

Abstract
The assembly of mitochondrial respiratory chain complex IV (cytochrome c oxidase) involves the coordinated action of several assembly chaperones. In Saccharomyces cerevisiae, at least 30 different assembly chaperones have been identified. To date, pathogenic mutations leading to a mitochondrial disorder have been identified in only seven of the corresponding human genes. One of the genes for which the relevance to human pathology is unknown is C2orf64, an ortholog of the S. cerevisiae gene PET191. This gene has previously been shown to be a complex IV assembly factor in yeast, although its exact role is still unknown. Previous research in a large cohort of complex IV deficient patients did not support an etiological role of C2orf64 in complex IV deficiency. In this report, a homozygous mutation in C2orf64 is described in two siblings affected by fatal neonatal cardiomyopathy. Pathogenicity of the mutation is supported by the results of a complementation experiment, showing that complex IV activity can be fully restored by retroviral transduction of wild-type C2orf64 in patient-derived fibroblasts. Detailed analysis of complex IV assembly intermediates in patient fibroblasts by 2D-BN PAGE revealed the accumulation of a small assembly intermediate containing subunit COX1 but not the COX2, COX4, or COX5b subunits, indicating that C2orf64 is involved in an early step of the complex IV assembly process. The results of this study demonstrate that C2orf64 is essential for human complex IV assembly and that C2orf64 mutational analysis should be considered for complex IV deficient patients, in particular those with hypertrophic cardiomyopathy.
Main Text
Identification of the disease-causing mutation in patients with a mitochondrial disorder due to cytochrome c oxidase (complex IV) deficiency (MIM 220110) is complicated by the sheer number of candidate genes. Mutations in mtDNA-encoded genes MT-CO1, MT-CO2, and MT-CO3, as well as MT-TS1 and MT-TL1, have been identified, although these are relatively rare.1 Remarkably, mutations in the ten nuclear-encoded structural complex IV subunits appear to be even rarer because only one case has been described to date.2 The majority of deficiencies are caused by mutations in nuclear genes encoding proteins involved in the synthesis and assembly of complex IV. These include TACO1 (MIM 612958), a translational activator of COX1;3 LRPPRC (MIM 607544) that stabilizes COX1 and 3 mRNA;4,5 COX10 (MIM 602125) and COX15 (MIM 603646) that are involved in heme A biosynthesis;6,7 SCO1 (MIM 603644) and SCO2 (MIM 604272) that catalyze copper insertion into COX2;8–10 and SURF1 (MIM 185620) that is involved in early maturation of COX1.11,12 Despite the progress that has been made in understanding the mechanism of complex IV assembly, many complex IV deficient cases remain unsolved at the molecular genetic level. Thus, in yeast, many more assembly factors that have human orthologs have been identified, including Cox11p, Cox16p, Cox17p, Cox19p, and Pet191p. Some of the human orthologs of the genes encoding these proteins have been screened for the presence of mutations in complex IV deficient patients. However, no mutations have been found thus far, suggesting that these genes might not be relevant to human pathology.13,14
The study described here has been carried out in the Netherlands in accordance with the applicable rules concerning the review of research ethics committees of the Radboud University Nijmegen Medical Center and informed consent. This report describes two patients from a consanguineous family of Turkish origin. The first child was born by Caesarian section because of fetal stress after 39 weeks of an uneventful pregnancy. The patient died on day 8 after birth because of hypertrophic cardiomyopathy of the left and right ventricles. During the next pregnancy, an ultrasound examination performed at week 34 revealed signs of fetal cardiomyopathy. The girl was born at week 36 by caesarean section because of fetal stress. On day 10 after birth, this patient also died because of hypertrophic restrictive cardiomyopathy. For both patients, functional impairment of brain or skeletal muscle, often observed in complex IV deficiency,15 were not documented. Postmortem microscopic investigations showed accumulation of lipid droplets in cardiomyocytes and mitochondrial proliferation. Measurements of the mitochondrial respiratory chain enzyme activities were performed by previously described methods16,17 in cultured fibroblasts of patient P1 revealed a severe complex IV deficiency (Table 1). The activity of complex IV in cultured fibroblasts and in heart muscle was reduced as well (Table 2). In the same family, two subsequent pregnancies were tested for the presence of complex IV deficiency by measuring enzyme activities in native chorionic villi.18 The activities in both the chorionic villi and in cultured fibroblasts obtained from skin biopsies collected after birth revealed (near-) normal complex IV activities (Table 2). Genetic analysis did not reveal mutations in the mtDNA and in the nuclear genes known to cause complex IV deficiency, including SCO1, SCO2, COX10, and COX15. Subsequently, homozygosity mapping was performed on the index case patient P1 (VI-1 in Figure 1A), patient P2 (VI-2 in Figure 1A), and the two healthy siblings (VI-3 and VI-4 in Figure 1A), revealing a large candidate region on chromosome 2 (73.28–145.07 Mb). This region, containing a total of 710 genes, was screened for known human complex IV genes as well as for human orthologs of yeast genes encoding known complex IV assembly and maintenance proteins. Only one candidate gene is present in this region, C2orf64, the human ortholog of PET191. It has been shown that complex IV activity is deficient in yeast pet191Δ mutant strain, whereas the activity of succinate:cytochrome c oxidoreductase (complex II+III) is elevated, and complex III and V protein levels are unaffected.19 This indicates that mitochondrial translation in pet191Δ cells is normal, yet levels of Cox1p, Cox2p, and Cox3p protein were found to be reduced. From these findings, it was concluded that Pet191p is a complex IV assembly protein in yeast.19 In a previously published study of this gene in a large cohort of complex IV deficient patients, no mutations were observed.14 We found a homozygous mutation at c.157G>C (p.Ala53Pro) in C2orf64 (NM_001008215.1) in the two affected children, whereas healthy sibling S3 was heterozygous for this mutation, and healthy sibling S4 carried two wild-type alleles (Figure 1B). This was in agreement with the homozygosity mapping data (Table S1). Both parents were heterozygous for the mutation. This mutation was not detected in 216 alleles of healthy control individuals of Turkish origin, nor is it present in EST databases, consistent with pathogenicity of the mutation. In order to assess whether the C2orf64 p.Ala53Pro mutation had an effect on complex IV assembly, we performed 1D and 2D blue native PAGE (BN-PAGE) analysis on fibroblasts of the two patients and their healthy siblings. One-dimensional BN-PAGE showed that both the activity and amount of holocomplex complex IV was strongly reduced in both patients compared to the siblings (Figure 2A). Two-dimensional BN-PAGE analysis subsequently confirmed the near absence of holocomplex IV and showed COX1 accumulation in subcomplexes (Figure 2C). Complex IV assembly is a stepwise process with three milestones in the form of subassemblies S1, S2, and S3 that are formed by the sequential addition of subunits and cofactors.20,21 The predominant subcomplex in the C2orf64 patients was similar in size to the smallest subcomplex observed in control cells (Figure 2C). This subcomplex appears to be similar to the previously described subcomplex S122 that has also been observed in complex IV deficiency due to mutations in the gene-encoding assembly factor SURF1,23,24 although these patients also show a varying degree of accumulation of subcomplex S2, the next subassembly in the complex IV assembly pathway. The levels of individual complex IV subunits COX1, COX2, COX4, and COX5a were also reduced (Figure 2B), which suggests that the very low levels of holocomplex IV and absence of higher order assembly intermediates beyond subcomplex S1 results in downregulation or destabilization of individual complex IV subunits. The reduced levels of COX1 and COX2 are compatible with the reduced levels of the yeast orthologs in Pet191p deficient yeast cells.19 Taken together, these observations suggest a role for C2orf64 in an early stage of the complex IV assembly process.
Table 1.
Activity of Respiratory Chain Enzymes in Fibroblasts from Patient P1
| Enzyme | Enzyme Activitya | Control Rangea |
|---|---|---|
| Complex I | 150 | 100–310 |
| Complex II | 528 | 520–845 |
| Complex III | 1523 | 1320–2610 |
| Complex IV | 150 | 680–1190 |
| Citrate synthase | 206 | 144–257 |
Patient P1 is homozygous for the c.157G>C mutation in C2orf64.
Activities of complex I, complex II, complex III, and complex IV are expressed as milliunits per unit citrate synthase (CS); activity of citrate synthase is expressed as milliunits per milligram protein.
Table 2.
Activity of Complex IV in Tissues and Cultured Fibroblasts from Patients and Healthy Sibling
| Patient or Healthy Sibling | Genotypea | Tissue | Complex IV Activityb | Control Rangeb |
|---|---|---|---|---|
| Patient P2 | mut/mut | heart | 4 | 114–424 |
| fibroblasts | 16 | 680–1190 | ||
| Healthy sibling S3 | wt/mut | chorionic villi | 269 | 271–922 |
| fibroblasts | 623 | 680–1190 | ||
| Healthy sibling S4 | wt/wt | chorionic villi | 557 | 271–922 |
| fibroblasts | 714 | 680–1190 |
Genotype refers to the presence of wild-type (wt) or c.157G>C mutated (mut) C2orf64 alleles.
The activity of complex IV is expressed as milliunits per unit citrate synthase.
Figure 1.
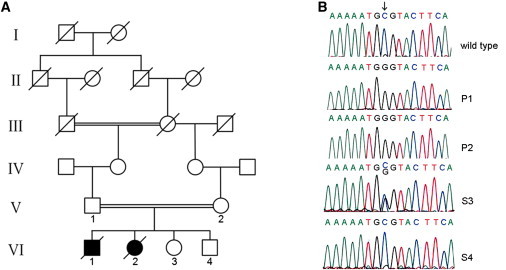
Family Pedigree and Molecular Genetic Analysis of the C2orf64 cDNA
(A) Pedigree of the family of the two patients described in this report.
(B) Electropherograms showing the wild-type sequence of C2orf64 (top panel) and the nucleotide changes in the complex IV deficient patients P1 (VI-1 in A) and P2 (VI-2) and the healthy siblings S3 (VI-3) and S4 (VI-4). The arrow indicates the mutated nucleotide c.157G>C. P1 and P2 are homozygous for the c.157G>C mutation, whereas S3 is a heterozygous carrier and S4 carries the wild-type sequence. Please note that the reverse sequence is shown.
Figure 2.
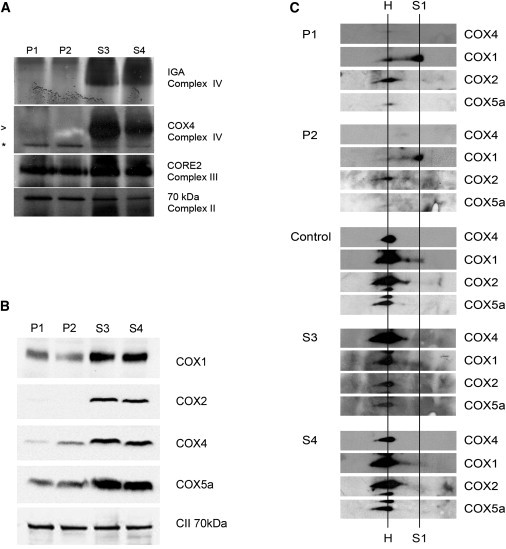
Reduced Complex IV Activity and Amount in Fibroblasts from the Patients with a C2orf64 Mutation
(A) Fibroblasts from patients P1 and P2 show a severely reduced in-gel activity of complex IV32 compared to the unaffected siblings S3 and S4 (top panel). The lower three panels show the results of immunoblots after nondenaturing BN-PAGE,33 revealing a severely reduced complex IV amount in patients P1 and P2. Complex IV was stained with anti-COX4 antibodies. Equal loading of the gel was tested by staining for complex III (by using anti-CORE2) and complex II (by using anti-70 kDa subunit). Holocomplex IV is indicated by the arrowhead; the asterisk indicates a nonspecific band.
(B) Immunoblot after SDS-PAGE of fibroblast extracts of patients P1 and P2 and healthy siblings S3 and S4 showing reduced expression levels of complex IV subunits COX1, COX2, COX4, and COX5a in fibroblasts of both patients. The complex II 70 kDa subunit (CII 70 kDa) was used as a loading control.
(C) Two dimensional BN-PAGE analysis of fibroblasts from patients P1 and P2 and the healthy siblings S3 and S4 was performed in accordance with standard procedures. Blots were stained by using antibodies against different complex IV subunits as indicated. Holocomplex IV is indicated (H). Patients P1 and P2 show accumulation of a small subcomplex (indicated by S1) that contains COX1 but not COX2, COX4, and COX5a. This indicates that mutation of C2orf64 results in accumulation of a COX1 containing early complex IV assembly intermediate. Note that blots of different fibroblasts and blots stained with different antibodies can only be compared qualitatively because exposure times are not the same.
To confirm that the mutation in C2orf64 caused the strong reduction of complex IV holocomplex amount and activity, we retrovirally transduced both index patient and healthy sibling fibroblasts with human full-length C2orf64. Complementation of patient fibroblasts with wild-type C2orf64 resulted in normalization of fully assembled complex IV (Figure 3). Moreover, normal in-gel activity of complex IV was present in fibroblasts from the healthy siblings and in the C2orf64-complemented patient cell line (Figure 3). Overexpression of wild-type C2orf64 in healthy siblings had no effect on complex IV levels or activity (Figure 3). These observations support the notion that the mutation in C2orf64 is responsible for the complex IV deficiency in our patients.
Figure 3.
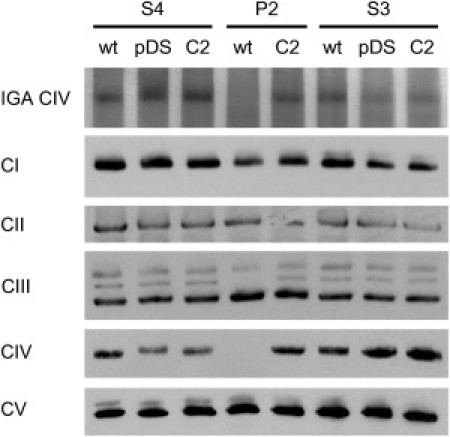
Complementation with Wild-Type C2orf64 Restores Complex IV
Fibroblasts from patient P2 and healthy siblings S3 and S4 were infected with retrovirus containing no insert (pDS) or C2orf64 (C2). The C2orf64 complementation was done by cloning full length human C2orf64 cDNA clone IOH26651 (GeneID: 493753; Imagenes, Berlin, Germany) into the Gateway retroviral destination vector pDS_FBneo (# MBA-295, LGC, Middlesex, UK) with the Gateway LR Clonase II enzyme mix (Invitrogen). Recombinant viruses were produced by using the amphotropic packaging cell line PA317 (#CRL-9078, LGC, Middlesex, UK) according to the manufacturer's protocol (Invitrogen, Breda, The Netherlands). P2, S3, and S4 fibroblasts were incubated for 24 hr with a 1:1 mixture of growth medium and virus-containing medium in the presence of 4 μg/ml polybrene,34 followed by 14 days selection with 500 μg/ml geneticin (G418, PAA, Pasching, Austria). G418-resistant cells were used for biochemical analyses within 5 passages after transduction. P2 fibroblasts grew very slowly and all cells failed to survive the selection procedure upon retroviral transduction of the empty vector. Mitochondrion-enriched fractions from all other transduced cell lines were tested for in-gel activity of complex IV after BN-PAGE. In addition, amounts of OXPHOS complexes were visualized with anti-NDUFA9 (complex I; CI), anti-70 kDa subunit of complex II (CII), anti-CORE2 for complex III (CIII), anti-COX1 for complex IV (CIV) and anti-complex Vα for complex V (CV). The results show a clear complex IV deficiency, consistent with the results in Figure 2A, which is rescued by complementation with wild-type C2orf64 (indicated by C2). Lanes indicated with wt are nontransduced fibroblasts, lanes with pDS indicate fibroblasts transduced with empty virus. Note that the complex III panel shows three bands, the lowest of which represents holo-complex III, whereas the two more slowly migrating bands most likely represent supercomplexes containing complex III. The middle band is absent in patient P2 and returns after complementation with wild-type C2orf64. This middle band therefore most likely represents a supercomplex containing complex III and complex IV.
Further support for the pathogenic role of the p.Ala53Pro mutation came from protein sequence analysis. The C2orf64 protein contains two Cx9C motifs that are highly conserved among all eukaryotes with complex IV (Figure 4A). In twin Cx9C motif proteins with a known 3D structure, COX17 and COX6b, the cysteines in these motifs form intramolecular disulfide bridges that stabilize two antiparallel α helices in a hairpin conformation.25,26 If C2orf64 adopts such a helical hairpin structure, the p.Ala53Pro mutation will have a significant influence on protein structure, because proline residues lack an amide proton, precluding the hydrogen bond required for the formation of α-helices.27 Additionally, sterical interference of the proline sidechain with the α-helix backbone28 restricts the presence of prolines to the first four positions of a helix.27 Indeed, analysis of eukaryotic orthologs of not only Pet191p (Figure 4A) but also all other 23 human mitochondrial twin Cx9C proteins shows that prolines appear exclusively within the first four residues of α helices (data not shown). In contrast, the p.Ala53Pro mutation is present at the seventh position and is likely to interfere with its conformation (Figure 4B). Consistent with the predicted structure of Pet191p, the first cysteine (C14) of the first Cx9C motif and the second cysteine (C57) of the second Cx9C motif, which are predicted to interact with each other, have been shown to be essential for respiration in yeast.19 Thus, the p.Ala53Pro mutation in C2orf64 is likely to interfere with the oxidative folding process of this twin Cx9C domain-containing protein that is linked to its mitochondrial inter membrane space localization.29–31
Figure 4.
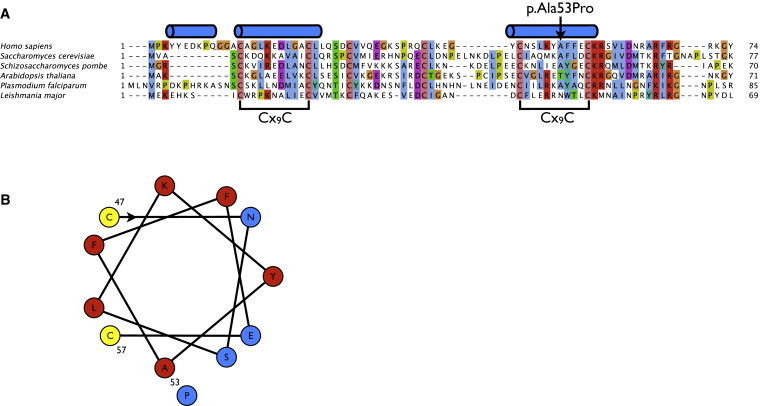
The p.Ala53Pro Mutation in the Protein Sequence of C2orf64
(A) Alignment of the C2orf64 protein with its eukaryotic orthologs. Predicted α helices35 are indicated above the alignment.
(B) Helical-wheel projection of the second Cx9C motif. Hydrophobic and hydrophilic residues are marked in red and blue, respectively. Cysteine residues (yellow) on the hydrophobic face of the helix could form disulfide bridges with the first cysteine motif.19
Acknowledgments
Part of this work was supported by a grant from the Innovatiegerichte Onderzoeksprogramma's Genomics program of Senternovem (Grant IGA05003 to M.H.) and the Netherlands Genomics Initiative (Horizon Program to R.S.). The authors thank Cindy Dieteren for assistance with the Gateway cloning, Hanka Venselaar for helpful suggestions on structure-function relationship, and the colleagues of the muscle and tissue culture labs of the Laboratory of Genetic, Endocrine, and Metabolic Diseases for excellent technical assistance.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank
NCBI database, http://www.ncbi.nlm.nih.gov
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
References
- 1.Shoubridge E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001;106:46–52. doi: 10.1002/ajmg.1378. [DOI] [PubMed] [Google Scholar]
- 2.Massa V., Fernandez-Vizarra E., Alshahwan S., Bakhsh E., Goffrini P., Ferrero I., Mereghetti P., D'Adamo P., Gasparini P., Zeviani M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008;82:1281–1289. doi: 10.1016/j.ajhg.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weraarpachai W., Antonicka H., Sasarman F., Seeger J., Schrank B., Kolesar J.E., Lochmüller H., Chevrette M., Kaufman B.A., Horvath R., Shoubridge E.A. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- 4.Mootha V.K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu F., Morin C., Mitchell G., Ackerley C., Robinson B.H. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: Mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem. J. 2004;382:331–336. doi: 10.1042/BJ20040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonicka H., Leary S.C., Guercin G.H., Agar J.N., Horvath R., Kennaway N.G., Harding C.O., Jaksch M., Shoubridge E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003;12:2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- 7.Valnot I., von Kleist-Retzow J.C., Barrientos A., Gorbatyuk M., Taanman J.W., Mehaye B., Rustin P., Tzagoloff A., Munnich A., Rötig A. A mutation in the human heme A: Farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000;9:1245–1249. doi: 10.1093/hmg/9.8.1245. [DOI] [PubMed] [Google Scholar]
- 8.Leary S.C., Sasarman F., Nishimura T., Shoubridge E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009;18:2230–2240. doi: 10.1093/hmg/ddp158. [DOI] [PubMed] [Google Scholar]
- 9.Papadopoulou L.C., Sue C.M., Davidson M.M., Tanji K., Nishino I., Sadlock J.E., Krishna S., Walker W., Selby J., Glerum D.M. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 10.Valnot I., Osmond S., Gigarel N., Mehaye B., Amiel J., Cormier-Daire V., Munnich A., Bonnefont J.P., Rustin P., Rötig A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 2000;67:1104–1109. doi: 10.1016/s0002-9297(07)62940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiranti V., Hoertnagel K., Carrozzo R., Galimberti C., Munaro M., Granatiero M., Zelante L., Gasparini P., Marzella R., Rocchi M. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 13.Horvath R., Lochmüller H., Stucka R., Yao J., Shoubridge E.A., Kim S.H., Gerbitz K.D., Jaksch M. Characterization of human SCO1 and COX17 genes in mitochondrial cytochrome-c-oxidase deficiency. Biochem. Biophys. Res. Commun. 2000;276:530–533. doi: 10.1006/bbrc.2000.3495. [DOI] [PubMed] [Google Scholar]
- 14.Tay S.K., Nesti C., Mancuso M., Schon E.A., Shanske S., Bonilla E., Davidson M.M., Dimauro S. Studies of COX16, COX19, and PET191 in human cytochrome-c oxidase deficiency. Arch. Neurol. 2004;61:1935–1937. doi: 10.1001/archneur.61.12.1935. [DOI] [PubMed] [Google Scholar]
- 15.Böhm M., Pronicka E., Karczmarewicz E., Pronicki M., Piekutowska-Abramczuk D., Sykut-Cegielska J., Mierzewska H., Hansikova H., Vesela K., Tesarova M. Retrospective, multicentric study of 180 children with cytochrome C oxidase deficiency. J. Pediatr. Res. 2006;59:21–26. doi: 10.1203/01.pdr.0000190572.68191.13. [DOI] [PubMed] [Google Scholar]
- 16.Janssen A.J., Trijbels F.J., Sengers R.C., Smeitink J.A., van den Heuvel L.P., Wintjes L.T., Stoltenborg-Hogenkamp B.J., Rodenburg R.J. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- 17.Rodenburg R.J. Biochemical diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 2010 doi: 10.1007/s10545-010-9081-y. in press. Published online May 4, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niers L., van den Heuvel L., Trijbels F., Sengers R., Smeitink J., Nijmegen Centre for Mitochondrial Disorders, The Netherlands Prerequisites and strategies for prenatal diagnosis of respiratory chain deficiency in chorionic villi. J. Inherit. Metab. Dis. 2003;26:647–658. doi: 10.1023/b:boli.0000005605.57420.b4. [DOI] [PubMed] [Google Scholar]
- 19.Khalimonchuk O., Rigby K., Bestwick M., Pierrel F., Cobine P.A., Winge D.R. Pet191 is a cytochrome c oxidase assembly factor in Saccharomyces cerevisiae. Eukaryot. Cell. 2008;7:1427–1431. doi: 10.1128/EC.00132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fornuskova D., Stiburek L., Wenchich L., Vinsova K., Hansikova H., Zeman J. Novel insights into the assembly and function of human nuclear-encoded cytochrome c oxidase subunits 4, 5a, 6a, 7a and 7b. Biochem. J. 2010;428:363–374. doi: 10.1042/BJ20091714. [DOI] [PubMed] [Google Scholar]
- 21.Mick D.U., Fox T.D., Rehling P. Inventory control: Cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 2011;12:14–20. doi: 10.1038/nrm3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nijtmans L.G., Taanman J.W., Muijsers A.O., Speijer D., Van den Bogert C. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 1998;254:389–394. doi: 10.1046/j.1432-1327.1998.2540389.x. [DOI] [PubMed] [Google Scholar]
- 23.Coenen M.J., van den Heuvel L.P., Nijtmans L.G., Morava E., Marquardt I., Girschick H.J., Trijbels F.J., Grivell L.A., Smeitink J.A. SURFEIT-1 gene analysis and two-dimensional blue native gel electrophoresis in cytochrome c oxidase deficiency. Biochem. Biophys. Res. Commun. 1999;265:339–344. doi: 10.1006/bbrc.1999.1662. [DOI] [PubMed] [Google Scholar]
- 24.Tiranti V., Galimberti C., Nijtmans L., Bovolenta S., Perini M.P., Zeviani M. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum. Mol. Genet. 1999;8:2533–2540. doi: 10.1093/hmg/8.13.2533. [DOI] [PubMed] [Google Scholar]
- 25.Arnesano F., Balatri E., Banci L., Bertini I., Winge D.R. Folding studies of Cox17 reveal an important interplay of cysteine oxidation and copper binding. Structure. 2005;13:713–722. doi: 10.1016/j.str.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Longen S., Bien M., Bihlmaier K., Kloeppel C., Kauff F., Hammermeister M., Westermann B., Herrmann J.M., Riemer J. Systematic analysis of the twin cx(9)c protein family. J. Mol. Biol. 2009;393:356–368. doi: 10.1016/j.jmb.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 27.Woolfson D.N., Williams D.H. The influence of proline residues on α-helical structure. FEBS Lett. 1990;277:185–188. doi: 10.1016/0014-5793(90)80839-b. [DOI] [PubMed] [Google Scholar]
- 28.Richardson J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 29.Chacinska A., Pfannschmidt S., Wiedemann N., Kozjak V., Sanjuán Szklarz L.K., Schulze-Specking A., Truscott K.N., Guiard B., Meisinger C., Pfanner N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004;23:3735–3746. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofmann S., Rothbauer U., Mühlenbein N., Baiker K., Hell K., Bauer M.F. Functional and mutational characterization of human MIA40 acting during import into the mitochondrial intermembrane space. J. Mol. Biol. 2005;353:517–528. doi: 10.1016/j.jmb.2005.08.064. [DOI] [PubMed] [Google Scholar]
- 31.Herrmann J.M., Köhl R. Catch me if you can! Oxidative protein trapping in the intermembrane space of mitochondria. J. Cell Biol. 2007;176:559–563. doi: 10.1083/jcb.200611060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calvaruso M.A., Smeitink J., Nijtmans L. Electrophoresis techniques to investigate defects in oxidative phosphorylation. Methods. 2008;46:281–287. doi: 10.1016/j.ymeth.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 33.Nijtmans L.G., Henderson N.S., Holt I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- 34.Miller A.D., Miller D.G., Garcia J.V., Lynch C.M. Use of retroviral vectors for gene transfer and expression. Methods Enzymol. 1993;217:581–599. doi: 10.1016/0076-6879(93)17090-r. [DOI] [PubMed] [Google Scholar]
- 35.Cole C., Barber J.D., Barton G.J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36(Web Server issue) doi: 10.1093/nar/gkn238. W197-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.