi: 10.1016/j.bbr.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 15.Green SM, Cote CJ. Ketamine and neurotoxicity: clinical perspectives and implications for emergency medicine. Ann Emerg Med. 2009;54:181–90. doi: 10.1016/j.annemergmed.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Haberny KA, Paule MG, Scallet AC, Sistare FD, Lester DS, Hanig JP, Slikker W., Jr Ontogeny of the N-methyl-D-aspartate (NMDA) receptor system and susceptibility to neurotoxicity. Toxicol Sci. 2002;68:9–17. doi: 10.1093/toxsci/68.1.9. [DOI] [PubMed] [Google Scholar]
- 17.Hall RW, Shbarou RM. Drugs of choice for sedation and analgesia in the neonatal ICU. Clin Perinatol. 2009;36:215–26. doi: 10.1016/j.clp.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi H, Dikkes P, Soriano SG. Repeated administration of ketamine may lead to neuronal degeneration in the developing rat brain. Paediatr Anaesth. 2002;12:770–4. doi: 10.1046/j.1460-9592.2002.00883.x. [DOI] [PubMed] [Google Scholar]
- 19.Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110:813–25. doi: 10.1097/ALN.0b013e31819b602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hotchkiss CE, Wang C, Slikker W., Jr Effect of prolonged ketamine exposure on cardiovascular physiology in pregnant and infant rhesus monkeys (Macaca mulatta) J Am Assoc Lab Anim Sci. 2007;46:21–8. [PubMed] [Google Scholar]
- 21.Huang EP, Stevens CF. The matter of mind: molecular control of memory. Essays Biochem. 1998;33:165–78. doi: 10.1042/bse0330165. [DOI] [PubMed] [Google Scholar]
- 22.Ikonomidou C, Bittigau P, Koch C, Genz K, Hoerster F, Felderhoff-Mueser U, Tenkova T, Dikranian K, Olney JW. Neurotransmitters and apoptosis in the developing brain. Biochem Pharmacol. 2001;62:401–5. doi: 10.1016/s0006-2952(01)00696-7. [DOI] [PubMed] [Google Scholar]
- 23.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 24.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jevtovic-Todorovic V, Olney JW. PRO: Anesthesia-induced developmental neuroapoptosis: status of the evidence. Anesth Analg. 2008;106:1659–63. doi: 10.1213/ane.0b013e3181731ff2. [DOI] [PubMed] [Google Scholar]
- 26.Kohrs R, Durieux ME. Ketamine: teaching an old drug new tricks. Anesth Analg. 1998;87:1186–93. doi: 10.1097/00000539-199811000-00039. [DOI] [PubMed] [Google Scholar]
- 27.Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science. 1993;260:95–7. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- 28.Liang G, Ward C, Peng J, Zhao Y, Huang B, Wei H. Isoflurane causes greater neurodegeneration than an equivalent exposure of sevoflurane in the developing brain of neonatal mice. Anesthesiology. 2010;112:1325–34. doi: 10.1097/ALN.0b013e3181d94da5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Littel R, Milliken G, Stroup W, Wolfinger R, Schabenber O. SAS for Mixed Models. Second Edition. SAS Press; 2006. [Google Scholar]
- 30.Loepke AW, McGowan FX, Jr, Soriano SG. CON: The toxic effects of anesthetics in the developing brain: the clinical perspective. Anesth Analg. 2008;106:1664–9. doi: 10.1213/ane.0b013e3181733ef8. [DOI] [PubMed] [Google Scholar]
- 31.Loepke AW, Soriano SG. An assessment of the effects of general anesthetics on developing brain structure and neurocognitive function. Anesth Analg. 2008;106:1681–707. doi: 10.1213/ane.0b013e318167ad77. [DOI] [PubMed] [Google Scholar]
- 32.Ma D, Hossain M, Chow A, Arshad M, Battson RM, Sanders RD, Mehmet H, Edwards AD, Franks NP, Maze M. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann Neurol. 2005;58:182–93. doi: 10.1002/ana.20547. [DOI] [PubMed] [Google Scholar]
- 33.Ma D, Williamson P, Januszewski A, Nogaro MC, Hossain M, Ong LP, Shu Y, Franks NP, Maze M. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology. 2007;106:746–53. doi: 10.1097/01.anes.0000264762.48920.80. [DOI] [PubMed] [Google Scholar]
- 34.McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev. 1990;15:41–70. doi: 10.1016/0165-0173(90)90011-c. [DOI] [PubMed] [Google Scholar]
- 35.Mellon RD, Simone AF, Rappaport BA. Use of anesthetic agents in neonates and young children. Anesth Analg. 2007;104:509–20. doi: 10.1213/01.ane.0000255729.96438.b0. [DOI] [PubMed] [Google Scholar]
- 36.Olney JW, Brambrink AM, Avidan MS, Farber NB, Back SA. Isoflurane-induced oligoapoptosis in neonatal rhesus macaque brain. Am Soc Anesth Abs #A1598. 2010 doi: 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olney JW. New insights and new issues in developmental neurotoxicology. Neurotoxicology. 2002;23:659–68. doi: 10.1016/S0161-813X(01)00092-4. [DOI] [PubMed] [Google Scholar]
- 38.Olney JW, Farber NB, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C. Environmental agents that have the potential to trigger massive apoptotic neurodegeneration in the developing brain. Environ Health Perspect. 2000;108(Suppl 3):383–8. doi: 10.1289/ehp.00108s3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olney JW, Wozniak DF, Farber NB, Jevtovic-Todorovic V, Bittigau P, Ikonomidou C. The enigma of fetal alcohol neurotoxicity. Ann Med. 2002;34:109–19. doi: 10.1080/07853890252953509. [DOI] [PubMed] [Google Scholar]
- 40.Olney JW, Wozniak DF, Jevtovic-Todorovic V, Farber NB, Bittigau P, Ikonomidou C. Drug-induced apoptotic neurodegeneration in the developing brain. Brain Pathol. 2002;12:488–98. doi: 10.1111/j.1750-3639.2002.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olney JW, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C. Glutamate signaling and the fetal alcohol syndrome. Ment Retard Dev Disabil Res Rev. 2001;7:267–75. doi: 10.1002/mrdd.1037. [DOI] [PubMed] [Google Scholar]
- 42.Olney JW, Young C, Wozniak DF, Ikonomidou C, Jevtovic-Todorovic V. Anesthesia-induced developmental neuroapoptosis. Does it happen in humans? Anesthesiology. 2004;101:273–5. doi: 10.1097/00000542-200408000-00004. [DOI] [PubMed] [Google Scholar]
- 43.Olney JW, Young C, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C. Do pediatric drugs cause developing neurons to commit suicide? Trends Pharmacol Sci. 2004;25:135–9. doi: 10.1016/j.tips.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Patel P, Sun L. Update on neonatal anesthetic neurotoxicity: insight into molecular mechanisms and relevance to humans. Anesthesiology. 2009;110:703–8. doi: 10.1097/ALN.0b013e31819c42a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paule MG. Chronic drug exposures during development in nonhuman primates: models of brain dysfunction in humans. Front Biosci. 2005;10:2240–9. doi: 10.2741/1693. [DOI] [PubMed] [Google Scholar]
- 46.Paule MG. Developmental exposure to drugs of abuse: Alterations in nonhuman primate development as models of adverse consequences. In: S GP, Burbacher TM, Grant KS, editors. Nonhuman Primate Models of Children’s Health and Developmental Disabilities. Elsevier; 2008. pp. 301–324. [Google Scholar]
- 47.Paule MG, Li M, Zou X, Hotchkiss C, Hanig JP, Slikker W, Jr, Wang C. Early postnatal ketamine anesthesia causes persistent behavioral effects in rhesus monkeys. Soc Neurosci. 2009 Abst. #413.7. [Google Scholar]
- 48.Paule MG. Using identical behavioral tasks in children, monkeys and rats to study the effects of drugs. Curr Therap Res. 2001;62:820–833. [Google Scholar]
- 49.Paule MG. Validation of a behavioral test battery for monkeys. In: Buccafusco JJ, editor. Methods of Behavioral Analysis in Neuroscience. CRC Press LLC; 2001. pp. 281–294. [Google Scholar]
- 50.Paule MG, Chelonis JJ, Buffalo EA, Blake DJ, Casey PH. Operant test battery performance in children: correlation with IQ. Neurotoxicol Teratol. 1999;21:223–30. doi: 10.1016/s0892-0362(98)00045-2. [DOI] [PubMed] [Google Scholar]
- 51.Paule MG, Cranmer JM, Wilkins JD, Stern HP, Hoffman EL. Quantitation of complex brain function in children: preliminary evaluation using a nonhuman primate behavioral test battery. Neurotoxicology. 1988;9:367–78. [PubMed] [Google Scholar]
- 52.Paule MG, Forrester TM, Maher MA, Cranmer JM, Allen RR. Monkey versus human performance in the NCTR Operant Test Battery. Neurotoxicol Teratol. 1990;12:503–7. doi: 10.1016/0892-0362(90)90014-4. [DOI] [PubMed] [Google Scholar]
- 53.Popke EJ, Allen RR, Pearson EC, Hammond TG, Paule MG. Differential effects of two NMDA receptor antagonists on cognitive--behavioral performance in young nonhuman primates II. Neurotoxicol Teratol. 2001;23:333–47. doi: 10.1016/s0892-0362(01)00138-6. [DOI] [PubMed] [Google Scholar]
- 54.Popke EJ, Allen RR, Pearson EC, Hammond TG, Paule MG. Differential effects of two NMDA receptor antagonists on cognitive-behavioral development in nonhuman primates I. Neurotoxicol Teratol. 2001;23:319–32. doi: 10.1016/s0892-0362(01)00156-8. [DOI] [PubMed] [Google Scholar]
- 55.Reiprich P, Kilb W, Luhmann HJ. Neonatal NMDA receptor blockade disturbs neuronal migration in rat somatosensory cortex in vivo. Cereb Cortex. 2005;15:349–58. doi: 10.1093/cercor/bhh137. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez JS, Morris SM, Hotchkiss CE, Doerge DR, Allen RR, Mattison DR, Paule MG. The effects of chronic methylphenidate administration on operant test battery performance in juvenile rhesus monkeys. Neurotoxicol Teratol. 2010;32:142–51. doi: 10.1016/j.ntt.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rovnaghi CR, Garg S, Hall RW, Bhutta AT, Anand KJ. Ketamine analgesia for inflammatory pain in neonatal rats: a factorial randomized trial examining long-term effects. Behav Brain Funct. 2008;4:35–47. doi: 10.1186/1744-9081-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanders RD, Xu J, Shu Y, Januszewski A, Halder S, Fidalgo A, Sun P, Hossain M, Ma D, Maze M. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110:1077–85. doi: 10.1097/ALN.0b013e31819daedd. [DOI] [PubMed] [Google Scholar]
- 59.Satomoto M, Satoh Y, Terui K, Miyao H, Takishima K, Ito M, Imaki J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–37. doi: 10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- 60.Scallet AC, Schmued LC, Slikker W, Jr, Grunberg N, Faustino PJ, Davis H, Lester D, Pine PS, Sistare F, Hanig JP. Developmental neurotoxicity of ketamine: morphometric confirmation, exposure parameters, and multiple fluorescent labeling of apoptotic neurons. Toxicol Sci. 2004;81:364–70. doi: 10.1093/toxsci/kfh224. [DOI] [PubMed] [Google Scholar]
- 61.Scheetz AJ, Constantine-Paton M. Modulation of NMDA receptor function: implications for vertebrate neural development. FASEB J. 1994;8:745–52. doi: 10.1096/fasebj.8.10.8050674. [DOI] [PubMed] [Google Scholar]
- 62.Schulze GE, McMillan DE, Bailey JR, Scallet A, Ali SF, Slikker W, Jr, Paule MG. Acute effects of delta-9-tetrahydrocannabinol in rhesus monkeys as measured by performance in a battery of complex operant tests. J Pharmacol Exp Ther. 1988;245:178–86. [PubMed] [Google Scholar]
- 63.Shi Q, Guo L, Patterson TA, Dial S, Li Q, Sadovova N, Zhang X, Hanig JP, Paule MG, Slikker W, Jr, et al. Gene expression profiling in the developing rat brain exposed to ketamine. Neuroscience. 2010;166:852–63. doi: 10.1016/j.neuroscience.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shu Y, Patel SM, Pac-Soo C, Fidalgo AR, Wan Y, Maze M, Ma D. Xenon pretreatment attenuates anesthetic-induced apoptosis in the developing brain in comparison with nitrous oxide and hypoxia. Anesthesiology. 2010;113:360–8. doi: 10.1097/ALN.0b013e3181d960d7. [DOI] [PubMed] [Google Scholar]
- 65.Singer JD, Willet JB. Applied Longitudinal Data Analysis: Modeling Change and Event Occurrence, Editoin. Oxford University Press; 2003. [Google Scholar]
- 66.Sircar R. Developmental maturation of the N-methyl-D-aspartic acid receptor channel complex in postnatal rat brain. Int J Dev Neurosci. 2000;18:121–31. doi: 10.1016/s0736-5748(99)00069-6. [DOI] [PubMed] [Google Scholar]
- 67.Slikker W, Jr, Zou X, Hotchkiss CE, Divine RL, Sadovova N, Twaddle NC, Doerge DR, Scallet AC, Patterson TA, Hanig JP, Paule MG, Wang C. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–58. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- 68.Straiko MM, Young C, Cattano D, Creeley CE, Wang H, Smith DJ, Johnson SA, Li ES, Olney JW. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology. 2009;110:862–8. doi: 10.1097/ALN.0b013e31819b5eab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stratmann G, May LD, Sall JW, Alvi RS, Bell JS, Ormerod BK, Rau V, Hilton JF, Dai R, Lee MT, et al. Effect of hypercarbia and isoflurane on brain cell death and neurocognitive dysfunction in 7-day-old rats. Anesthesiology. 2009;110:849–61. doi: 10.1097/ALN.0b013e31819c7140. [DOI] [PubMed] [Google Scholar]
- 70.Stratmann G, Sall JW, May LD, Bell JS, Magnusson KR, Rau V, Visrodia KH, Alvi RS, Ku B, Lee MT, Dai R. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 2009;110:834–48. doi: 10.1097/ALN.0b013e31819c463d. [DOI] [PubMed] [Google Scholar]
- 71.Stratmann G, Sall JW, May LD, Loepke AW, Lee MT. Beyond Anesthetic Properties: The Effects of Isoflurane on Brain Cell Death, Neurogenesis, and Long-Term Neurocognitive Function. Anesth Analg. 2009;110:431–7. doi: 10.1213/ANE.0b013e3181af8015. [DOI] [PubMed] [Google Scholar]
- 72.Tomita H, Shibata Y, Sakurai T, Okada Y. Involvement of a protein kinase C-dependent process in long-term potentiation formation in guinea pig superior colliculus slices. Brain Res. 1990;536:146–52. doi: 10.1016/0006-8993(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 73.Turner CP, Debenedetto D, Ware E, Walburg C, Lee A, Stowe R, Swanson J, Lambert A, Lyle M, Desai P, et al. MK801-induced activated caspase-3 exhibits selective co-localization with GAD67. Neurosci Lett. 2009;462:152–6. doi: 10.1016/j.neulet.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verbeke G, Molenberghs G. Linear Mixed Models for Longitudinal Data, Edition. Springer; 2009. [Google Scholar]
- 75.Wang C, Sadovova N, Fu X, Schmued L, Scallet A, Hanig J, Slikker W. The role of the N-methyl-D-aspartate receptor in ketamine-induced apoptosis in rat forebrain culture. Neuroscience. 2005;132:967–77. doi: 10.1016/j.neuroscience.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 76.Wang C, Sadovova N, Hotchkiss C, Fu X, Scallet AC, Patterson TA, Hanig J, Paule MG, Slikker W., Jr Blockade of N-methyl-D-aspartate receptors by ketamine produces loss of postnatal day 3 monkey frontal cortical neurons in culture. Toxicol Sci. 2006;91:192–201. doi: 10.1093/toxsci/kfj144. [DOI] [PubMed] [Google Scholar]
- 77.Wang C, Sadovova N, Patterson TA, Zou X, Fu X, Hanig JP, Paule MG, Ali SF, Zhang X, Slikker W., Jr Protective effects of 7-nitroindazole on ketamine-induced neurotoxicity in rat forebrain culture. Neurotoxicology. 2008;29:613–20. doi: 10.1016/j.neuro.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 78.Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–27. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 79.Young C, Jevtovic-Todorovic V, Qin YQ, Tenkova T, Wang H, Labruyere J, Olney JW. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146:189–97. doi: 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zacharias R, Schmidt M, Kny J, Sifringer M, Bercker S, Bittigau P, Buhrer C, Felderhoff-Muser U, Kerner T. Dose-dependent effects of erythropoietin in propofol anesthetized neonatal rats. Brain Res. 2010;1343:14–9. doi: 10.1016/j.brainres.2010.04.081. [DOI] [PubMed] [Google Scholar]
- 81.Zerhouni EA. Prematurity research at the NIH. http://www.nichd.nih.gov/publications/pubs/upload/Prematurity_Research_at_NIH_02_2008.pdf.
- 82.Zhang X, Paule MG, Newport GD, Zou X, Sadovova N, Berridge MS, Apana SM, Hanig JP, Slikker W, Jr, Wang C. A minimally invasive, translational biomarker of ketamine-induced neuronal death in rats: microPET Imaging using 18F-annexin V. Toxicol Sci. 2009;111:355–61. doi: 10.1093/toxsci/kfp167. [DOI] [PubMed] [Google Scholar]
- 83.Zhang X, Paule MG, Newport GD, Sadovova N, Berridge MS, Apana SM, Kabalka G, Miao M, Slikker W, Jr, Wang C. MicroPET imaging of ketamine-induced neuronal apoptosis with radiolabeled DFNSH. J Neural Trans. 2010 doi: 10.1007/s00702-010-0499-z. In press. [DOI] [PubMed] [Google Scholar]
- 84.Zou X, Patterson TA, Divine RL, Sadovova N, Zhang X, Hanig JP, Paule MG, Slikker W, Jr, Wang C. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int J Dev Neurosci. 2009;27:727–31. doi: 10.1016/j.ijdevneu.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 85.Zou X, Patterson TA, Sadovova N, Twaddle NC, Doerge DR, Zhang X, Fu X, Hanig JP, Paule MG, Slikker W, et al. Potential neurotoxicity of ketamine in the developing rat brain. Toxicol Sci. 2009;108:149–58. doi: 10.1093/toxsci/kfn270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zou X, Sadovova N, Patterson TA, Divine RL, Hotchkiss CE, Ali SF, Hanig JP, Paule MG, Slikker W, Jr, Wang C. The effects of L-carnitine on the combination of, inhalation anesthetic-induced developmental, neuronal apoptosis in the rat frontal cortex. Neuroscience. 2008;151:1053–65. doi: 10.1016/j.neuroscience.2007.12.013. [DOI] [PubMed] [Google Scholar]